
Abstract
Terpene synthases (TPS) are responsible for the terminal biosynthetic step of terpenoid production. They are encoded by a highly diverse gene family believed to evolve by tandem duplication in response to adaptive pressures. Taxa in the Myrtaceae family are renowned for their diversity of terpenoid-rich essential oils, and among them, the tribe Eucalypteae has the largest TPS gene family found in any plant (> 100 TPS). In this study, comparative analysis of Melaleuca alternifolia (tea tree), from the related tribe Melaleuceae, revealed some Myrtaceae have smaller TPS families, as a total of 58 putatively functional full-length TPS genes, and 21 pseudogenes were identified by manual annotation of a newly released long-read assembly of the genome. The TPS-a and TPS-b2 subfamilies that synthesise secondary compounds often mediating plant-environment interactions were more diminutive than those in eucalypts, probably reflecting key differences in the evolutionary histories of the two lineages. Of the putatively functional TPS-b1, 13 clustered into a region of around 400 kb on one scaffold. The organisation of these TPS suggested that tandem duplication was instrumental in the evolution and diversity of terpene chemistry in Melaleuca. Four TPS-b1 likely to catalyse the synthesis of the three monoterpenoid components that are used to classify tea tree chemotypes were encoded within a single small region of 87 kb in the larger cluster of TPS-b1, raising the possibility that coregulation and linkage may lead to their behaviour as a single locus, providing an explanation for the categorical inheritance of complex multiple-component chemotypes in the taxon.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Terpenes and terpenoids are hydrocarbon-based substances produced by plants, fungi, bacteria, and insects (Chen et al. 2011; Schmidt-Dannert 2015; Yamada et al. 2015; Beran et al. 2016). In plants, all terpenoids are derived from the isomeric 5-carbon precursors isopentenyl diphosphate (IPP) and dimethylallyl diphosphate (DMAPP), which can be synthesised in two distinct pathways that are both well understood (see Online Resource 1 and Vranová et al. (2013) and Tholl (2015)). They are a structurally diverse group of compounds that can be categorised based on the number of carbon atoms they contain, i.e. hemiterpenoids (C5), monoterpenoids (C10), sesquiterpenoids (C15), and diterpenoids (C20) (Sell 2010). Many terpenoids are primary metabolites such as sterols, which are incorporated into membranes, abscisic acid, or gibberellins, which play essential roles as phytohormones, carotenoids that are pigments for photosynthesis (Yu and Utsumi 2009), and phytol, which is a component of chlorophyll (Gutbrod et al. 2019). Other mono- and sesquiterpenoids are secondary metabolites, which are not vital for survival and are only represented in some groups of plants. Nonetheless, these volatile terpenoids are important as mediators between a plant and its biotic and abiotic environment, facilitating direct and indirect defences against herbivores or pathogens (Block et al. 2019), or attracting pollinators (Pichersky and Gershenzon 2002). They have been characterised extensively in many plant species and are the largest group of plant secondary metabolites (Dudareva et al. 2004). The structures for more than 40,000 terpenoids are already known (Fürstenberg-Hägg et al. 2013), and large variation in the terpenoid composition is observed among and within plant species (Moore et al. 2014). The main driver for the high diversification of terpenoid substances is most likely herbivore and pathogen specialisation. In 1964, Ehrlich and Raven proposed plant and insect coevolution as the main factor contributing to the development of highly diverse plant secondary substances (Ehrlich and Raven 1964). Hence, the continuous interaction between herbivores and plants led to stepwise adaptations by selection, resulting in the vast array of plant secondary compounds we see today and a similarly wide array of insect mechanisms to deal with these plant defences (Ryan and Byrne 1988; Keane and Ryan 1999; Yu 2008). Particularly long-lived plants, such as trees or woody perennials, are believed to be rich in terpenoids because they have to defend themselves against rapidly evolving pathogens and insect herbivores over a long period of time.
Comparative studies among species within plant families have provided insight into the importance of terpenoids in the evolution and chemical ecology of plants. The final steps of terpenoid biosynthesis are primarily catalysed by a class of enzymes called terpene synthases (TPS). They can produce an extensive variety of terpenoids from a few common substrates, with many enzymes being capable of synthesising different products from a single substrate (Schwab 2003). Within genomic sequences, the enzymes are coded by members of the TPS gene family. Comparative genomics allowed both the identification of conserved regions in genes among species that are suggestive of shared functional importance, as well as regions of divergence that may indicate rapidly evolving sequences subject to adaptative evolution (Strachan and Read 2011). It has been shown, for example that TPS genes can be classified based on their intron–exon organisation on the genomic level (Trapp and Croteau 2001), and that all angiosperm and gymnosperm terpene synthases have highly similar structural features (Cao et al. 2010; Chen et al. 2011). This coincides with the observation that overall amino acid sequences in TPS are well conserved, while their catalytic pockets are highly variable (Bohlmann et al. 1998). Current evidence suggests that despite the high diversification of TPS gene family, they most likely all evolved from one ancestral gene (Trapp and Croteau 2001; Keeling et al. 2010).
