
Abstract
The receptor for advanced glycation end-products (RAGE) has been implicated in several pathophysiological processes relevant to Alzheimer’s disease (AD), including transport and synaptotoxicity of AD-associated amyloid β (Aβ) peptides. A recent Chinese study (Li et al. in J Neural Transm 117:97–104, 2010) suggested an association between the 82S allele of the functional single nucleotide polymorphism (SNP) G82S (rs2070600) in the RAGE-encoding gene AGER and risk of AD. The present study aimed to investigate associations between AGER, AD diagnosis, cognitive scores and cerebrospinal fluid AD biomarkers in a European cohort of 316 neurochemically verified AD cases and 579 controls. Aside from G82S, three additional tag SNPs were analyzed to cover the common genetic variation in AGER. The 82S allele was associated with increased risk of AD (P c = 0.04, OR = 2.0, 95% CI 1.2–3.4). There was no genetic interaction between AGER 82S and APOE ε4 in producing increased risk of AD (P = 0.4), and none of the AGER SNPs showed association with Aβ42, T-tau, P-tau181 or mini-mental state examination scores. The data speak for a weak, but significant effect of AGER on risk of AD.
Similar content being viewed by others


Introduction
Alzheimer’s disease (AD) is the most frequent form of dementia, accounting for 50–60% of all cases (Blennow et al. 2006). It is a genetically heterogeneous disease with both familial and sporadic forms. The familial forms of AD are very rare, but the few cases that exist all carry mutations that in some way affect the processing of the amyloid precursor protein (APP) in a manner that accelerates the formation of synaptotoxic amyloid β (Aβ) (Blennow et al. 2006). Pathologically, AD is characterized by senile plaques containing aggregates of Aβ, and neurofibrillary tangles consisting of hyperphosphorylated tau protein. These pathological hallmarks generally debut in the hippocampus, a brain structure that is necessary for explicit learning (Salmon and Bondi 2009). Consistent with hippocampal pathology, the main clinical feature in patients with AD is anterograde amnesia, and as the disease progresses, the patients also develop other cognitive symptoms (Salmon and Bondi 2009). In line with these clinical findings, animal research has shown that long-term potentiation (LTP), a process that is widely believed to constitute the synaptic substrate for learning and memory (Martin et al. 2000), is inhibited by low-n oligomers of Aβ (Shankar et al. 2008; Townsend et al. 2006; Walsh et al. 2002; Klyubin et al. 2008).
The mechanisms by which Aβ inhibit LTP have been elusive and several suggestions have been made (Lauren et al. 2009; Li et al. 2009; Origlia et al. 2009; Townsend et al. 2007). It has, for example, been suggested that the LTP-inhibiting effect of Aβ depend on the receptor for advanced glycation end-products (RAGE) (Arancio et al. 2004; Origlia et al. 2008, 2009). In addition, this protein has been implicated in several other aspects of AD pathology, such as inflammation, oxidative stress and transport of Aβ across the blood–brain barrier (Deane et al. 2003; Schmidt et al. 2009; Yan et al. 1996). Together, these findings make the gene encoding RAGE (AGER) an interesting candidate gene for AD. Indeed, a recent Chinese case–control study presented evidence for an association between the G82S single nucleotide polymorphism (SNP) in AGER and AD (Li et al. 2010).
The RAGE receptor is a multi-ligand receptor, and one of its ligands is Aβ (Yan et al. 1996). Several intracellular pathways are activated by RAGE (Lue et al. 2009), and aside from inhibiting LTP, RAGE activation can stimulate the expression of β-site APP-cleaving enzyme 1 (BACE1) (Cho et al. 2009), an enzyme that is necessary for the production of Aβ. Accordingly, Aβ might exert positive feedback on its own production. Moreover, RAGE can induce its own expression through activation of the transcription factor NF-κB, and thereby initiate yet another potentially hazardous positive feedback loop (Lue et al. 2009).
