
Abstract
Reliable biomarkers that can be used for early diagnosis and tracking disease progression are the cornerstone of the development of disease-modifying treatments for Parkinson’s disease (PD). The German Society of Experimental and Clinical Neurotherapeutics (GESENT) has convened a Working Group to review the current status of proposed biomarkers of neurodegeneration according to the following criteria and to develop a consensus statement on biomarker candidates for evaluation of disease-modifying therapeutics in PD. The criteria proposed are that the biomarker should be linked to fundamental features of PD neuropathology and mechanisms underlying neurodegeneration in PD, should be correlated to disease progression assessed by clinical rating scales, should monitor the actual disease status, should be pre-clinically validated, and confirmed by at least two independent studies conducted by qualified investigators with the results published in peer-reviewed journals. To date, available data have not yet revealed one reliable biomarker to detect early neurodegeneration in PD and to detect and monitor effects of drug candidates on the disease process, but some promising biomarker candidates, such as antibodies against neuromelanin, pathological forms of α-synuclein, DJ-1, and patterns of gene expression, metabolomic and protein profiling exist. Almost all of the biomarker candidates were not investigated in relation to effects of treatment, validated in experimental models of PD and confirmed in independent studies.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction and aims of the present review
A biomarker (or biological marker) is defined as a characteristic that can be objectively measured and evaluated as an indicator of normal biological processes, pathogenic processes or pharmacologic responses to a therapeutic intervention (Biomarkers Definitions Working Group 2001). According to the type of information they provide, biomarkers for central nervous system (CNS) diseases can be classified as clinical, neuroimaging, biochemical, genetic or proteomic biomarkers. Biomarkers serve a wide range of purposes, including confirmation of diagnosis, epidemiological screening, predictive testing, monitoring of disease progression after diagnosis, drug development and response to treatment, and studies of brain–behaviour relationship.
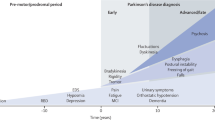
There is a growing need for biomarkers of Parkinson’s disease (PD, synonyms: idiopathic Parkinson syndrome, paralysis agitans) pathology to improve drug development related to the disease (Eller and Williams 2009; Gerlach et al. 2008; Halperin et al. 2009; Maetzler et al. 2009a; Marek et al. 2008; Michell et al. 2004; Morgan et al. 2010). Current therapeutic strategies for PD focus primarily on reducing the severity of its symptoms using dopaminergic medications. Although these strategies significantly improve motor symptoms and the quality of life for patients suffering from this neurodegenerative disease, treatment does not slow or halt the underlying pathologic processes. The goal of finding such a therapy (i.e., a neuroprotective or disease-modifying therapy) or one that could reverse pathologic damage (i.e., a neurorestorative therapy) is a major drive for preclinical research in PD. Despite 25 years of work dedicated to this goal, success has remained elusive. Several promising candidates for a disease-modifying therapy have failed in human studies, although they showed neuroprotective effects in experimental models of PD. Problems with establishing a disease-modifying therapy arise from the complexity of the disease process as well as the limitations of clinical tools available to monitor the progression of the disease and to observe the effects of an intervention. Major issues of the complexity of the disease, which become frequently evident in clinical studies are the long duration and slow progressive course of the disease, the variability and heterogeneity of symptoms and signs, cyclic episodes in severity of the symptoms during the day related to the time of medication (wearing-off and on/off fluctuations) and polypharmacy. In addition, misdiagnosis, co-morbidity and co-medication add to the heterogeneity of the patient population.
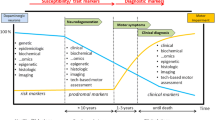
Since a disease-modifying therapy is likely to be most effective early in the course of disease, early diagnosis is highly desirable before neurodegeneration becomes severe and widespread. Thus, there is a great need for biomarkers that can be used for early diagnosis and tracking disease progression to monitor a disease-modifying therapy.
The German Society of Experimental and Clinical Neurotherapeutics (GESENT) has convened a Working Group to develop a position paper and, if possible, a consensus statement on biomarker candidates of neurodegeneration in PD for evaluation of disease-modifying therapeutics. In June 2010, the Working Group met to define the criteria for evaluation biomarkers of neurodegeneration in PD, to review the current status of all proposed biomarkers of neurodegeneration according to the defined criteria and to develop this consensus statement. This paper is planned as a basis for further discussion to finally reach the goal of a comprehensive evaluation of biomarkers for progression in PD.
Criteria for the development of biomarkers of neurodegeneration in PD for proof of disease-modifying therapeutics
Driven in part by Alzheimer’s disease (AD) drug discovery research, AD is at the forefront of biomarker development for CNS diseases, and many current concepts about ideal biomarkers for PD have come from AD research (Frank et al. 2003; Hampel et al. 2004, 2010; Shaw et al. 2007; The Ronald and Nancy Reagan Research Institute of the Alzheimer’s Association and the National Institute on Aging Working Group 1998). We propose the following criteria for an ideal biomarker to be useful to assess neurodegeneration in PD and to evaluate disease-modifying therapeutics: The biomarker should be
-
linked to fundamental features of PD neuropathology and mechanisms underlying neurodegeneration in PD,
-
correlated to disease progression assessed by clinical rating scales,
-
able to monitor the actual disease status,
-
pre-clinically validated,
-
confirmed by at least two independent studies conducted by qualified investigators with the results published in peer-reviewed journals.
In addition, an ideal biomarker of neurodegeneration should be inexpensive, non-invasive, simple to use, and technically validated (e.g., reliable, sensitive to change).
Clinical biomarker candidates
Symptoms associated with motor function
The most widely used scale currently available for the clinical evaluation of motor dysfunction in PD is the Unified Parkinson’s Disease Rating Scale III (UPDRS-III) (Goetz et al. 2007). This scale is subjective, has suboptimal sensitivity, and it is widely accepted that more objective and shorter assessments are needed. A promising tool is the timed motor test, and, in particular, the pegboard test (Haaxma et al. 2008). At group level, a trial using “change from baseline” as endpoint and applying these tests would require only 57–75% of the patients needed with the UPDRS-III (Haaxma et al. 2008).
In addition, there is a relevant and growing body of literature which reports about objective, quantitative and mobile assessments of movement disturbances in PD using technical devices, such as accelerometers and gyroscopes. A definite advantage of such methods is the possibility to focus on cardinal motor disturbances, i.e. bradykinesia, rigidity, tremor and postural instability, but also on problems of sensorimotor integration which is also a key symptom associated with PD. So far, studies carried out have mainly focused on the usefulness of the parameters in differentiating PD from controls, but not on correlation aspects, e.g. with disease duration. In addition, most of them have not been put into context to clinical scales and are thus not validated with regard to measuring disease progression. Nevertheless, as these methods are generally easy to perform (e.g. in an ambulatory setting), cheap, unobtrusive, focus on mechanisms underlying the neurodegeneration in PD (e.g. cardinal motor symptoms), and based on a well-investigated pathophysiological background (many of the investigated symptoms have been—directly of indirectly—validated in pre-clinical models in an extensive way), they should be seriously considered when defining e.g. an assessment panel for future progression studies in PD.
Mobile quantitative assessment of bradykinesia
According to the definition of Berardelli et al. (2001), “bradykinesia” encompasses problems of slowness or absence of movement (including increased gait variability and freezing): Promising quantitative markers are sit-to-stand and stand-to-sit procedures (Bloem et al. 1997; Hausdorff 2008; Najafi et al. 2002; Weiss et al. 2010), anticipatory postural adjustment (i.e. the attempt to voluntarily initiate the first step to begin walking) (Carpinella et al. 2007; Mancini et al. 2009), gait variability (Plotnik et al. 2007, 2009), and peak arm swing velocity (Zampieri et al. 2010).
Mobile quantitative assessment of rigidity
Rigidity is defined as an increase in muscle tone leading to a resistance to passive movement throughout the range of motion. Promising quantitative markers are turning procedures when walking or receiving rotational perturbations (Carpenter et al. 2004; Carpinella et al. 2007; Huxham et al. 2008; Visser et al. 2007; Zampieri et al. 2010) and straight walking (pelvic oscillations) (Huxham et al. 2008).