One plant group renowned for its terpenoid diversity is the angiosperm family of Myrtaceae, which consists of trees and shrubs that are mainly distributed in forests and woodlands of the southern hemisphere (Wilson 2011). Myrtaceae are a major element of the Australian flora and are renowned for producing high concentrations of volatile terpenoids usually stored in schizogenous secretory cavities in their leaves (Brophy et al. 2013). The oils of these species are mostly dominated by mono- and sesquiterpenoids and display terpenoid diversity at a taxonomic, population, as well as individual level (Keszei et al. 2010a). Within the family, the three tribes Melaleuceae, Eucalypteae, and Myrteae have the highest diversity regarding unique monoterpenoid and sesquiterpenoid compounds in their leaves (Padovan et al. 2014). The Eucalypteae tribe, which consists of the genera Eucalyptus, Corymbia, and Angophora, is one of the best-known groups within the Myrtaceae, with some species of worldwide economic importance for the timber and essential oil industries (Grattapaglia et al. 2012).
The terpenoid diversity of eucalypt oils is matched by corresponding high numbers of TPS genes. Using the reference genome for the model Myrtaceae Eucalyptus grandis, Külheim et al. (2015) identified 113 TPS genes, which is the highest number of TPS genes known for any plant. Relatively high numbers of TPS have subsequently been found in other members of the eucalypt group, with 106 putatively functional TPS genes in E. globulus (Külheim et al. 2015) and 102 in Corymbia citriodora (Butler et al. 2018). In terpenoid-rich species, such as eucalypts, the TPS genes, as well as TPS pseudogenes, are usually found in genomic clusters with high sequence similarities (Külheim et al. 2015; Butler et al. 2018). Like other gene families involved in plant—environment interactions, TPS genes are thought to expand via lineage-specific tandem duplications in response to adaptive pressures (Tholl 2006; Hanada et al. 2008; Moore et al. 2014). The mono- and sesquiterpenoid producing TPS families, in particular, often undergo large expansions in gene numbers in some higher plants (Martin et al. 2010; Külheim et al. 2015). The large TPS gene families in eucalypts are thought to be a consequence of the need to evolve complex plant defences to allow these long-lived woody species to persist in a diverse range of often adverse habitats. Whether this lineage-specific expansion is a feature of eucalypts, or more broadly of the Myrtaceae is unknown.
The Melaleuceae provide an outgroup for phylogenetic and evolutionary studies of the Eucalypteae, and are arguably a model taxon for terpene biochemistry and genetics in the Myrtaceae. The essential oil distilled from the leaf of Melaleuca alternifolia (medicinal tea tree) is renowned for its therapeutic properties, and used in medicinal and cosmetic products throughout the world (Carson et al. 2006). The Australian tea tree industry produces around 1000 metric tonnes of oil per annum, of which 90% are exported to international markets (Larkman 2020). Australian plantations are based on genetically improved populations from a long-term breeding program (Doran et al. 1996; Baker et al. 2010), and a wealth of studies has investigated the chemistry, biochemistry, and genetics of the leaf oils in tea tree, including the identification and characterisation of key TSP (e.g. Shelton et al. 2002; Keszei et al. 2010b; Bustos-Segura et al. 2017; Padovan et al. 2017a). Genomic resources for the taxon were further enhanced with the first draft genome assembly for tea tree in 2018 and the first attempt to characterise its TPS gene family (Calvert et al 2018). Despite the highly diverse terpenoid compounds (~ 100) produced by this species (Brophy et al. 1989), and in contrast with the extraordinarily high number of TPS genes found in eucalypts, only 37 putatively functional TPS were found in tea tree (Calvert et al. 2018). But this first estimate used an assembly created from short Illumina paired-end reads and therefore may have underestimated the number of TPS, as some genes may have been overlooked due to genome fragmentation and incomplete gene models.
To more reliably characterise the TPS gene family for tea tree, a new draft assembly was generated using long reads as well as short reads to enhance completeness (Voelker et al. 2021a). This 362 Mb assembly was close in size to a flow cytometry estimate of 357 Mb (Calvert et al. 2018, 2021), and the scaffold N50 was increased by a factor of 214 in comparison with the earlier draft. Hence, this new draft was expected to permit a more reliable detection of genes and a more detailed analysis of gene organisation due to the larger scaffold size (N50 = 1.9 Mb). Although increased relative to the earlier estimate, our manual annotation of this new assembly revealed that the TPS gene family in M. alternifolia was still around half that of the average eucalypt TPS count, mainly due to reduction in the secondary metabolite producing subfamilies. New insights into the organisation of TPS in M. alternifolia indicated that, like other plants, the TPS in tea tree evolve in clusters via tandem duplication.