There is, however, another facet of RAGE; a soluble form, which is formed by alternative splicing (Ding and Keller 2005) or proteolytic cleavage by the proteinase ADAM 10 (Raucci et al. 2008). Soluble RAGE (sRAGE) contains the ligand-binding site, but does not have the signaling properties of full-length RAGE (flRAGE). As a result, it could act as a decoy receptor by competing with flRAGE for ligands, and thus have protective properties. Indeed, studies have shown that sRAGE can reduce the accumulation of Aβ in the brains of mice (Deane et al. 2003), and that it can inhibit Aβ aggregation (Chaney et al. 2005). In addition, it has been shown that sRAGE is present at lower levels in the blood and brain of AD patients (Emanuele et al. 2005; Nozaki et al. 2007). Whatever role is most important for the development of AD, be it the sword or the shield, remains to be seen. Either way these findings highlight AGER as a most interesting candidate gene for AD.
Although several susceptibility genes for AD have been identified, few have been confirmed in independent populations (Bertram and Tanzi 2010). Thus, independent evaluation of the finding by Li et al. (2010) is warranted. A distinguishing feature of our study is that all AD cases were neurochemically verified, i.e., had low cerebrospinal fluid (CSF) levels of the 42 amino acid isoform of Aβ (Aβ1–42) as a sign of brain amyloid pathology and high levels of total tau (T-tau) as a sign of cortical axonal degeneration (Blennow et al. 2010). This approach reduces the risk of including patients with other dementing illnesses in the AD group.
Subjects and methods
Subjects
The case–control material consisted of 316 AD cases and 579 controls (Table 1). All individuals were of Caucasian origin. All diagnoses were set according to the National Institute of Neurological and Communicative Disorders and Stroke—Alzheimer’s Disease and Related Disorders Association (NINCDS-ADRDA) criteria (McKhann et al. 1984) after detailed clinical investigation including medical history, physical, neurological and psychiatric examination, screening laboratory tests, and computed tomography of the brain. No patient had a family history of autosomal dominant dementia. For all 316 cases, previously determined AD CSF biomarker levels of T-tau and Aβ1-42 were available, and for a subgroup of cases (n = 111) P-tau181 levels were available as well (Table 1). CSF biomarker concentrations were determined using INNOTEST ELISAs (Innogenetics, Ghent, Belgium) as described elsewhere (Blennow et al. 1995; Vanderstichele et al. 2000; Vanmechelen et al. 2000). Clinical diagnoses were set without knowledge of the results from the biochemical and genetic analyses and vice versa. The ambition was to study a pure AD group and minimize the risk of including other dementias in the AD group. Therefore, we only included cases with a typical AD CSF biomarker profile, i.e. Aβ42 < 400 pg/mL and T-tau > 400 pg/mL (Hansson et al. 2006; Zetterberg et al. 2003). Controls did not have dementia and did not show any signs of psychiatric illness, malignant disease or systematic disorder. Mini-mental state examination (MMSE) scores (Folstein et al. 1975) were available for most AD cases (n = 277) and all controls (Table 1). Potential controls with MMSE scores below 28 were excluded to minimize the risk of including incipient AD cases. APOE ε4 carrier status was known for all cases and controls.
Tag SNP selection
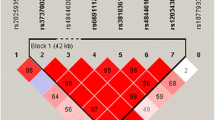
Single nucleotide polymorphism genotyping data covering AGER (gene ID: 177) for the European population CEU (Utah residents with ancestry from northern and western Europe) were downloaded from the HapMap Genome Browser (Phases 1 and 2—full dataset) at the International Haplotype Mapping Project web site (http://www.hapmap.org) (HapMap-Consortium 2003) and processed in the Haploview software (Barrett et al. 2005). Tag SNPs were assigned using the Tagger function (Barrett et al. 2005). A minor allele frequency of ≥5% and pairwise tagging with a minimum r 2 of 0.80 were applied to capture the common variations within the whole gene. The functional SNP G82S (rs2070600) was included using the “force include” option. The complete common genetic variation of AGER was tagged for by G82S and three additional tag SNPs: rs1800684, rs3131300 and rs1035798 (Table 2).