Mobile quantitative assessment of tremor
Although tremor is an obvious sign of PD, and clinically easily to diagnose, the quantification of this symptom remains a technical challenge. In a study with PD patients using electromyography, tremor amplitude and burst duration increased, whereas frequency decreased with longer disease duration (Milanov 2002). The first results with acceptable accuracy in detecting the severity of resting tremor using tri-axial accelerometers have been published (Mamorita et al. 2009; Rigas et al. 2009; Schlesinger et al. 2009).
Mobile quantitative assessment of postural instability
Accurate assessment of postural instability in PD remains difficult with currently available clinical measurement tools, but may be quantifiable with ambulatory devices which focus on anterior–posterior and medial–lateral angular velocity deviations, e.g. at the trunk (Adkin et al. 2005). In addition, prospective detection of frequency of near-falls and falls may be a promising approach to detect PD progression.
Mobile quantitative assessment of sensorimotor integration deficits
There is an increasing awareness of sensorimotor integration deficits in PD patients, and it is highly probable that this feature also declines with increasing disease duration. Promising markers may be the switch from kinaesthetic-dependent to vision-dependent balance control (De Nunzio et al. 2007), and overestimation of (balance) limits (Kamanli et al. 2008).
Biomarker candidates of cognition and neuropsychiatric symptoms
Cognitive symptoms
There is an increasing awareness of the high prevalence of cognitive dysfunction in the course of PD. Independent studies found a higher incidence of dementia in PD patients as compared to healthy persons of the same age (Aarsland et al. 2001; de Lau et al. 2004). There is compelling evidence that dementia prevalence increases with disease duration (Maetzler et al. 2009a). Deterioration of cognitive decline was most often assessed with the MMSE and the cognitive section of the Cambridge Examination for Mental Disorders (CAMCOG), two assessment tools validated for AD (Aarsland et al. 2004; Athey and Walker 2006). As cognitive symptoms in PD clearly differ from AD symptoms, it was more and more realised that AD-related assessment tools have relevant flaws in determining cognitive dysfunction in PD, and effort has been put into the development of sensitive and reliable PD-relevant measurement tools. One of the most promising tools is the “Scales for outcomes in Parkinson’s disease-cognition (SCOPA-COG)” having advantages as compared to the MMSE, the most important having a greater discriminative capacity (Serrano-Duenas et al. 2010); however, longitudinal studies are not yet available.
Hallucinations and depression
Based on the retrospective pathologically confirmed (Williams and Lees 2005) and prospective studies (Forsaa et al. 2008; Goetz et al. 2005) visual hallucinations are regularly observed in PD patients in particular at later disease stages, and frequency increases with longer disease duration.
Depression has been shown to occur with higher incidence in PD as compared to the general population, but incidence and severity of symptoms may not relevantly change during disease course (Karlsen et al. 1999; Rojo et al. 2003; Schrag et al. 2007).
Sleep disturbances
Among a number of sleep disturbances which are associated with PD rapid eye movement (REM) sleep behaviour disorder (RBD) may be the most promising biomarker candidate for detecting disease progression. A prospective longitudinal study investigating patients with questionnaire and polysomnography found an increase of RBD-associated features from baseline (6–11% after 3, 24% after 6, and 39 percent after 8 years) (Onofrj et al. 2002). This increase in occurrence could be confirmed in an evidence level II study with mid- to late-stage PD patients using a semi-structured interview and a sleep questionnaire (Gjerstad et al. 2007).
Biomarker candidates of autonomic and sensory dysfunction
Based on a controlled prospective study of 3 years duration (Mesec et al. 1999) and a cross-sectional study (Linden et al. 1997) heart rate variability decreases, and orthostatic dysfunction probably increases with longer disease duration. A reduction in sympathetic skin response with increasing disease duration has been demonstrated in two cross-sectional studies (Orimo et al. 1999; Schestatsky et al. 2006). Prevalence and severity of urinary and gastrointestinal symptoms most probably also increase during PD course (Wullner et al. 2007); however, it may be difficult to quantify these changes adequately.
There is compelling evidence from prospective longitudinal studies (Diederich et al. 2002; Katsarou et al. 1998) that visuospatial and colour discrimination deteriorate with longer PD duration. These symptoms may be influenced by medication status (Onofrj et al. 2002). There is no evidence that olfactory dysfunction progresses significantly during PD course (Maetzler et al. 2009a).
Myocardial [123I]metaiodobenzylguanidine (MIBG) scintigraphy and [18F]fluorodopamine positron emission tomography (PET) are used to detect local sympathetic nerve damage in the heart, which regularly occurs in PD, but rarely in healthy older people and in other forms of parkinsonism. However, it is unlikely that cardiac sympathetic innervation decreases with PD duration in the clinical phase (Orimo et al. 1999; Shibata et al. 2009; Suzuki et al. 2007).
Biomarker candidates of brain imaging
Presynaptic imaging of dopaminergic neurons is part of clinical diagnostics of PD and appears to be a useful progression marker (Table 1). Disadvantages of this approach are that the subjects are exposed to radioactivity, that the costs are relatively high, and that the method is only available at specialised centres. The imaging of the dopaminergic system is possible by measuring aromatic amino acid decarboxylase activity (e.g. with [18F]-DOPA) or by visualisation of synaptic membrane dopamine transporter (e.g. [123I]β-CIT, [123I]FP-CIT, [123I]IPT, [18F]CFT). In longitudinal studies of PD progression, PET and single photon emission computed tomography (SPECT) studies using these tracers have shown an annualised striatal rate of reduction in tracer uptake of about 4–13% in PD patients versus 0–2.5% change in healthy controls (Marek et al. 2008; Nurmi et al. 2001). This decline may rather be exponentially (Hilker et al. 2005). With regard to using these functional imaging techniques for measuring disease progression, it needs to be considered that correlations of changes in imaging and clinical findings are rather inconsistent, probably, because different aspects of the disease are reflected (Marek et al. 2008).
[18F]-2-F-Deoxyglucose-PET may have some potential in detecting metabolic changes associated with motor (Huang et al. 2007) and cognitive decline (Huang et al. 2007; Liepelt et al. 2009), but these preliminary data should be confirmed in prospective longitudinal studies.
There is only limited evidence that magnetic resonance imaging is of added value in detecting disease progression in PD: Two cross-sectional studies with advanced PD patients showed a positive correlation between T2 relaxation time in the putamen and disease duration which indicates a progressive loss of iron (Graham et al. 2000; Ryvlin et al. 1995). However, a recent study in PD patients and controls measuring quantitative magnetic resonance parameters sensitive to complementary tissue characteristics (i.e. volume atrophy, iron deposition and microstructural damage) in six subcortical structures including the SN and the putamen showed no relation of the relaxation rates such as R 2* as an indirect measure of the iron level to disease progression (Peran et al. 2010).
Genetic and biochemical biomarker candidates
Genetic markers
PD-associated DNA variants (including mutations and polymorphisms) are by definition predictive markers and are not suitable for measuring progression. However, gene expression profiling may be a promising approach for defining valuable progression markers as human SN pars compacta of PD patients showed down-regulation of 68, and up-regulation of 69 genes, as compared to control SN (Grunblatt et al. 2004). Based on the recent publications particularly interesting targets are pyridoxal kinase and pyruvate metabolism (Ahmed et al. 2009; Elstner et al. 2009).
A recent study (Grunblatt et al. 2010) examined the profiling of 12 transcripts via quantitative RT-PCR in RNA originating from peripheral blood samples that were chosen based on the previous postmortem brain profiling (Grunblatt et al. 2004). Multiple analyses resulted in four significant genes: proteasome (prosome, macropain) subunit-α type-2 (PSMA2), laminin, β-2 (laminin S) (LAMB2), aldehyde dehydrogenase 1 family-member A1 (ALDH1A1), and histone cluster-1 H3e (HIST1H3E) differentiating between medicated PD subjects versus controls. Using the combination of these four gene profiles for PD diagnosis, a sensitivity and specificity of more than 80% was achieved. In AD subjects, no significant results were observed. Therefore, the authors concluded that this combination is specific for PD.
A transcriptome-wide scan using RNA microarrays in whole blood of patients with early-stage PD (Scherzer et al. 2007) identified a molecular multigene marker that is associated with risk of PD in 66 samples of the training set comprising healthy and disease controls. This was further validated in 39 independent test samples. Insights into disease-linked processes detectable in peripheral blood are offered by 22 unique genes differentially expressed in patients with PD versus healthy individuals (Scherzer et al. 2007). These include the co-chaperone ST13, which stabilises heat-shock protein 70, a modifier of α-synuclein misfolding and toxicity. ST13 messenger RNA copies are lower in patients with PD than in controls in two independent populations.