Methods
A brief description of methods is given below. A more detailed description of the methodology, including the use of software settings and commands, is available as protocol on protocols.io (Voelker 2023).
Manual TPS annotation
Genes in the tea tree genome assembly of reference genotype SCU01 (BioProject PRJNA702189) were predicted with Fgenesh++ v7.2.2 (Solovyev et al. 2006, RRID:SCR_018928) and GenemarkET+ v4.59 (Lomsadze et al. 2014, RRID:SCR_011930). The methods for the Fgenesh++ prediction are explained in Voelker et al. (2021a). For both prediction tools, the NCBI nr-protein plant database and RNA-seq reads (BioProject PRJNA388506) were aligned to the genome sequence and included as input in the gene prediction process. As further evidence for exon–intron borders of terpene synthases, TPS protein sequences were aligned to the genome assembly with Exonerate v2.4.0 (RRID:SCR_016088). First, protein sequences matching the ‘terpene synthase’ keyword search were retrieved from Phytozome v13 (accessed 20/05/2021) for Arabidopsis thaliana ARAPORT11 (Cheng et al. 2017), Corymbia citriodora v2.1 (Healey et al. 2021), Eucalyptus grandis v2.0 (Myburg et al. 2014), Populus trichocarpa v4.1 (Tuskan et al. 2006), and Vitis vinifera v2.1 (Jaillon et al. 2007). These sequences were then aligned to the genome using the protein2genome mode of Exonerate.
All lines of gene evidence, the Fgenesh++ prediction, GenemarkET+ prediction, spliced RNAseq alignments, and Exonerate TPS alignments, were visualised together with the genome assembly in the genome annotation editor Apollo (Dunn et al. 2019, RRID:SCR_001936). Manual adjustments of exon–intron borders were carried out where appropriate evidence for canonical splice sites was found in the alignments (Online Resource 2). Potential assembly errors were identified by examining alignments of the raw sequencing reads to the regions of interest. After finishing the manual annotation of TPS, they were named consecutively (MaltTPS001-MaltTPS73) based on their location on a scaffold, from lowest to highest scaffold number, and their location within the scaffold. Some putative TPS were later removed due to potential scaffold duplications, which were assessed by sequence identity of predicted TPS, and all-vs-all alignment of TPS-containing scaffolds. The final predicted protein sequences were screened for the presence of conserved amino acid motifs (RRx8W, DDxxD, RxR, and NSE/DTE), conserved C- and N-terminal TPS Pfam domains (PF03936, PF01397), as well as potential N-terminal chloroplast transit peptides (cTPs). Visualisations of gene structures and TPS organisation on a scaffold were created with the R-packages ggplot2 (Wickham 2016), gggenomes (Hackl and Ankenbrand 2022), gggenes (Wilkins 2020), and ggrepel (Slowikowski 2021).
Other genes potentially involved in terpenoid synthesis were identified by pathway mapping of the Fgenesh++ predicted protein sequences using Mercator4 v5.0 (Schwacke et al. 2019).
Phylogenetic tree construction
The predicted M. alternifolia TPS protein sequences were manually assessed in Unipro UGENE v39.0 (Okonechnikov et al. 2012, RRID:SCR_005579) and where possible, trimmed to the start of the RRx8W motif. Multiple sequence alignment (MSA) was carried out with the MUSCLE tool (RRID:SCR_011812) in UGENE, and columns with gaps > 75% were removed from the alignment. The trimmed MSA output was used as input for phylogenetic tree construction using PhyML v3.3 (Guindon et al. 2010, RRID:SCR_014629). The resulting gene family tree was visualised in iTOL v6 (Letunic and Bork 2021) and manually rooted at the node that separates subfamilies c and e/f (class I primary metabolite TPS) from a, b, and g (class III secondary metabolite TPS).