Genotyping
Tag SNPs were genotyped using genomic DNA extracted from blood tissue. TaqMan® Pre-Designed SNP genotyping assays (Applied Biosystems, Foster City, CA, USA) were used (Table 2) according to the TaqMan Allelic Discrimination technology (Livak 1999) on the ABI PRISM 7900HT Sequence Detection System (Applied Biosystems, Foster City, CA, USA).
Statistical analyses
Statistical analyses comparing the demographics in AD cases and controls were performed with SYSTAT11 (SYSTAT Software GmbH, Erkrath, Germany) using χ2-test for categorical parameters and using Mann–Whitney U test for continuous parameters. The effects of known risk factors, e.g., sex and APOE ε4 were taken into consideration by identifying significantly relevant covariates for each outcome variable (disease risk, MMSE and levels of AD CSF biomarkers) using forward stepwise logistic/linear regression.
The genetic association analyses were performed using HelixTree 6.3 (Golden Helix, Bozeman, MT, USA; available at: http://www.goldenhelix.com). All tag SNPs were analysed for deviation from Hardy–Weinberg equilibrium. Single marker associations were performed using logistic or linear regression including relevant covariates in an additive model (SNP coded as minor allele count). Haplotype analysis was carried out using stepwise forward logistic or linear haplotype regression always keeping the identified covariates in the model. Haplotype frequencies were estimated using the expectation–maximization algorithm (Excoffier and Slatkin 1995) and only haplotypes with a frequency of ≥5% were included in the analysis. Detailed description of the haplotype analyses have been given elsewhere (von Otter et al. 2010). Difference in OR according to APOE ε4 status was tested by adding an interaction term between APOE ε4 and AGER. Corrected P values (P c) of ≤0.05 were considered statistically significant. To correct for multiple testing, Bonferroni correction for the number of studied SNPs was used in all single marker analyses (n = 4) and permutation tests with 10,000 permutations were performed in the haplotype analyses.
Ethics
The study was approved by the ethics committees at the University of Gothenburg and the University of Lund in Sweden. The cases (or their closest relatives) and the controls gave informed consent to participate in the study, which was conducted in accordance with the provisions of the Helsinki Declaration.
Results
Demographics
Mean age and sex distribution differed between cases and controls (Table 1). As expected, the APOE ε4 allele was strongly overrepresented in the AD group when compared with the control group (Table 1). Identified covariates were sex and APOE ε4 for the risk association, and APOE ε4 alone for analysis of association with Aβ42 and MMSE.
SNP genotyping
All markers were in Hardy–Weinberg equilibrium and had a genotyping call rate >98%.
Association analyses
A significant association between the 82S allele (encoding amino acid Ser of the non-synonymous SNP rs2070600) and risk of AD was observed [P c = 0.04, OR = 2.0 (95% CI 1.2–3.4) per allele, Table 3]. When stratifying for APOE ε4 status, the carriers showed a significant association between the 82S allele and risk of AD [P = 0.01, OR = 2.6 (95% CI 1.2–5.3], while the non-carriers showed a weaker non-significant association [P = 0.37, OR = 1.5 (95% CI 0.6–3.6) per allele, Table 4]. This OR difference was, however, not significant (P = 0.4) according to an interaction test between G82S and APOE ε4 status in the logistic regression. None of the four studied SNPs showed association with Aβ42, T-tau, P-tau181 or MMSE (data not shown). Haplotype analysis revealed no additional association with risk of AD or with any of the other studied outcomes (data not shown).
Discussion
In the present European case–control study, we show that the G82S SNP (rs2070600) in AGER, the gene encoding RAGE, is associated with increased risk of AD (P c = 0.04, OR = 2.0, 95% CI 1.2–3.4, n = 893). This finding corroborates the finding of Li et al. (2010), who showed that the G82S SNP in AGER was associated with AD diagnosis in a Chinese case–control study. They reported an OR of approximately 1.5, which is slightly lower than reported here. These results reinforce each other and suggest that AGER indeed is a susceptibility gene for AD. The lack of association of the synonymous SNP (rs1800684) is in line with previously published data (Blomqvist et al. 2006; Emahazion et al. 2001) while the other SNPs have not been previously analysed regarding risk of AD.