Biochemical markers
A summary of the most thoroughly investigated biochemical biomarker candidates that may be used in the diagnostics of PD was published previously (Fasano et al. 2008; Halperin et al. 2009; Morgan et al. 2010; Nyhlen et al. 2010). Here we focus on biomarker candidates with a particular reference to their potential for monitoring neurodegeneration and disease-modifying therapeutics.
PD is neuropathologically characterised at the cellular level by a relative selective destruction of neuromelanin (NM)-containing dopaminergic cells in the SN pars compacta (Hirsch et al. 1988). When melaninised dopaminergic neurons die, NM is released from the degenerating cell body and removed from the brain by the cells of the immune system (Beach et al. 2007; Depboylu et al. 2011; Orr et al. 2005). It was hypothesised that the removal of NM from the brain by immune cells might stimulate an antibody response that could be measured in blood. Indeed, a novel enzyme-linked immunosorbent assay (ELISA) to measure levels of antibodies against NM in human blood sera (NM-ELISA) demonstrated an increased antibody response in the sera of PD patients when compared with age-matched controls (Double et al. 2009). The immune response was not affected by disease severity assessed by Hoehn and Yahr staging and the UPDRS. However, there was a negative correlation with disease duration.
α-Synuclein is the major component of Lewy bodies, one of the pathological hallmarks of PD, and mutations and multiplications of the α-synuclein-encoding gene, SNCA, have been found to cause familial forms of PD. Aberrant metabolism of the protein has been suggested as a possible driving force in the degenerative process of PD in a manner similar to Aβ1–42 in AD. Cerebrospinal fluid (CSF) Aβ1–42 levels in PD tend to be lower with longer disease duration and cognitive decline (Maetzler et al. 2009b; Mollenhauer et al. 2006). A recent study demonstrated that the CSF fractalkine (an inflammatory marker)/Aβ1–42 ratio was positively correlated with PD severity in cross-sectional samples as well a with PD progression in longitudinal samples (Shi et al. 2011).
Increased concentrations of soluble α-synuclein oligomers in plasma appear to have good specificity (85%) for detecting PD when compared with controls in some studies (El-Agnaf et al. 2006). The most consisting finding is decreased α-synuclein concentrations in the CSF from PD when compared with controls (see for a review, Morgan et al. 2010; Nyhlen et al. 2010), but there is still no convincing evidence that these levels change over disease course (Hong et al. 2010). There is increasing evidence that α-synuclein can be used to distinguish PD and related synucleinopathies (dementia with Lewy bodies and multiple system atrophy) from other movement disorders and dementia (Mollenhauer et al. 2011); however, discriminatory power is limited. In addition, the current α-synuclein assays are limited because they do no not attempt to discriminate between normal and pathological (phosphorylated and/or aggregated) forms of this protein.
The major hypotheses believed to contribute to the eventual demise of nigral dopamine producing cells include protein aggregation, oxidative stress, mitochondrial dysfunction, dysfunction of proteasomal pathways and neuroinflammation (Alvarez-Erviti et al. 2010; Chu et al. 2009; Double et al. 2010; Gerlach et al. 2006; Hatano et al. 2009; Schiesling et al. 2008; Yang et al. 2009). Complex I and IV mitochondrial activity has been shown to be lower in PD patients than in controls, and at very early disease stages a negative correlation between complex I and IV activity in platelet mitochondria, and disease duration has been demonstrated (Benecke et al. 1993).
Markers of oxidative stress showed that in the blood of PD patients there is either an increased production of free radicals, reactive oxygen and nitrogen species or a disturbed defence mechanisms against oxidative damage (Gerlach et al. 2008; Morgan et al. 2010; Younes-Mheni et al. 2007). However, these markers are not specific for PD because similar results in other neurodegenerative diseases, including AD were found. In addition, a variety of conditions alter oxidative stress in a given patient (for example normal ageing, smoking, vigorous exercise, antioxidants, food, drugs, cancer, and chemotherapy), and these may be hard to control for. Interestingly, some markers of oxidative stress appear to be useful for tracking disease progression in PD. For example, concentrations of 8-hydroxydeoxyguanosine, a product of oxidised DNA, were shown to be stage-dependently increased in the urine of PD patients (Sato et al. 2005). Surprisingly, this increase was not influenced by l-DOPA (3,4-dihydroxyphenylalanine, levodopa) therapy. Multiple large epidemiological studies have demonstrated a reduced risk of developing PD with higher concentrations of uric acid (a potent antioxidant and free radical scavenger in the blood) in serum (Schlesinger and Schlesinger 2008), but recent evidence also indicates a potential for slower progression of PD with higher uric acid levels (Schwarzschild et al. 2008). Clinical use for uric acid as a biomarker is not supported by existing knowledge, since the studies conclude that it is a risk marker rather than a diagnostic marker. In addition, to date there are no data available which make hope that uric acid is a potential progression marker.
DJ-1 is a part of the cellular defence against oxidative stress (Kahle et al. 2009), and mutations in its gene are responsible for some forms of familial PD (Klein et al. 2009). A study has also found elevated DJ-1 levels in CSF from patients with multiple sclerosis (Hirotani et al. 2008), suggesting a link between secreted DJ-1, neuroinflammation and oxidative stress. Studies using CSF of PD patients have demonstrated both increased (Waragai et al. 2006) and decreased values compared with controls (Hong et al. 2010), thus warranting further investigations. In the study by Hong et al. (2010), no association between DJ-1 and the severity of PD was demonstrated. The results obtained from studies using serum of PD patients are also inconsistent, showing no change (Maita et al. 2008) or elevated concentrations compared with controls (Waragai et al. 2006).
The complement system is part of the non-specific immune system. Using 2D-gel-electrophoresis, Goldknopf et al. (2006) found differences in serum levels of nine complement factors between PD and controls. Osteopontin is a molecule with multiple functions, including modulation of inflammatory response of microglia, and shows much higher levels in CSF than in serum. Higher CSF and serum levels are detectable in PD as compared to controls, and there is some evidence that CSF osteopontin levels increase with disease duration (Maetzler et al. 2007).
Several studies suggest that elevated plasma total homocysteine, an endogenous product of methionine metabolism, is a risk factor for cognitive impairment and AD (Clarke et al. 1998; McCaddon et al. 2003). However, other studies did not detect significant associations with AD or cognitive status (Miller et al. 2002). In agreement with these studies, it was recently reported that plasma total homocysteine concentrations did not differ across AD, mild cognitive impairment, cerebral amyloid angiopathy, and non-demented control subjects, but were increased in the PD group (Irizarry et al. 2005). The elevated levels within the PD group were the result of high concentrations of plasma total homocysteine in PD patients treated with l-DOPA. Two cross-sectional studies found also increased homocysteine plasma levels in PD compared with controls, these levels correlated positively with disease duration (Dos Santos et al. 2009; Hassin-Baer et al. 2006). In one study, in addition, homocysteine levels were associated with l-DOPA treatment duration, but not with l-DOPA dose (Hassin-Baer et al. 2006). In the other study (Dos Santos et al. 2009), l-DOPA treatment did not significantly correlate with plasma homocysteine levels.
There are some interesting first results which may potentially reflect very early disease activity. Serum insulin-like growth factor 1 (IGF-1) levels have been shown to be higher in PD patients compared with controls, with high levels in particular at early PD disease stages, and a negative correlation between serum IGF-1 levels and disease duration (Godau et al. 2010). In addition, using rapidly processed CSF samples, we recently found lowered levels of reduced glutathione in the CSF of Lewy body disease patients as compared to controls (Maetzler et al. 2011)—which basically confirms neuropathological findings in the brainstem of PD patients (Sian et al. 1994)—and these levels were negatively associated with age but not with disease-associated parameters. Thus, it is tempting to speculate that changes of the glutathione system, similar to IGF-1, may be an early event in the disease course.
Several recent studies have used hypothesis-unrelated, explorative proteomic and metabolomic techniques to find novel biomarker candidates for PD in brain tissue and CSF (see for a review Fasano et al. 2008; Morgan et al. 2010; Nyhlen et al. 2010). Generally, these studies may be considered promising. However, these techniques require considerable technical expertise and have not been well tested for PD versus other neurodegenerative diseases and the link to disease progression. In addition, the biomarker candidates found in these studies need to be validated in a greater and statistically significant universe of individual samples employing distinct methodologies, such as Western blot, ELISA or single and multiple reaction monitoring (Martins-de-Souza 2010).