For comparison with other species, the TPS amino acid sequences for E. grandis and E. globulus (Külheim et al. 2015), C. citriodora (Butler et al. 2018), and V. vinifera (Martin et al. 2010) were retrieved from the respective published resources. The data from Külheim et al. (2015) also included sequences for A. thaliana and P. trichocarpa; however, many of these sequences contained gaps and seemed to be incomplete. Hence, it was decided to use the TPS identified by Jiang et al. (2019) for these species. Their methodology was similar to the method chosen for M. alternifolia, with TPS being classified as putatively functional when they contain both TPS Pfam domains. The gene names for these TPS were retrieved and used to extract the TPS amino acid sequences from the proteomes of A. thaliana ARAPORT11 and P. trichocarpa v3.0 from Phytozome v13. The sequences for all species were loaded into Unipro UGENE and where possible, trimmed to the start of the RRx8W motif, as already done for M. alternifolia. Next, MSA was carried out for all species, using MUSCLE and trimming the resulting alignment to remove all columns with > 75% gap representation. Phylogenetic tree construction was carried out with 100 bootstraps and the same settings as for the M. alternifolia tree. After the whole tree was visualised and annotated, different clades were exported into separated trees for a clearer visualisation of each subfamily.
Orthogroup analysis
In addition to a comparative study of TPS, the overall similarity of protein sequences among tea tree was assessed in comparison with other long-lived members of the rosids clade. Tea tree was expected to share more orthologues with eucalypts than the other rosids, with phylogenetic relationships mirroring the overall ordering of species in the TPS gene analysis. Protein sequences for primary transcripts of E. grandis v2, C. citriodora v2.1, P. trichocarpa v4, Salix purpurea v1 (Zhou et al. 2018), and V. vinifera v2.1 were obtained from Phytozome v13. For M. alternifolia, protein sequences predicted by Fgenesh + + and the manually predicted TPS sequences were used. All sequences were used to identify single-copy orthologues, i.e. orthogroups containing one orthologous gene per species, with all species present in the group. After this analysis in OrthoFinder v2.5.2 (Emms and Kelly 2019, RRID:SCR_017118), all single-copy orthologues were aligned, translated into coding sequences (CDS) using pal2nal v14 (Suyama et al. 2006), and pairwise substitution rate calculation within orthogroups was carried out with PAML codeml v4.10.3 (Yang 2007, RRID:SCR_014932). Pairwise synonymous substitution rates among species were visualised with ggplot2 (Wickham 2016) in R (R Core Team 2022). The synonymous substitution (Ks) mutation rate/site/year (R) between species was calculated with R = Ks/(2*divergence age) following the method by Healey et al. (2021).
Results
Manual TPS annotation
After manual annotation of the TPS in M. alternifolia, 73 gene models were initially declared as putatively functional and full length, plus a further 21 pseudogenes. However, upon further investigation of TPS sequence identities, and all-vs-all alignment of the scaffolds containing TPS, 15 full-length genes were removed as redundant sequences due to overlapping scaffolds or potential assembly errors. After removal of these sequences, a set of 58 putatively functional TPS sequences and 21 pseudogenes remained, with the following classification: I) 29 complete genes containing both TPS Pfam domains and RNAseq alignment evidence, II) 25 complete genes containing both TPS Pfam domains but no RNAseq evidence, III) four genes with both TPS Pfam domains and one premature stop codon or missing start codon, which might be a point mutation or assembly error, and IV) 21 pseudogenes containing only one TPS Pfam domain or multiple stop codons and frameshifts (see Online Resource 2 for gene annotation examples of all four classes).
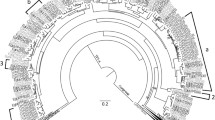
A phylogenetic tree was constructed from the aligned amino acid sequences of the final 58 TPS in class I, II, or III, which resulted in high bootstrap support values for clade separations (Fig. 1). The putatively functional TPS were classified into the subfamilies TPS-a (25), TPS-b (24), TPS-c (1), TPS-e/f (1 TPS-e and 3 TPS-f), and TPS-g (4). A second phylogenetic tree including translated sequences for the 21 pseudogenes was also generated (Online Resource 3). The pseudogenes were found to be similar to members of all subfamilies except TPS-c. The largest numbers of pseudogenes were classed into the secondary metabolite producing subfamilies TPS-a and TPS-b (Table 1a). The increased sequence variation due to the inclusion of pseudogenes introduced more uncertainty; hence, the bootstrap values were low for many clades. The grouping of genes into subfamilies, however, remained unchanged (Online Resource 3).

Maximum likelihood phylogenetic tree of the Melaleuca alternifolia TPS gene family. Nodepoints indicate bootstrap evidence ranging from 19 to 95. The larger the point, the stronger the bootstrap support, whereas nodes without point have a bootstrap support > 95. TPS subfamilies are labelled on the branches
As shown in Table 1a, more putatively functional TPS were identified in comparison with the previous study by Calvert et al. (2018). The number of members in the TPS-b2, TPS-c, TPS-e/f, and TPS-g subfamily remained the same, but the count of TPS-a and TPS-b1 TPS was increased by eleven and eight, respectively. Two TPS amino acid sequences, MaltTPS018 and MaltTPS070, were identical to TPS identified by Calvert et al. (2018) (MelG011320 and MelG016462, respectively). One further TPS, MaltTPS040, had only one amino acid differing from MelG016338 identified in the earlier study.