In the study by Li et al. (2010) the risk association of 82S carriers with AD (OR = 1.6) was found significant among the APOE ε4 non-carriers, but slightly weaker (OR = 1.4) and not significant among the smaller group of APOE ε4 carriers. When analyzing the difference in these two ORs by adding an interaction term in a logistic regression model, no significant deviation was found. The G82S effect on risk of AD differed slightly according to APOE ε4 status in our study as well. However, we had a significant effect (OR = 2.6) among the APOE ε4 carriers, but a weaker non-significant effect (OR = 1.5) in the APOE ε4 non-carrier group. The difference between our two ORs was also not significant. Taken together, the two studies indicate that the risk associated with G82S is independent of APOE ε4 status. The lack of association of AGER SNPs with CSF biomarkers for neuropathological changes in AD and MMSE scores may be considered reasonable in the light of the small effect size of AGER G82S on disease risk.
What biological effects of a single amino acid substitution in RAGE may explain the association with AD? First, RAGE may affect both production and accumulation of Aβ in the brain (Chaney et al. 2005; Cho et al. 2009; Deane et al. 2003). Because the G82S SNP is located in an exon that codes for the ligand-binding site, it is possible that the observed effect is related to ligand binding. The 82S variant has been shown to increase the ligand-binding affinity of the receptor (Hofmann et al. 2002; Osawa et al. 2007). This could potentially lead to an increased signaling, which in turn, would accelerate APP processing through BACE1 [since BACE1 has been shown to be positively regulated by RAGE (Cho et al. 2009)], and thereby increase Aβ production. In addition, an increased transport of circulating Aβ into the brain would be expected because RAGE has been shown to transport Aβ across the blood–brain barrier into the brain (Deane et al. 2003). It seems unlikely that this is how the 82S variant increases the risk for AD though, since we found no association between G82S genotype and Aβ levels. The 82S variant may on the other hand be more effective in mediating the LTP-inhibiting effect of Aβ (Arancio et al. 2004; Origlia et al. 2008, 2009), a hypothesis that remains to be tested.
It is not entirely obvious that a higher affinity for ligand binding should lead to an increased risk for AD since sRAGE should also be affected, and hence, have an increased propensity to scavenge Aβ, thus increased protective properties. Nevertheless, this is not the case and an explanation could be that flRAGE is engaged in positive feedback mechanisms, thereby enhancing its own production, thus giving little room for sRAGE to exert its proposed protective mechanisms. This notion is supported by the finding that flRAGE expression is increased in AD brains (Lue et al. 2001; Miller et al. 2008). In addition, it has been shown that 82S carriers have roughly half as much circulating sRAGE when compared with 82G carriers (Jang et al. 2007; Li et al. 2010), implying that the increased ligand affinity of the receptor leads to a dysregulation of RAGE isoforms. In this context, it is interesting to note that we found no association between G82S genotype and Aβ levels in CSF, since it implies that the protective properties of sRAGE observed in animal models of AD may be of limited clinical relevance.
It was recently shown that AGER deletion leads to lower levels of Aβ in 6 months old mice carrying the familial Arctic (Arc) and Swedish (Swe) AD mutations. Interestingly, this was not the case for 12 months old animals, and surprisingly neither the 6 months nor 12 months old knockout animals performed better than the control Arc/Swe animals in a Y-maze memory test (Vodopivec et al. 2009). This is in conflict with the results from Fang et al. (2010) showing that deletion of the intracellular signaling domain in RAGE (dominant negative, DN-RAGE) has a protective effect on memory function. One explanation for these contradicting results could be that the two studies used different memory testing paradigms. Another explanation could be that DN-RAGE still has the possibility to form sRAGE. A third piece in this knockout puzzle is the study by Takuma et al. (2009) which showed that deletion of AGER reduced the amount of intracellular Aβ, p38 MAPK phosphorylation and Aβ-induced mitochondrial dysfunction in cultured cortical neurons. To summarize, it seems like deletion of AGER leads to reduction in Aβ and its related pathology. The results on Aβ levels by Vodopivec et al. might be due to the highly pathogenic Arc/Swe mutations, and might also suggest that RAGE is mainly a contributing factor in the disease process highlighting the importance of RAGE in the acquisition of sporadic AD. If this is the case, one would expect age at onset to be affected. Unfortunately, this analysis was not possible to perform in our material, but it would be a valuable addition in future replication studies.