A study using a multiplex quantitative proteomics method for detecting biomarkers in the CSF of patients with neurodegenerative diseases, including AD, dementia with Lewy body and PD, suggests as potential candidates for the clinical diagnosis of PD and monitoring disease progression chromogranins, amyloid precursor protein-like protein 1 and the prion protein (Abdi et al. 2006), but more studies are needed to confirm or refute the findings and to assess the specificity of the protein profiles against other neurodegenerative diseases (Zetterberg et al. 2008). Recent research has identified an eight-protein CSF multi-analyte profile using proteomics that fully differentiate PD patients from controls, with the profile designation agreeing with an expert clinical diagnosis of PD 95% of the time (Zhang et al. 2008).
The idea that the whole metabolism, regulated by genes, exogenous substances and proteins, might be affected in diseased patients, and that these affected molecules could together, form a distinct profile, underlies metabolomics (Kaddurah-Daouk and Krishnan 2005). This approach has been tested in PD and some results appear promising, such as the confirmatory finding of reduced concentrations of uric acid in plasma of idiopathic PD patients and PD patients with LRRK2 mutations (Johansen et al. 2009). However, both idiopathic and LRRK2 PD subjects involved in this study were taking anti-parkinsonian medications, and no samples from the un-medicated patients were available. Therefore, it is possible that the observed separation could be related to drug effects, which could involve unknown drug metabolites and drug-induced changes in metabolism.
Bogdanov et al. (2008) were able to accurately categorise 25 controls and 66 un-medicated PD patients based only on their metabolic profiles in blood, obtaining complete separation between the two groups. Interestingly, concentrations of 8-hydroxydeoxyguanosine, a marker of oxidative DNA damage, were significantly increased in PD patients (confirming results obtained in urine), but overlapped controls. In addition, concentrations of two other markers of oxidative stress, uric acid and glutathione were significantly reduced and significantly increased in PD, respectively.
Review of some putative biomarkers for the use in clinical trials of disease-modifying therapeutics
Table 1 summarises the qualification of biomarker candidates for the use in studies to proof disease-modifying therapeutics by considering the criteria for the development of biomarkers of neurodegeneration in PD as defined above. Qualification is used to mean the establishment of the credibility of a biomarker assay in its application to questions relevant to drug treatment (Hampel et al. 2010). Validation is usually applied to mean the determination of the performance characteristics of an assay such as for example sensitivity and specificity in measuring a specific analyte. Qualification requires specific patient populations and a specific therapeutic intervention. For example, a validated assay may be qualified as a PD biomarker to detect and monitor effects of drug candidates on the disease process by intervention in the α-synuclein aggregation, but not in non-α-synuclein mechanisms. It could be said therefore that the assay which was validated for quantification of α-synuclein fibrillisation in the brain or CSF is “qualified for use” as a biomarker in α-synucleinopathies such PD and Lewy body dementia for drugs that inhibit the aggregation of α-synuclein. The ultimate use of a biomarker is a surrogate end point, which requires that a biomarker has been qualified to substitute for a well-established clinical endpoint such that the biomarker reasonably predicts the clinical outcome and therefore can serve as a surrogate (Hampel et al. 2010).
The first criterion means that the biomarker is linked to the neuropathology of PD and/or mirror fundamental pathogenetic events in PD. The validity of a biomarker with respect to a supposed pathogenetic mechanism will be relevant for the evaluation of disease-modifying treatments. Pathologically, PD is characterised by a preferential loss of NM-containing dopamine neurons in the pars compacta of the SN, with intracellular proteinaceous inclusions named Lewy bodies in the SN and other brain regions, and a reduction in striatal dopamine (Bernheimer et al. 1973; Braak et al. 1995; Jellinger 1991). This ongoing loss of nigral dopaminergic neurons mainly leads to clinical diagnosis due to occurrence of motor symptoms such as rigidity, tremor and bradykinesia, which results from a reduction of about 70% of striatal dopamine (Bernheimer et al. 1973; Riederer and Wuketich 1976).
To mirror a pathological feature it would be helpful to know the cause of the disease. However, despite numerous attempts, the cause of PD remains unclear. It is hypothesised that the cause of neurodegeneration in PD is multi-factorial in terms of both aetiology and pathogenesis. Genetic factors are known to cause PD in small numbers of patients with a familial form of the disorder. Mutations in different genes (for example, SNCA-synuclein, LRRK2, parkin, DJ1 and PINK1) have been identified, and PD subtypes have been linked in addition to different chromosomal loci (for example, Hatano et al. 2009; Schiesling et al. 2008; Yang et al. 2009).These Mendelian forms of PD are relatively rare. However, high-throughput genotyping and sequencing technologies have more recently provided evidence that low-penetrance variants in some of these and other genes may also contribute significantly to the aetiology of the common sporadic disease. Moreover, rare variants in further genes, such as the glucocerebrosidase A gene associated with Gaucher’s disease, have been found to be important risk factors in a subgroup of patients (Gasser 2010). Therefore, an increasingly complex interplay of different genes seems to contribute in distinct ways to disease risk and progression. Hence, current thinking favours the hypothesis that most sporadic cases are caused by a complex interplay between different genetic and environmental factors. This interplay may result in alterations of biochemical cascades. Altered biochemical pathways involved in the pathogenetic cascade of events leading to cell dysfunction and neuronal cell death in PD result among others in measurable mitochondrial complex I deficiency, a disturbed iron metabolism, free radicals, excitotoxicity, disturbed calcium homeostasis, microglia activation and protein aggregation (Alvarez-Erviti et al. 2010; Chu et al. 2009; Double et al. 2010; Gerlach et al. 2006; Hatano et al. 2009; Schiesling et al. 2008; Yang et al. 2009).
The second criterion for the development of a biomarker of neurodegeneration and to detect and monitor effects of drug candidates on the disease process is that the biomarker must track disease progression. We defined a progression marker as a disease-associated feature that changes in the frequency of occurrence or severity, or both, over time. The definitions used in our evaluation of progression of features have been published previously (Maetzler et al. 2009a). For the qualification of a biomarker as a surrogate endpoint, there should be a link between a treatment-induced change in the biomarker and the desired clinical outcome measure, as well as a link between the treatment-induced change in the biomarker and change of disease process (Hampel et al. 2010).
Although imaging techniques measuring the presynaptic nigrostriatal system with, for example [18F]-DOPA or [123I]β-CIT, can readily distinguish subjects with early PD from controls, and abnormalities can be observed even before motor symptoms and signs are apparent, these studies have been failed in monitoring both disease-modifying (Ponsen et al. 2009) and neurorestorative therapies (Freed et al. 2001; Whone et al. 2003). This was discussed to be due, in part, to potential pharmacological modulation or regulation of presynaptic proteins that may not relate to the actual disease status. Therefore, we postulated as a third criterion that the biomarker is monitoring the disease status. This would require that the drug aimed to proof a disease-modifying effect does not pharmacologically influence the biomarker. To date, trials to proof disease-modifying drugs have largely evaluated subjects in the early clinical stages of the disease (generally untreated), using clinical endpoints that involve either the change in a classical clinical measure of the disease over time or progression to the point of reaching a disease milestone (for example, need for a dopaminergic therapy). The greatest concern in these studies has been the potential for the study intervention to cause symptomatic benefit that precludes the determination of disease-modifying effects. Approaches developed to overcome this problem, such as the washout and delayed-start design, however, have either failed to adequately overcome this problem or is not without its own potential problems (Lang, 2010). Therefore, clinical biomarkers that are changed by symptomatic drug therapy cannot be considered sufficient surrogate biomarkers for the evaluation of disease-modifying therapeutics.
The fourth criterion for the development of a biomarker of neurodegeneration for proof of disease-modifying therapeutics in PD is that the biomarker candidate is validated in experimental models of PD. This means that in an in vivo model of PD there should be a correlation between the degree of neurodegeneration and the change of the biomarker candidate. In addition, based on the assumed mechanism of action of a given compound, the proposed mechanism underlying neurodegeneration should be modified. This validation will be relevant for predicting the response of PD patients to putative disease-modifying therapies.
The fifth criterion, validation of the biomarker in independent studies is essential but also trivial. Only a progression marker that mirrors progression independent from the investigator can be used as a biomarker for disease-modifying therapeutics.