In addition to finding a higher number of TPS genes in the latest genome assembly, the overall completeness of the TPS sequences was also improved with the new predictions. Of the previous protein sequences predicted from the short-read assembly (Calvert et al. 2018), 15 of the 37 TPS were shorter than 400 amino acids (aa), and five of these short protein sequences contained only one of the two TPS Pfam domains. In contrast, all protein sequences from the newly annotated TPS were longer than 400 aa and had the N- and C-terminal Pfam domains. Three of the manually annotated TPS were still relatively short with a length of around 400–420 aa, but since they also contained the conserved amino acid motifs of class III TPS, we considered them as putatively functional.
Besides classifying the TPS based on the presence of Pfam domains, the predicted protein sequences were screened for the conserved N-terminal motif RRx8W and the C-terminal motifs RxR, DDxxD, and NSE/DTE. The highly conserved DDxxD motif and the less conserved NSE/DTE motif are usually located on opposite sides of the catalytic pocket of the enzyme (Christianson 2006; Degenhardt et al. 2009). Based on our annotation, all tea tree TPS-a members contained a conserved RxR and DDxxD motif, separated by exactly 34 amino acids in all but two predicted sequences. The RRx8W motif was found in all but one sequence, with the first two positions showing variable amino acids in 13 TPS-a. In all 24 TPS-b, the DDxxD motif was located exactly 34 amino acids upstream of the RxR motif. The RRx8W motif was encoded in all TPS-b but interrupted by a premature stop codon in one gene (Fig. 2).

Gene structure representation of putatively functional Melaleuca alternifolia TPS. Grey boxes show exons, and black lines indicate intron positions. Differently coloured shapes, as depicted in the legend, represent the conserved motifs RRx8W, RxR, and DDxxD. Premature stop codons are also shown for three genes, whereby sequence alignments suggested that these could be assembly errors. The TPS subfamily classification was determined by phylogenetic analysis. The TPS-a, TPS-b, and TPS-g subfamilies have six to eight exons, while the TPS-c and TPS-e/f have 12–14 exons. The RRx8W motif is usually encoded within the first exon. The gene visualisation was created with the R-package gggenomes (Hackl and Ankenbrand 2022)
Each of the TPS-g genes showed sequence similarities to one of the four shorter and most likely incomplete TPS-g identified by Calvert et al. (2018). Two TPS-g genes contained seven exons and the other two had six. All four TPS possessed both the conserved DDxxD motif, and the RRx8W motif, with the first two arginines being less conserved. In the sequence of one TPS-g protein, MaltTPS067, the distance of the RxR to the DDxxD motif was 55 aa instead of the usual 34 aa, and the motifs were encoded on the second exon of the gene. The gene might have lost some exons after duplication, leading to a shorter protein length of 426 aa. Nevertheless, all required motifs were present, and the encoding gene was still considered putatively functional.
The NSE/DTE motif was identified in 49 of the 53 TPS-a, TPS-b, and TPS-g amino acid sequences. The sequence pattern in the tea tree TPS was [LF][MNCTWGI][ND]D[LMIV]x[TGSM]xxxE (see sequence logo in Online Resource 4), which differed slightly from the reported consensus sequence of [LV][VLA][ND]D[LIV]x[ST]xxxE (Christianson 2006), and was similar to variations reported in Vitis vinifera (Martin et al. 2010).
The predicted TPS-f genes contained 12–13 exons and a resulting protein length of 725–845 aa. MaltTPS065 was predicted to be TPS-e and had 14 exons and a protein length of 784 aa. The DDxxD motif was encoded in the TPS-e/f members, but not in the TPS-c gene, which does not require this domain (Martin et al. 2010). The RRx8W motif was not well conserved in the genes of these three TPS clades, as can be seen in the gene structure visualisation in Fig. 2.
The location of TPS genes on scaffolds was also assessed. In 12 cases, a single TPS was predicted on a scaffold with no other TPS present. Some scaffolds, however, were long enough to provide evidence for TPS clustering. For example all TPS-g genes were located on one scaffold, together with two incomplete pseudogenes, and all three TPS-f genes were encoded on a single scaffold, together with two TPS-b genes further downstream. Another scaffold had 13 predicted putatively functional TPS-b1 genes (i.e. > 50% of all TPS-b), along with six additional TPS pseudogenes, arranged in tandem in a cluster of around 400 kb on a scaffold with an overall length of 720 kb (see later for further discussion). Overall, ten scaffolds were found to contain combinations of between two and five TPS genes.