Aside from the importance of identifying genetic risk factors, which might aid future therapeutic strategies, and early diagnosis, gene association studies constitute a bridge between experimental animal research aiming in understanding the mechanisms of disease, and clinical research on the actual patients whom the experimentalists aim to aid, thereby lending further support to the findings made in animal models of the disease. This is of great importance since a lot of effort is put into understanding the etiology of different diseases. By showing associations between disease and a gene under intense study, we can confirm that the clues found in experimental model systems are relevant to the disease under study, and thus, should be further pursued.
In conclusion, our data, together with the findings by Li et al. speak for a weak, but significant effect of AGER on risk of AD. Further studies on independent and larger cohorts are highly desired, as is the exploration of a possible association of AGER G82S with age at onset of AD.
References
Arancio O, Zhang HP, Chen X, Lin C, Trinchese F, Puzzo D, Liu S, Hegde A, Yan SF, Stern A, Luddy JS, Lue LF, Walker DG, Roher A, Buttini M, Mucke L, Li W, Schmidt AM, Kindy M, Hyslop PA, Stern DM, Du Yan SS (2004) RAGE potentiates Abeta-induced perturbation of neuronal function in transgenic mice. EMBO J 23:4096–4105
Barrett JC, Fry B, Maller J, Daly MJ (2005) Haploview: analysis and visualization of LD and haplotype maps. Bioinformatics 21:263–265
Bertram L, Tanzi RE (2010) Alzheimer disease: new light on an old CLU. Nat Rev Neurol 6:11–13
Blennow K, Wallin A, Agren H, Spenger C, Siegfried J, Vanmechelen E (1995) Tau protein in cerebrospinal fluid: a biochemical marker for axonal degeneration in Alzheimer disease? Mol Chem Neuropathol 26:231–245
Blennow K, De leon MJ, Zetterberg H (2006) Alzheimer’s disease. Lancet 368:387–403
Blennow K, Hampel H, Weiner M, Zetterberg H (2010) Cerebrospinal fluid and plasma biomarkers in Alzheimer disease. Nat Rev Neurol 6:131–144
Blomqvist MEL, Reynolds C, Katzov H, Feuk L, Andreasen N, Bogdanovic N, Blennow K, Brookes AJ, Prince JA (2006) Towards compendia of negative genetic association studies: an example for Alzheimer disease. Hum Genet 119:29–37
Chaney MO, Stine WB, Kokjohn TA, Kuo Y-M, Esh C, Rahman A, Luehrs DC, Schmidt AM, Stern D, Yan SD, Roher AE (2005) RAGE and amyloid beta interactions: atomic force microscopy and molecular modeling. Biochim Biophys Acta 1741:199–205
Cho HJ, Son SM, Jin SM, Hong HS, Shin DH, Kim SJ, Huh K, Mook-Jung I (2009) RAGE regulates BACE1 and Abeta generation via NFAT1 activation in Alzheimer’s disease animal model. FASEB J 23:2639–2649
Deane R, Du Yan S, Submamaryan RK, Larue B, Jovanovic S, Hogg E, Welch D, Manness L, Lin C, Yu J, Zhu H, Ghiso J, Frangione B, Stern A, Schmidt AM, Armstrong DL, Arnold B, Liliensiek B, Nawroth P, Hofman F, Kindy M, Stern D, Zlokovic B (2003) RAGE mediates amyloid-beta peptide transport across the blood–brain barrier and accumulation in brain. Nat Med 9:907–913
Ding Q, Keller JN (2005) Splice variants of the receptor for advanced glycosylation end products (RAGE) in human brain. Neurosci Lett 373:67–72