Conclusions and future perspectives
Reliable biomarkers that can be used for early diagnosis and tracking disease progression are the cornerstone of the development of disease-modifying treatments for PD. To date, available data have not yet revealed one reliable biomarker to detect early neurodegeneration in PD and to detect and monitor effects of drug candidates on the disease process, but some promising biomarker candidates, such as antibodies against NM, pathological forms of α-synuclein, DJ-1, and patterns of gene expression, metabolomic and protein profiling exist (Table 1). Most of the reported disease-associated changes are relatively small, with a clinically problematic overlap between patients and controls. Almost all of the biomarker candidates were not investigated in relation to effects of treatment, validated in experimental models of PD and confirmed in independent studies.
To solve some of the problems associated with the development of biomarkers that can be used for early diagnosis and tracking disease progression, the Parkinson’s Progression Markers Initiative (PPMI) was founded (http://www.ppmi-info.org). This public–private partnership leaded by the Michael J. Fox Foundation aims to identify clinical, imaging, and biological markers of disease progression. The emphasis will initially be on fluid markers including α-synuclein, DJ-1, amyloid β, and tau in CSF and urate in blood. The initiative will enrol 400 newly diagnosed patients who are not yet on medication and who have evidence of dopamine transporter loss on dopamine transporter imaging with SPECT and 200 healthy age-matched controls. As with the AD Neuroimaging Initiative (ADNI), a crucial aspect of the PPMI is that all data and biological specimens, stored in a central repository, will be available for the research community. The PPMI will thus provide a valuable resource to fuel further academic and industry-initiated studies and innovations, and promising biomarker candidates identified through such efforts could be validated and qualified against the large, prospective PPMI dataset.
However, to fulfil the promise of the PPMI for delivering objective biomarkers that can be used for early diagnosis and tracking disease progression, specimen collection, processing, and storage methods have to be standardised. Further, quality-control mechanisms should be in place to ensure data are acceptable before they are made publicly available. Finally, well-defined quantitative biomarker outcomes that are consistent among many research sites and laboratories should be established.
References
Aarsland D, Andersen K, Larsen JP, Lolk A, Nielsen H, Kragh-Sorensen P (2001) Risk of dementia in Parkinson’s disease: a community-based, prospective study. Neurology 56:730–736
Aarsland D, Andersen K, Larsen JP, Perry R, Wentzel-Larsen T, Lolk A, Kragh-Sorensen P (2004) The rate of cognitive decline in Parkinson disease. Arch Neurol 61:1906–1911
Abdi F, Quinn JF, Jankovic J, McIntosh M, Leverenz JB, Peskind E, Nixon R, Nutt J, Chung K, Zabetian C, Samii A, Lin M, Hattan S, Pan C, Wang Y, Jin J, Zhu D, Li GJ, Liu Y, Waichunas D, Montine TJ, Zhang J (2006) Detection of biomarkers with a multiplex quantitative proteomic platform in cerebrospinal fluid of patients with neurodegenerative disorders. J Alzheimers Dis 9:293–348
Adkin AL, Bloem BR, Allum JH (2005) Trunk sway measurements during stance and gait tasks in Parkinson’s disease. Gait Posture 22:240–249
Ahmed SS, Santosh W, Kumar S, Christlet HT (2009) Metabolic profiling of Parkinson’s disease: evidence of biomarker from gene expression analysis and rapid neural network detection. J Biomed Sci 16 (article no. 63) (doi:10.1186/1423-0127-16-63)
Alvarez-Erviti L, Rodriguez-Oroz MC, Cooper JM, Caballero C, Ferrer I, Obeso JA, Schapira AHV (2010) Chaperone-mediated autophagy markers in Parkinson disease brains. Arch Neurol 67:1464–1472
Athey RJ, Walker RW (2006) Demonstration of cognitive decline in Parkinson’s disease using the Cambridge Cognitive Assessment (Revised) (CAMCOG-R). Int J Geriatr Psychiatry 21:977–982
Beach TG, Sue LI, Walker DG, Lue LF, Connor DJ, Caviness JN, Sabbagh MN, Adler CH (2007) Marked microglial reaction in normal aging human substantia nigra: correlation with extraneuronal neuromelanin pigment deposits. Acta Neuropathol 114:419–424
Benecke R, Strumper P, Weiss H (1993) Electron transfer complexes I and IV of platelets are abnormal in Parkinson’s disease but normal in Parkinson-plus syndromes. Brain 116:1451–1463
Berardelli A, Rothwell JC, Thompson PD, Hallett M (2001) Pathophysiology of bradykinesia in Parkinson’s disease. Brain 124:2131–2146
Bernheimer H, Birkmayer W, Hornykiewicz O, Jellinger K, Seitelberger F (1973) Brain dopamine and the syndromes of Parkinson and Huntington: clinical, morphological and neurochemical correlations. J Neurol Sci 20:415–455
Biomarkers Definitions Working Group (2001) Biomarkers and surrogate endpoints: preferred definitions and conceptual framework. Clin Pharmacol Ther 69:89–95
Bloem BR, Roon KI, Delleman NJ, van Dijk JG, Roos RA (1997) Prolonged duration of standing up is an early dopa-sensitive abnormality in Parkinson’s disease. J Neurol Sci 146:41–44
Bogdanov M, Matson WR, Wang L, Matson T, Saunders-Pullman R, Bressman SS, Beal MF (2008) Metabolomic profiling to develop blood biomarkers for Parkinson’s disease. Brain 131:389–396
Braak H, Braak E, Yilmazer D, Schulz C, de Vos RAI, Jansen ENH (1995) Nigral and extranigral pathology in Parkinson’s disease. J Neural Transm (Suppl 46):15–31
Carpenter MG, Allum JH, Honegger F, Adkin AL, Bloem BR (2004) Postural abnormalities to multidirectional stance perturbations in Parkinson’s disease. J Neurol Neurosurg Psychiatry 75:1245–1254
Carpinella I, Crenna P, Calabrese E, Rabuffetti M, Mazzoleni P, Nemni R, Ferrarin M (2007) Locomotor function in the early stage of Parkinson’s disease. IEEE Trans Neural Syst Rehabil Eng 15:543–551
Chu YP, Dodiya H, Aebischer P, Olanow CW, Kordower JH (2009) Alterations in lysosomal and proteasomal markers in Parkinson’s disease: relationship to alpha-synuclein inclusions. Neurobiol Dis 35:385–398
Clarke R, Smith AD, Jobst KA, Refsum H, Sutton L, Ueland PM (1998) Folate, vitamin B-12, and serum total homocysteine levels in confirmed Alzheimer disease. Arch Neurol 55:1149–1455
de Lau LM, Giesbergen PC, de Rijk MC, Hofman A, Koudstaal PJ, Breteler MM (2004) Incidence of parkinsonism and Parkinson disease in a general population: the Rotterdam Study. Neurology 63:1240–1244
De Nunzio AM, Nardone A, Schieppati M (2007) The control of equilibrium in Parkinson’s disease patients: delayed adaptation of balancing strategy to shifts in sensory set during a dynamic task. Brain Res Bulletin 74:258–270
Depboylu C, Schafer MKH, Arias-Carrion O, Oertel WH, Weihe E, Hoglinger GU (2011) Possible involvement of complement factor C1q in the clearance of extracellular neuromelanin from the substantia nigra in Parkinson disease. J Neuropathol Exp Neurol 70:125–132
Diederich NJ, Raman R, Leurgans S, Goetz CG (2002) Progressive worsening of spatial and chromatic processing deficits in Parkinson disease. Arch Neurol 59:1249–1252