A further quality control to test the completeness of the TPS gene models was a screen for chloroplast transit peptides (cTPs) in the predicted protein sequences. In plants, sesquiterpenes are produced in the cytosol by TPS-a proteins, while all remaining TPS syntheses occur in the chloroplast (Chen et al. 2011). Hence, a cTP should be present in most of the predicted TPS sequences, especially the monoTPS. However, cTPs display a lot of variation in their sequence length and structure (Lee et al. 2008), leading to difficulties in cTP predictions. In the tea tree TPS, 20 sequences were found to contain a cTP. However, seven TPS-b proteins were putatively allocated to the cytoplasm, indicating that the program either did not recognise the cTP target signal, or that these protein sequences are still incomplete or contain assembly errors. Calvert et al. (2018) reported only five predicted TPS containing a cTP in M. alternifolia, so in order to compare results based on the same methods, the 37 previously predicted tea tree TPS were also analysed, and only nine TPS could be located to the plastids. This implies that even though advancements in predicting transit peptides have been made, the notable increase in the number of predicted cTP sequences compared to the previous studies can be attributed to the improved gene models reported in our study. A more detailed description of the cTP screening results can be found in the supplementary material (Online Resource 5).
Identification of other genes involved in terpenoid biosynthesis
Genes that might be involved in the cytosolic mevalonate (MVA) pathway and plastidic methylerythritol phosphate (MEP) pathway, which both lead to the production of IPP and DMAPP, were also identified in this study (see Online Resource 1a and b for pathway information). Three genes that mapped to the MVA pathway were located on scaffolds with TPS annotations. Two genes encoding potential isopentenyl pyrophosphate isomerases (IPPI) were found. The encoded protein sequences had the same length as isopentenyl diphosphate isomerase (IDI) 1 (291 aa) and IDI2 (235 aa) that were identified for M. alternifolia from previously isolated cDNA clones by Shelton et al. (2004a). The sequences had high similarities, with 226 aa and 223 aa being identical with IDI1 and IDI2, respectively. Furthermore, the predicted protein with high similarity to IDI1 also contained a chloroplast targeting signal, confirming the findings by Shelton et al. (2004a) that IDI1 is responsible for the reversible isomerisation from IPP to DMAPP in the chloroplasts, while IDI2 has the same function in the cytosol. The IDI1-like gene was located on the same scaffold as the TPS-c gene MaltTPS071.
Comparative genomics
Melaleuca and eucalypts are angiosperms and belong to the family Myrtaceae. Molecular phylogenetic studies date the divergence of Myrtaceae from their sister group Vochysiaceae to 86.7–96.4 Ma, and the genera Eucalyptus and Melaleuca are thought to have a shared ancestry until 68 Ma (Thornhill et al. 2015). Based on characteristics of the least specialised members of the genus, Barlow (1988) inferred that Melaleuca originated as a tree in 'seasonally drowned habitats at the margins of tropical rainforests'. This hypothetical ancestor is believed to have evolved scleromorphous traits in the early to mid-Tertiary (23–65 Ma), which resulted in the radiation of the genus and expansion to other habitats. Nowadays, the majority of the genus encompasses small trees and shrubs that grow in wetland or periodically waterlogged habitats, with most species not being well adapted to dry conditions (Brophy et al. 2013).
Within the eucalypt group, the genus Eucalyptus is estimated to have diverged from the Corymbia and Angophora lineage around 52 Ma (Thornhill et al. 2015). In contrast with melaleucas, eucalypts dominate sclerophyllous forests and are specially adapted to survive in arid environments with frequently occurring fires (Hill et al. 2016). This distinction in their habitat may have evoked unique adaptive responses in eucalypts compared to the melaleucas and led to a divergence in the TPS gene family evolution between these genera. Whole genome sequences and manually annotated TPS gene families are available for Eucalyptus grandis (Myburg et al. 2014; Külheim et al. 2015) and Corymbia citriodora (Butler et al. 2017, 2018; Healey et al. 2021), which provides a good foundation for comparative genomic studies with M. alternifolia. Based on the estimated divergence times and phylogenies, M. alternifolia should still share a relatively high sequence conservation with the two reference eucalypts when compared to other woody angiosperms.
Prior to conducting phylogenetic analyses of the TPS family in different species, the overall similarity of sequences among tea tree and the eucalypts was assessed in comparison with other long-lived members of the rosids clade. The pairwise synonymous substitution rates (Ks) among single-copy orthologous genes that are shared by E. grandis, C. citriodora, M. alternifolia, Populus trichocarpa, Salix purpurea, and Vitis vinifera were investigated. In total, a set of 2488 orthogroups contained a single-copy orthologue from each species, and the pairwise Ks values among species were calculated for the sequences in each orthogroup before being summarised in a density plot.