Emahazion T, Feuk L, Jobs M, Sawyer SL, Fredman D, St Clair D, Prince JA, Brookes AJ (2001) SNP association studies in Alzheimer’s disease highlight problems for complex disease analysis. Trends Genet 17:407–413
Emanuele E, D’angelo A, Tomaino C, Binetti G, Ghidoni R, Politi P, Bernardi L, Maletta R, Bruni AC, Geroldi D (2005) Circulating levels of soluble receptor for advanced glycation end products in Alzheimer disease and vascular dementia. Arch Neurol 62:1734–1736
Excoffier L, Slatkin M (1995) Maximum-likelihood estimation of molecular haplotype frequencies in a diploid population. Mol Biol Evol 12:921–927
Fang F, Lue LF, Yan S, Xu H, Luddy JS, Chen D, Walker DG, Stern DM, Schmidt AM, Chen JX, Yan SS (2010) RAGE-dependent signaling in microglia contributes to neuroinflammation, Abeta accumulation, and impaired learning/memory in a mouse model of Alzheimer’s disease. FASEB J 24:1043–1055
Folstein MF, Folstein SE, Mchugh PR (1975) Mini-mental state: a practical method for grading the cognitive state of patients for the clinician. J Psychiatr Res 12:189–198
Hansson O, Zetterberg H, Buchhave P, Londos E, Blennow K, Minthon L (2006) Association between CSF biomarkers and incipient Alzheimer’s disease in patients with mild cognitive impairment: a follow-up study. Lancet Neurol 5:228–234
Hapmap-Consortium (2003) The International Hapmap Project. Nature 426:789–796
Hofmann MA, Drury S, Hudson BI, Gleason MR, Qu W, Lu Y, Lalla E, Chitnis S, Monteiro J, Stickland MH, Bucciarelli LG, Moser B, Moxley G, Itescu S, Grant PJ, Gregersen PK, Stern DM, Schmidt AM (2002) RAGE and arthritis: the G82S polymorphism amplifies the inflammatory response. Genes Immun 3:123–135
Jang Y, Kim JY, Kang S-M, Kim J-S, Chae JS, Kim OY, Koh SJ, Lee HC, Ahn CW, Song YD, Lee JH (2007) Association of the Gly82 Ser polymorphism in the receptor for advanced glycation end products (RAGE) gene with circulating levels of soluble RAGE and inflammatory markers in nondiabetic and nonobese Koreans. Metab Clin Exp 56:199–205
Klyubin I, Betts V, Welzel AT, Blennow K, Zetterberg H, Wallin A, Lemere CA, Cullen WK, Peng Y, Wisniewski T, Selkoe DJ, Anwyl R, Walsh DM, Rowan MJ (2008) Amyloid beta protein dimer-containing human CSF disrupts synaptic plasticity: prevention by systemic passive immunization. J Neurosci 28:4231–4237
Lauren J, Gimbel DA, Nygaard HB, Gilbert JW, Strittmatter SM (2009) Cellular prion protein mediates impairment of synaptic plasticity by amyloid-beta oligomers. Nature 457:1128–1132
Li S, Hong S, Shepardson NE, Walsh DM, Shankar GM, Selkoe D (2009) Soluble oligomers of amyloid beta protein facilitate hippocampal long-term depression by disrupting neuronal glutamate uptake. Neuron 62:788–801
Li K, Dai D, Zhao B, Yao L, Yao S, Wang B, Yang Z (2010) Association between the RAGE G82S polymorphism and Alzheimer’s disease. J Neural Transm 117:97–104
Livak KJ (1999) Allelic discrimination using fluorogenic probes and the 5′ nuclease assay. Genet Anal 14:143–149
Lue LF, Walker DG, Brachova L, Beach TG, Rogers J, Schmidt AM, Stern DM, Yan SD (2001) Involvement of microglial receptor for advanced glycation endproducts (RAGE) in Alzheimer’s disease: identification of a cellular activation mechanism. Exp Neurol 171:29–45