Dos Santos EF, Busanello EN, Miglioranza A, Zanatta A, Barchak AG, Vargas CR, Saute J, Rosa C, Carrion MJ, Camargo D, Dalbem A, da Costa JC, de Sousa Miguel SR, de Mello Rieder CR, Wajner M (2009) Evidence that folic acid deficiency is a major determinant of hyperhomocysteinemia in Parkinson’s disease. Metab Brain Dis 24:257–269
Double KL, Rowe DB, Carew-Jones FM, Hayes M, Chan DKY, Blackie J, Corbett A, Joffe R, Fung VS, Morris J, Riederer P, Gerlach M, Halliday GM (2009) Anti-melanin antibodies are increased in sera in Parkinson’s disease. Exp Neurol 217:297–301
Double KL, Reyes S, Werry EL, Halliday GM (2010) Selective cell death in neurodegeneration: why are some neurons spared in vulnerable regions? Prog Neurobiol 92:316–329
El-Agnaf OM, Salem SA, Paleologou KA, Curran MD, Gibson MJ, Court JA, Schlossmacher MG, Allsop D (2006) Detection of oligomeric forms of alpha-synuclein protein in human plasma as a potential biomarker for Parkinson’s disease. FASEB J 20:419–425
Eller M, Williams DR (2009) Biological fluid biomarkers in neurodegenerative parkinsonism. Nat Rev Neurol 5:561–570
Elstner M, Morris CM, Heim K, Lichtner P, Bender A, Mehta D, Schulte C, Sharma M, Hudson G, Goldwurm S, Giovanetti A, Zeviani M, Burn DJ, McKeith IG, Perry RH, Jaros E, Kruger R, Wichmann HE, Schreiber S, Campbell H, Wilson JF, Wright AF, Dunlop M, Pistis G, Toniolo D, Chinnery PF, Gasser T, Klopstock T, Meitinger T, Prokisch H, Turnbull DM (2009) Single-cell expression profiling of dopaminergic neurons combined with association analysis identifies pyridoxal kinase as Parkinson’s disease gene. Ann Neurol 66:792–798
Fasano M, Alberio T, Lopiano L (2008) Peripheral biomarkers of Parkinson’s disease as early reporters of central neurodegeneration. Biomarkers Med 2:465–478
Forsaa EB, Larsen JP, Wentzel-Larsen T, Herlofson K, Alves G (2008) Predictors and course of health-related quality of life in Parkinson’s disease. Mov Disord 23:1420–1427
Frank RA, Galasko D, Hampel H, Hardy J, de Leon MJ, Mehta PD, Rogers J, Siemers E, Trojanowski JQ (2003) Biological markers for therapeutic trials in Alzheimer’s disease. Proceedings of the biological markers working group; NIA initiative on neuroimaging in Alzheimer’s disease. Neurobiol Aging 24:521–536
Freed CR, Greene PE, Breeze RE, Tsai WY, DuMouchel W, Kao R, Dillon S, Winfield H, Culver S, Trojanowski JQ, Eidelberg D, Fahn S (2001) Transplantation of embryonic dopamine neurons for severe Parkinson’s disease. N Engl J Med 344:710–719
Gasser T (2010) Identifying PD-causing genes and genetic susceptibility factors: current approaches and future prospects. Prog Brain Res 183:2–20
Gerlach M, Double K, Götz ME, Youdim MBH, Riederer P (2006) The role of iron in the pathogenesis of Parkinson’s disease. In: Sigel A, Sigel H, Sigel RKO (eds) Neurodegenerative diseases and metal ions. Metal ions in life sciences, vol 1. Wiley, Chichester, pp 125–149
Gerlach M, Hendrich A, Hueber R, Jost W, Winkler J, Woitalla D, Riederer P (2008) The early detection of Parkinson’s disease: unmet needs. Neurodegener Dis 5:137–139
Gjerstad MD, Wentzel-Larsen T, Aarsland D, Larsen JP (2007) Insomnia in Parkinson’s disease: frequency and progression over time. J Neurol Neurosurg Psychiatry 78:476–479
Godau J, Herfurth M, Kattner B, Gasser T, Berg D (2010) Increased serum insulin-like growth factor 1 in early idiopathic Parkinson’s disease. J Neurol Neurosurg Psychiatry 81:536–538
Goetz CG, Wuu J, Curgian LM, Leurgans S (2005) Hallucinations and sleep disorders in PD: six-year prospective longitudinal study. Neurology 64:81–86
Goetz CG, Fahn S, Martinez-Martin P, Poewe W, Sampaio C, Stebbins GT, Stern MB, Tilley BC, Dodel R, Dubois B, Holloway R, Jankovic J, Kulisevsky J, Lang AE, Lees A, Leurgans S, LeWitt PA, Nyenhuis D, Olanow CW, Rascol O, Schrag A, Teresi JA, Van Hilten JJ, LaPelle N (2007) Movement Disorder Society-sponsored revision of the Unified Parkinson’s Disease Rating Scale (MDS-UPDRS): Process, format, and clinimetric testing plan. Mov Disord 22:41–47
Goldknopf IL, Sheta EA, Bryson J, Folsom B, Wilson C, Duty J, Yen AA, Appel SH (2006) Complement C3c and related protein biomarkers in amyotrophic lateral sclerosis and Parkinson’s disease. Biochem Biophys Res Commun 342:1034–1039
Graham JM, Paley MN, Grunewald RA, Hoggard N, Griffiths PD (2000) Brain iron deposition in Parkinson’s disease imaged using the PRIME magnetic resonance sequence. Brain 123:2423–2431
Grunblatt E, Mandel S, Jacob-Hirsch J, Zeligson S, Amariglo N, Rechavi G, Li J, Ravid R, Roggendorf W, Riederer P, Youdim MB (2004) Gene expression profiling of parkinsonian substantia nigra pars compacta; alterations in ubiquitin-proteasome, heat shock protein, iron and oxidative stress regulated proteins, cell adhesion/cellular matrix and vesicle trafficking genes. J Neural Transm 111:1543–1573
Grunblatt E, Zehetmayer S, Jacob CP, Muller T, Jost WH, Riederer P (2010) Pilot study: peripheral biomarkers for diagnosing sporadic Parkinson’s disease. J Neural Transm 117:1387–1393
Haaxma CA, Bloem BR, Borm GF, Horstink MW (2008) Comparison of a timed motor test battery to the Unified Parkinson’s Disease Rating Scale-III in Parkinson’s disease. Mov Disord 23:1707–1717
Halperin I, Morelli M, Korczyn AD, Youdim MBH, Mandel SA (2009) Biomarkers for evaluation of clinical efficacy of multipotential neuroprotective drugs for Alzheimer’s and Parkinson’s diseases. Neurotherapeutics 6:128–140
Hampel H, Mitchell A, Blennow K, Frank RA, Brettschneider S, Weller L, Moller HJ (2004) Core biological marker candidates of Alzheimer’s disease—perspectives for diagnosis, prediction of outcome and reflection of biological activity. J Neural Transm 111:247–272
Hampel H, Frank R, Broich K, Teipel SJ, Katz RG, Hardy J, Herholz K, Bokde ALW, Jessen F, Hoessler YC, Sanhai WR, Zetterberg H, Woodcock J, Blennow K (2010) Biomarkers for Alzheimer’s disease: academic industry and regulatory perspectives. Nature Rev Drug Discov 9:560–574
Hassin-Baer S, Cohen O, Vakil E, Sela BA, Nitsan Z, Schwartz R, Chapman J, Tanne D (2006) Plasma homocysteine levels and Parkinson disease: disease progression, carotid intima-media thickness and neuropsychiatric complications. Clin Neuropharmacol 29:305–311
Hatano T, Kubo S, Sato S, Hattori N (2009) Pathogenesis of familial Parkinson’s disease: new insights based on monogenic forms of Parkinson’s disease. J Neurochem 111:1075–1193
Hausdorff JM (2008) The timed up and go test: more than meets the eye. Smolenice, Castle
Hilker R, Schweitzer K, Coburger S, Ghaemi M, Weisenbach S, Jacobs AH, Rudolf J, Herholz K, Heiss WD (2005) Nonlinear progression of Parkinson disease as determined by serial positron emission tomographic imaging of striatal fluorodopa F 18 activity. Arch Neurol 62:378–382
Hirotani M, Maita C, Niino M, Iguchi-Ariga AM, Hamada S, Ariga H, Sasaki H (2008) Correlation between DJ-1 levels in the cerebrospinal fluid and the progression of disabilities in multiple sclerosis patients. Mult Scler 14:1056–1060
Hirsch E, Graybiel A, Agid Y (1988) Melanized dopamine neurons are differentially susceptible to degeneration in Parkinson’s disease. Nature 334:345–348