Figure 3 shows the distribution of synonymous substitution rates among pairwise orthologue comparisons. C. citriodora and E. grandis orthologues have the lowest Ks values with a peak maximum of Ks = 0.1379. The Ks rates for orthologues among M. alternifolia and C. citriodora (peak max = 0.1839), as well as among M. alternifolia and E. grandis (peak max = 0.1891) show a highly similar distribution, which is consistent with expectations, since Melaleuca should share a common ancestor with Eucalyptus and Corymbia from before the eucalypt genera diverged. As expected, the pairwise Ks rates among the Myrtaceae are comparatively low relative to M. alternifolia and V. vinifera, which share increased synonymous substitutions. The highest Ks rates were observed when M. alternifolia was compared to P. trichocarpa and S. purpurea, which both belong to the Salicaceae and displayed corresponding Ks distributions. Overall, these observations corresponded with previous Ks rate investigations for C. citriodora (Healey et al. 2021). On the basis of the peak maxima of Ks distributions and the estimated divergence of melaleucas and eucalypts 68 Ma (Thornhill et al. 2015), the synonymous mutation rate per site per year among orthologous genes of M. alternifolia and E. grandis was predicted to be 1.390 × 10–9. Between M. alternifolia and C. citriodora, the same calculation led to an estimate of 1.3522 × 10–9 synonymous mutations (site/year). These values were in agreement with estimates for Salicaceae (Dai et al. 2014) and the eucalypts (Healey et al. 2021). The Ks peak maximum of 0.1379 for C. citriodora and E. grandis was slightly lower than the value of 0.1585 reported by Healey et al. (2021) but methodological differences in the selection of candidate genes might have led to this small discrepancy.

Pairwise synonymous substitution rates among single-copy orthologous genes. CC Corymbia citriodora; EG Eucalyptus grandis; MA Melaleuca alternifolia; PT Populus trichocarpa; SP Salix purpurea; and VV Vitis vinifera
In order to confirm the species’ relationships that should also be expected when investigating the TPS gene relatedness, a phylogenetic tree was created based on the sequence similarities of orthologous genes, which showed anticipated relationships among species. Melaleuca split from a common ancestor shared with Eucalyptus and Corymbia. The remaining rosids are on a separate clade with Salix and Populus being closely related, and Vitis located on a branch that splits from a common ancestor of the Salicaceae (Online Resource 6).
TPS subfamily representation among species
To assess the expansion or contraction of the M. alternifolia TPS subfamilies in comparison with other species, phylogenetic trees were constructed from the multiple sequence alignment of predicted tea tree TPS sequences and TPS proteins of A. thaliana, C. citriodora, E. globlulus, E. grandis, P. trichocarpa, and V. vinifera. The observed numbers of TPS in each subfamily (Table 1b) were consistent with the numbers reported in the previous studies for V. vinifera (Martin et al. 2010), E. grandis and E. globulus (Külheim et al. 2015), C. citriodora (Butler et al. 2018), and A. thaliana (Jiang et al. 2019). For P. trichocarpa (Jiang et al. 2019), however, the classification into subfamilies deviated from the previous findings. The difference was small, with two previous TPS-a members being classified as TPS-b in our study. Since these two subfamilies are structurally similar, a difference in alignment methodology might have led to these results.
Since the TPS in M. alternifolia were only classified as putatively functional if they contained both TPS Pfam domains (C– and N-terminal domain), it was also examined whether the TPS amino acid sequences published by Butler et al. (2018), Külheim et al. (2015), and Martin et al. (2010) contain these domains. Two TPS proteins in C. citriodora, three in E. globulus, and three in E. grandis were found to contain only one of the two TPS Pfam domains, and one further E. grandis protein contained no TPS Pfam domain at all. In V. vinifera, the number was considerably higher, with 16 proteins only encompassing one of the two TPS Pfam domains. For A. thaliana and P. trichocarpa, the methodology by Jiang et al. (2019) already took into account that a functional TPS should have both the C– and N-terminal Pfam domains, but since their study only annotated TPS from published proteomes for the respective species, some TPS might have been missed.
Phylogenetic tree construction and species comparison
The M. alternifolia TPS were found to be closely related to the other three Myrtaceae, C. citriodora, E. globulus, and E. grandis. No TPS cluster was found to be unique for tea tree, on each clade of the phylogenetic tree, the M. alternifolia TPS were closely related to TPS of the other Myrtaceae. Overall, the ordering of TPS in the phylogenetic tree showed the same relationships as the species tree inferred from Orthofinder (Online Resource 6). Melaleuca alternifolia TPS were more divergent to all eucalypt TPS than any among eucalypt comparisons. Within the eucalypt set, E. grandis and E. globulus were closer related, while Eucalyptus and Corymbia citriodora were more divergent (see Figs. 4, 5, and 6). These findings, although expected, confirmed the high quality of the manual TPS annotations for tea tree. Phylogenetic relationships for M. alternifolia TPS pseudogenes were also assessed and can be viewed in Online Resource 7 a–c.