Lue L-F, Walker DG, Jacobson S, Sabbagh M (2009) Receptor for advanced glycation end products: its role in Alzheimer’s disease and other neurological diseases. Future Neurol 4:167–177
Martin SJ, Grimwood PD, Morris RG (2000) Synaptic plasticity and memory: an evaluation of the hypothesis. Annu Rev Neurosci 23:649–711
Mckhann G, Drachman D, Folstein M, Katzman R, Price D, Stadlan EM (1984) Clinical diagnosis of Alzheimer’s disease: report of the NINCDS-ADRDA Work Group under the auspices of Department of Health and Human Services Task Force on Alzheimer’s Disease. Neurology 34:939–944
Miller MC, Tavares R, Johanson CE, Hovanesian V, Donahue JE, Gonzalez L, Silverberg GD, Stopa EG (2008) Hippocampal RAGE immunoreactivity in early and advanced Alzheimer’s disease. Brain Res 1230:273–280
Nozaki I, Watanabe T, Kawaguchi M, Akatsu H, Tsuneyama K, Yamamoto Y, Ohe K, Yonekura H, Yamada M, Yamamoto H (2007) Reduced expression of endogenous secretory receptor for advanced glycation endproducts in hippocampal neurons of Alzheimer’s disease brains. Arch Histol Cytol 70:279–290
Origlia N, Righi M, Capsoni S, Cattaneo A, Fang F, Stern DM, Chen JX, Schmidt AM, Arancio O, Yan SD, Domenici L (2008) Receptor for advanced glycation end product-dependent activation of p38 mitogen-activated protein kinase contributes to amyloid-beta-mediated cortical synaptic dysfunction. J Neurosci 28:3521–3530
Origlia N, Capsoni S, Cattaneo A, Fang F, Arancio O, Yan SD, Domenici l (2009) Abeta-dependent inhibition of LTP in different intracortical circuits of the visual cortex: the role of RAGE. J Alzheimers Dis 17:59–68
Osawa M, Yamamoto Y, Munesue S, Murakami N, Sakurai S, Watanabe T, Yonekura H, Uchigata Y, Iwamoto Y, Yamamoto H (2007) De-N-glycosylation or G82S mutation of rage sensitizes its interaction with advanced glycation endproducts. Biochim Biophys Acta 1770:1468–1474
Raucci A, Cugusi S, Antonelli A, Barabino SM, Monti L, Bierhaus A, Reiss K, Saftig P, Bianchi ME (2008) A soluble form of the receptor for advanced glycation endproducts (RAGE) is produced by proteolytic cleavage of the membrane-bound form by the sheddase a disintegrin and metalloprotease 10 (ADAM10). FASEB J 22:3716–3727
Salmon DP, Bondi MW (2009) Neuropsychological assessment of dementia. Annu Rev Psychol 60:257–282
Schmidt AM, Sahagan B, Nelson RB, Selmer J, Rothlein R, Bell JM (2009) The role of RAGE in amyloid-beta peptide-mediated pathology in Alzheimer’s disease. Curr Opin Investig Drugs 10:672–680
Shankar GM, Li S, Mehta TH, Garcia-Munoz A, Shepardson NE, Smith I, Brett FM, Farrell MA, Rowan MJ, Lemere CA, Regan CM, Walsh DM, Sabatini BL, Selkoe DJ (2008) Amyloid-beta protein dimers isolated directly from Alzheimer’s brains impair synaptic plasticity and memory. Nat Med 14:837–842
Takuma K, Fang F, Zhang W, Yan S, Fukuzaki E, Du H, Sosunov A, Mckhann G, Funatsu Y, Nakamichi N, Nagai T, Mizoguchi H, Ibi D, Hori O, Ogawa S, Stern DM, Yamada K, Yan SS (2009) RAGE-mediated signaling contributes to intraneuronal transport of amyloid-beta and neuronal dysfunction. Proc Natl Acad Sci USA 106:20021–20026
Townsend M, Shankar GM, Mehta T, Walsh DM, Selkoe DJ (2006) Effects of secreted oligomers of amyloid beta-protein on hippocampal synaptic plasticity: a potent role for trimers. J Physiol 572:477–492