Hong Z, Shi M, Chung KA, Quinn JF, Peskind ER, Galasko D, Jankovic J, Zabetian CP, Leverenz JB, Baird G, Montine TJ, Hancock AM, Hwang H, Pan C, Bradner J, Kang UJ, Jensen PH, Zhang J (2010) DJ-1 and alpha-synuclein in human cerebrospinal fluid as biomarkers of Parkinson’s disease. Brain 133:713–726
Huang C, Tang C, Feigin A, Lesser M, Ma Y, Pourfar M, Dhawan V, Eidelberg D (2007) Changes in network activity with the progression of Parkinson’s disease. Brain 130:1834–1846
Huxham F, Baker R, Morris ME, Iansek R (2008) Head and trunk rotation during walking turns in Parkinson’s disease. Mov Disord 23:1391–1397
Irizarry M, Gurol ME, Raju S, Diaz-Arrastia R, Locascio JJ, Tennis MRN, Hyman BT, Growdon JH, Greenberg SM, Bottiglieri T (2005) Association of homocysteine with plasma amyloid [beta] protein in aging and neurodegenerative disease. Neurology 65:1402–1408
Jellinger K (1991) Pathology of Parkinson’s disease. Changes other than the nigrostriatal pathway. Mol Chem Neuropathol 14:153–197
Johansen KK, Wang L, Aasly JP, White LR, Marson WR, Henchcliffe C, Beal MF, Bogdanov M (2009) Metabolomic profiling in LRRK2-related Parkinson’s disease. PLoS ONE 4:e7551
Kaddurah-Daouk R, Krishnan KR (2005) Metabolomics: a global biochemical approach to the study of central nervous system diseases. Neuropsychopharmacology 34:173–186
Kahle PJ, Waak J, Gasser T (2009) DJ-1 and prevention of oxidative stress in Parkinson’s disease and other age-related disorders. Free Radic Biol Med 47:1354–1361
Kamanli A, Ardicoglu O, Ozgocmen S, Yoldas TK (2008) Bone mineral density in patients with Parkinson’s disease. Aging Clin Exp Res 20:277–279
Karlsen KH, Larsen JP, Tandberg E, Maeland JG (1999) Influence of clinical and demographic variables on quality of life in patients with Parkinson’s disease. J Neurol Neurosurg Psychiatry 66:431–435
Katsarou Z, Bostantjopoulou S, Alevriadou A, Mentenopoulos G, Avraam X, Kiosseoglou G (1998) A longitudinal study of visuospatial discrimination in parkinsonian patients. Percept Mot Skills 86:171–180
Klein C, Schneider SA, Lang AE (2009) Hereditary parkinsonism: Parkinson’s disease look-alikes—an algorithm for clinicians to “Park” Genes and beyond. Mov Disord 24:2042–2058
Lang AE (2010) Clinical trials of disease-modifying therapies for neurodegenerative diseases: the challenges and the future. Nature Med 16:1223–1226
Liepelt I, Reimold M, Maetzler W, Godau J, Reischl G, Gaenslen A, Herbst H, Berg D (2009) Cortical hypometabolism assessed by a metabolic ratio in Parkinson’s disease primarily reflects cognitive deterioration-[18F]FDG-PET. Mov Disord 24:1504–1511
Linden D, Diehl RR, Berlit P (1997) Sympathetic cardiovascular dysfunction in long-standing idiopathic Parkinson’s disease. Clin Auton Res 7:311–314
Maetzler W, Berg D, Schalamberidze N, Melms A, Schott K, Mueller JC, Liaw L, Gasser T, Nitsch C (2007) Osteopontin is elevated in Parkinson’s disease and its absence leads to reduced neurodegeneration in the MPTP model. Neurobiol Dis 25:473–482
Maetzler W, Liepelt I, Berg D (2009a) Progression of Parkinson’s disease in the clinical phase: potential markers. Lancet Neurol 8:1158–1171
Maetzler W, Liepelt I, Reimold M, Reischl G, Solbach C, Becker C, Schulte C, Leyhe T, Keller S, Melms A, Gasser T, Berg D (2009b) Cortical PIB binding in Lewy body disease is associated with Alzheimer-like characteristics. Neurobiol Dis 34:107–112
Maetzler W, Schmid SP, Wurster I, Liepelt I, Gaenslen A, Gasser T, Berg D (2011) Reduced but not oxidized cerebrospinal fluid glutathione levels are lowered in Lewy body diseases. Mov Disord 26:176–181
Maita C, Tsuji S, Yabe I, Hamada S, Ogata A, Maita H, Iguchi-Ariga SMM, Sasaki H, Ariga H (2008) Secretion of DJ-1 into serum of patients with Parkinson’s disease. Neurosci Lett 431:86–89
Mamorita N, Iizuka T, Takeuchi A, Shirataka M, Ikeda N (2009) Development of a system for measurement and analysis of tremor using a three-axis accelerometer. Methods Inf Med 48:589–594
Mancini M, Zampieri C, Carlson-Kuhta P, Chiari L, Horak FB (2009) Anticipatory postural adjustments prior to step initiation are hypometric in untreated Parkinson’s disease: an accelerometer-based approach. Eur J Neurol 16:1028–1034
Marek K, Jennings D, Tamagnan G, Seibyl J (2008) Biomarkers for Parkinson’s disease: tools to assess Parkinson’s disease onset and progression. Ann Neurol (Suppl 64):S111–S121
Martins-de-Souza D (2010) Is the word “biomarker” being properly used by proteomics in neuroscience? Eur Arch Psychiatry Clin Neurosci 260:561–562
McCaddon A, Hudson B, Barber J, Lloyd A, Davies G, Regland B (2003) Alzheimer’s disease and total plasma aminothiols. Biol Psychiatry 53:254–260
Mesec A, Sega S, Trost M, Pogacnik T (1999) The deterioration of cardiovascular reflexes in Parkinson’s disease. Acta Neurol Scand 100:296–299
Michell AW, Lewis SJG, Foltynie T, Barker RA (2004) Biomarker and Parkinson’s disease. Brain 127:1693–1705
Milanov I (2002) Correlation between tremor parameters. Funct Neurol 17:19–23
Miller JW, Green R, Mungas DM, Reed BR, Jagust WJ (2002) Homocyteine, vitamin B-6, and vascular disease in AD patients. Neurology 58:1471–1475
Mollenhauer B, Trenkwalder C, von Ahsen N, Bibl M, Steinacker P, Brechlin P, Schindehuette J, Poser S, Wiltfang J, Otto M (2006) Beta-amyloid 1–42 and tau-protein in cerebrospinal fluid of patients with Parkinson’s disease dementia. Dement Geriatr Cogn Disord 22:200–208
Mollenhauer B, Locascio JJ, Schulz-Schaeffer W, Sixel-Doring F, Trenkwalder C, Schlossmacher MG (2011) alpha-Synuclein and tau concentrations in cerebrospinal fluid of patients presenting with parkinsonism: a cohort study. Lancet Neurol 10:230–240
Morgan JC, Mehta SH, Sethi KD (2010) Biomarkers in Parkinson’s disease. Curr Neurol Neurosci Rep 10:423–430
Najafi B, Aminian K, Loew F, Blanc Y, Robert PA (2002) Measurement of stand-sit and sit-stand transitions using a miniature gyroscope and its application in fall risk evaluation in the elderly. IEEE Trans Biomed Eng 49:843–851
Nurmi E, Ruottinen HM, Bergman J, Haaparanta M, Solin O, Sonninen P, Rinne JO (2001) Rate of progression in Parkinson’s disease: a 6-[18F]fluoro-L-dopa PET study. Mov Disord 16:608–615
Nyhlen J, Constantinescu R, Zetterberg H (2010) Problems associated with fluid biomarkers for Parkinson’s disease. Biomarkers Med 4:671–681
Onofrj M, Thomas A, D’Andreamatteo G, Iacono D, Luciano AL, Di Rollo A, Di Mascio R, Ballone E, Di Iorio A (2002) Incidence of RBD and hallucination in patients affected by Parkinson’s disease: 8-year follow-up. Neurol Sci 23(Suppl 2):S91–S94
Orimo S, Ozawa E, Nakade S, Sugimoto T, Mizusawa H (1999) (123)I-metaiodobenzylguanidine myocardial scintigraphy in Parkinson’s disease. J Neurol Neurosurg Psychiatry 67:189–194
Orr CF, Rowe DB, Mizuno Y, Mori H, Halliday GM (2005) A possible role for humoral immunity in the pathogenesis of Parkinson’s disease. Brain 128:2665–2674
Peran P, Cherubini A, Assogna F, Piras F, Quattrocchi C, Peppe A, Celsis P, Rascol O, Demonet JF, Stefani A, Pierantozzi M, Pontieri FE, Caltagirone C, Spalletta G, Sabatini U (2010) Magnetic resonance imaging markers of Parkinson’s disease nigrostriatal signature. Brain 133:3423–3433