Townsend M, Mehta T, Selkoe DJ (2007) Soluble Abeta inhibits specific signal transduction cascades common to the insulin receptor pathway. J Biol Chem 282:33305–33312
Vanderstichele H, van Kerschaver E, Hesse C, Davidsson P, Buyse MA, Andreasen N, Minthon L, Wallin A, Blennow K, Vanmechelen E (2000) Standardization of measurement of beta-amyloid(1–42) in cerebrospinal fluid and plasma. Amyloid 7:245–258
Vanmechelen E, Vanderstichele H, Davidsson P, Van kerschaver E, Van der perre B, Sjogren M, Andreasen N, Blennow K (2000) Quantification of tau phosphorylated at threonine 181 in human cerebrospinal fluid: a sandwich ELISA with a synthetic phosphopeptide for standardization. Neurosci Lett 285:49–52
Vodopivec I, Galichet A, Knobloch M, Bierhaus A, Heizmann CW, Nitsch RM (2009) RAGE does not affect amyloid pathology in transgenic ArcAbeta mice. Neurodegener Dis 6:270–280
von Otter M, Landgren S, Nilsson S, Zetterberg M, Celojevic D, Bergstrom P, Minthon L, Bogdanovic N, Andreasen N, Gustafson DR, Skoog I, Wallin A, Tasa G, Blennow K, Nilsson M, Hammarsten O, Zetterberg H (2010) Nrf2-encoding NFE2L2 haplotypes influence disease progression but not risk in Alzheimer’s disease and age-related cataract. Mech Ageing Dev 131:105–110
Walsh DM, Klyubin I, Fadeeva JV, Cullen WK, Anwyl R, Wolfe MS, Rowan MJ, Selkoe DJ (2002) Naturally secreted oligomers of amyloid beta protein potently inhibit hippocampal long-term potentiation in vivo. Nature 416:535–539
Yan SD, Chen X, Fu J, Chen M, Zhu H, Roher A, Slattery T, Zhao L, Nagashima M, Morser J, Migheli A, Nawroth P, Stern D, Schmidt AM (1996) RAGE and amyloid-beta peptide neurotoxicity in Alzheimer’s disease. Nature 382:685–691
Zetterberg H, Wahlund LO, Blennow K (2003) Cerebrospinal fluid markers for prediction of Alzheimer’s disease. Neurosci Lett 352:67–69
Acknowledgments
We thank Mrs Mona Seibt Palmér for excellent technical assistance and Prof. Eric Hanse for helpful comments on the manuscript. This work was supported by Grants from the Swedish Research Council, the Alzheimer’s Association (NIRG-08-90356), the Royal Swedish Academy of Sciences, the Sahlgrenska University Hospital, the Göteborg Medical Society, Swedish Brain Power, Stiftelsen för Gamla Tjänarinnor, Gun och Bertil Stohnes stiftelse, Handlanden Hjalmar Svensson Foundation, the Åhlén Foundation, Magn. Bergvall’s Foundation and the Alzheimer Foundation, Sweden. Funding sources did not influence the design and conduct of the study; collection, management, analyses, and interpretation of the data; or preparation, review, or approval of the manuscript.
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Author information
Authors and Affiliations
Corresponding author
Additional information
J. Daborg and M. von Otter contributed equally to this work.
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License (https://creativecommons.org/licenses/by-nc/2.0), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
Daborg, J., von Otter, M., Sjölander, A. et al. Association of the RAGE G82S polymorphism with Alzheimer’s disease. J Neural Transm 117, 861–867 (2010). https://doi.org/10.1007/s00702-010-0437-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00702-010-0437-0