Plotnik M, Giladi N, Hausdorff JM (2007) A new measure for quantifying the bilateral coordination of human gait: effects of aging and Parkinson’s disease. Exp Brain Res 181:561–570
Plotnik M, Giladi N, Hausdorff JM (2009) Bilateral coordination of gait and Parkinson’s disease: the effects of dual tasking. J Neurol Neurosurg Psychiatry 80:347–350
Ponsen MM, Stoffers D, Twisk JWR, Wolters EC, Berendse HW (2009) Hyposmia and executive dysfunction as predictors of future Parkinson’s disease: a prospective study. Mov Disord 24:1060–1065
Riederer P, Wuketich S (1976) Time course of nigrostriatal degeneration in Parkinson’s disease. A detailed study of influential factors in human brain amine analysis. J Neural Transm 38:277–301
Rigas G, Tzallas AT, Tsalikakis DG, Konitsiotis S, Fotiadis DI (2009) Real-time quantification of resting tremor in the Parkinson’s disease. Conf Proc IEEE Eng Med Biol Soc 1306–1309
Rojo A, Aguilar M, Garolera MT, Cubo E, Navas I, Quintana S (2003) Depression in Parkinson’s disease: clinical correlates and outcome. Parkinsonism Relat Disord 10:23–28
Ryvlin P, Broussolle E, Piollet H, Viallet F, Khalfallah Y, Chazot G (1995) Magnetic resonance imaging evidence of decreased putamenal iron content in idiopathic Parkinson’s disease. Arch Neurol 52:583–588
Sato S, Mizuno Y, Hattori N (2005) Urinary 8-hydroxyguanosine levels as a biomarker for progression of Parkinson’s disease. Neurology 64:1081–1083
Scherzer CR, Eklund AC, Morse LJ, Liao ZX, Locascio JJ, Fefer D, Schwarzschild MA, Schlossmacher MG, Hauser MA, Vance JM, Sudarsky LR, Standaert DG, Growdon JH, Jensen RV, Gullans SR (2007) Molecular markers of early Parkinson’s disease based on gene expression in blood. Proc Natl Acad Sci USA 104:955–960
Schestatsky P, Ehlers JA, Rieder CR, Gomes I (2006) Evaluation of sympathetic skin response in Parkinson’s disease. Parkinsonism Relat Disord 12:486–491
Schiesling C, Kieper N, Seidel K, Kruger R (2008) Familial Parkinson’s disease—genetics, clinical phenotype and neuropathology in relation to the common sporadic form of the disease. Neuropathol Appl Neurobiol 34:255–271
Schlesinger I, Schlesinger N (2008) Uric acid in Parkinson’s disease. Mov Disord 23:1653–1657
Schlesinger I, Benyakov O, Erikh I, Suraiya S, Schiller Y (2009) Parkinson’s disease tremor is diminished with relaxation guided imagery. Mov Disord 24:2059–2062
Schrag A, Dodel R, Spottke A, Bornschein B, Siebert U, Quinn NP (2007) Rate of clinical progression in Parkinson’s disease. A prospective study. Mov Disord 22:938–945
Schwarzschild MA, Schwid SR, Marek K, Watts A, Lang AE, Oakes D, Shoulson I, Ascherio A, and the Parkinson Study Group PRECEPT Investigators (2008) Serum urate as a predictor of clinical and radiographic progression in Parkinsons disease. Arch Neurol 65:716–723
Serrano-Duenas M, Calero B, Serrano S, Serrano M, Coronel P (2010) Psychometric attributes of the rating scale for gait evaluation in Parkinson’s disease. Mov Disord 25:2121–2127
Shaw LM, Korecka M, Clark CM, Lee VM-Y, Trojanowski JQ (2007) Biomarkers of neurodegeneration for diagnosis and monitoring therapeutics. Nature Rev 6:295–303
Shi M, Bradner J, Hancock AM, Chung KA, Quinn JF, Peskind ER, Galasko D, Jankovic J, Zabetian CP, Kim HM, Leverenz JB, Montine TJ, Ginghina C, Kang UJ, Cain KC, Wang Y, Aasly J, Goldstein D, Zhang J (2011) Cerebrospinal fluid biomarkers for Parkinson disease diagnosis and progression. Ann Neurol 69:570–580
Shibata M, Morita Y, Shimizu T, Takahashi K, Suzuki N (2009) Cardiac parasympathetic dysfunction concurrent with cardiac sympathetic denervation in Parkinson’s disease. J Neurol Sci 276:79–83
Sian J, Dexter DT, Lees AJ, Daniel S, Agid Y, Javoy-Agid F, Jenner P, Marsden CD (1994) Alterations in glutathione levels in Parkinson’s disease and other neurodegenerative disorders affecting basal ganglia. Ann Neurol 36:348–355
Suzuki M, Urashima M, Oka H, Hashimoto M, Taira K (2007) Cardiac sympathetic denervation in bradykinesia-dominant Parkinson’s disease. Neuroreport 18:1867–1870
The Ronald and Nancy Reagan Research Institute of the Alzheimer’s Association and the National Institute on Aging Working Group (1998) Consensus report of the Working Group on: “Molecular and Biochemical Markers of Alzheimer’s Disease”. Neurobiol Aging 19:109–116
Visser JE, Voermans NC, Oude Nijhuis LB, van der Eijk M, Nijk R, Munneke M, Bloem BR (2007) Quantification of trunk rotations during turning and walking in Parkinson’s disease. Clin Neurophysiol 118:1602–1606
Waragai M, Wei J, Fujita M, Nakai M, Ho GJ, Masliah E, Akatsu H, Yamada T, Hashimoto M (2006) Increased level of DJ-1 in the cerebrospinal fluids of sporadic Parkinson’s disease. Biochem Biophys Res Commun 345:967–972
Weiss A, Herman T, Plotnik M, Brozgol M, Maidan I, Giladi N, Gurevich T, Hausdorff JM (2010) Can an accelerometer enhance the utility of the Timed Up and Go Test when evaluating patients with Parkinson’s disease? Med Eng Phys 32:119–125
Whone AL, Watts RL, Stoessl AJ, Davis M, Reske S, Nahmias C, Lang AE, Rascol O, Ribeiro MJ, Remy P, Poewe WH, Hauser RA, Brooks DJ, for the REAL-PET Study Group (2003) Slower progression of Parkinson’s disease with ropinirole versus levodopa: the REAL-PET study. Ann Neurol 54:93–101
Williams DR, Lees AJ (2005) Visual hallucinations in the diagnosis of idiopathic Parkinson’s disease: a retrospective autopsy study. Lancet Neurol 4:605–610
Wullner U, Schmitz-Hubsch T, Antony G, Fimmers R, Spottke A, Oertel WH, Deuschl G, Klockgether T, Eggert K (2007) Autonomic dysfunction in 3414 Parkinson’s disease patients enrolled in the German Network on Parkinson’s disease (KNP e.V.): the effect of ageing. Eur J Neurol 14:1405–1408
Yang YX, Wood NW, Latchman DS (2009) Molecular basis of Parkinson’s disease. Neuroreport 20:150–156
Younes-Mheni S, Frih-Ayed M, Kerkeni A, Bost M, Chazot G (2007) Peripheral blood markers of oxidative stress in Parkinson’s disease. Eur Neurol 58:78–83
Zampieri C, Salarian A, Carlson-Kuhta P, Aminian K, Nutt JG, Horak FB (2010) The instrumented timed up and go test: potential outcome measure for disease modifying therapies in Parkinson’s disease. J Neurol Neurosurg Psychiatry 81:171–176
Zetterberg H, Ruetschi U, Portelius E, Brinkmalm G, Andreasson U, Blennow K, Brinkmalm A (2008) Clinical proteomics in neurodegenerative disorders. Acta Neurol Scand 118:1–11
Zhang J, Sokal I, Peskind ER, Quinn JF, Jankovic J, Kenney C, Chung KA, Millard SP, Nutt JG, Montine TJ (2008) CSF multianalyte profile distinguishes Alzheimer and Parkinson diseases. Am J Clin Pathol 129:526–529
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License (https://creativecommons.org/licenses/by-nc/2.0), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
Gerlach, M., Maetzler, W., Broich, K. et al. Biomarker candidates of neurodegeneration in Parkinson’s disease for the evaluation of disease-modifying therapeutics. J Neural Transm 119, 39–52 (2012). https://doi.org/10.1007/s00702-011-0682-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00702-011-0682-x