Phylogeny of the TPS-a subfamily for Melaleuca alternifolia, Eucalyptus grandis, E globulus, Corymbia citriodora, Populus trichocarpa, Vitis vinifera, and Arabidopsis thaliana. MaltTPS069 (TPS-g) was used as outgroup. Circles on nodes indicate bootstrap support values ranging from 13 to 95. The larger the circle, the higher the bootstrap values. Nodes without circle have a bootstrap support > 95. O1–O3 show examples of orthologous pairs, and a-c are examples discussed in the text

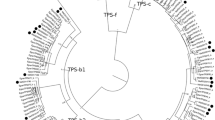
Phylogeny of the TPS-b and TPS-g subfamilies. MaltTPS004 (TPS-a) was used as outgroup. The clade labelled with a, as well as the orthologous groups O1–O4 are discussed further in the text. Circles on nodes indicate bootstrap support values ranging from 7 to 95. The larger the circle, the higher the bootstrap values. Nodes without circle have a bootstrap support > 95

Phylogeny of the TPS-c, TPS-e, and TPS-f subfamilies. MaltTPS036 (TPS-b) was used as outgroup. The labels O1–O3 refer to examples mentioned in the results. Circles on nodes indicate bootstrap support values ranging from 27 to 95. The larger the circle, the higher the bootstrap values. Nodes without circle have a bootstrap support > 95
Following the convention of Butler et al. (2018), the term orthologous gene pairs will be used in the subsequent results if the TPS gene of one species is more closely related to a gene of another species than to a gene within its own genome. In the TPS-a subfamily, M. alternifolia had only one orthologous pair between MaltTPS018 and CorciTPS036 (Fig. 4, O1). The two cases of orthologous pairing that were previously reported for C. citriodora and Eucalyptus (Butler et al. 2018) were also observed in this analysis, with no direct pairing to M. alternifolia genes (Fig. 4, O2 and O3). The notable TPS cluster containing EglobTPS022 and multiple C. citriodora TPS (Butler et al. 2018) was also displayed in the phylogenetic tree. While no orthology to any E. grandis TPS genes existed, three M. alternifolia TPS were found to belong to this clade (Fig. 4a). Some TPS-a clades have seen expansions in specific species. For example one cluster showed considerable expansion in C. citriodora (Fig. 4b). This expansion in comparison with the Eucalyptus species has already been mentioned by Butler et al. (2018). M. alternifolia TPS were also present in this clade, and even though they were not as numerous as in C. citriodora, they had two more members than Eucalyptus spp. However, in many cases, M. alternifolia TPS were underrepresented or missing completely from a clade (e.g. Figure 4c).
As previously reported by Butler et al. (2018), the E. globulus and E. grandis TPS genes are often in orthologous pairs (e.g. Figure 5, O4). In contrast, only two orthologous pairs were found between Eucalyptus and C. citriodora in the TPS-b subfamily and none for TPS-g. We observed the same orthologous genes that were already reported for C. citriodora, and M. alternifolia was included in these orthologous clusters (Fig. 5, O1 and O2). An additional orthologous pair was found between MaltTPS042 and CorciTPS080 (Fig. 5, O3), whereas the Eucalyptus TPS were expanded in this clade. A potential gene loss in E. grandis (Butler et al. 2018) was also detected (Fig. 5, a). Only one E. globulus gene was member of this clade, whereas M. alternifolia had two, and C. citriodora had three TPS. The TPS-b2 subfamily seems to be contracted in M. alternifolia compared to the other three Myrtaceae. While only two TPS-b2 members were found in M. alternifolia, which is the same as for P. trichocarpa, we observed nine TPS-b2 genes for E. grandis and ten for E. globulus and C. citriodora, respectively. The TPS-g family has also seen a larger expansion in the other three Myrtaceae than in M. alternifolia, and orthologous pairs were only observed between E. grandis and E. globulus.
In a concordance with previously reported findings, the single C. citriodora gene in the TPS-c subfamily formed an orthologous group with Eucalyptus (Fig. 6, O1) (Butler et al. 2018). The M. alternifolia TPS-c gene was located on the same clade. The Eucalyptus spp. TPS-c subfamily was slightly expanded, with an additional orthologous pair between E. globulus and E. grandis.
In the TPS-e subfamily, the phylogeny was similar to the results reported by Butler et al. (2018). MaltTPS065 occurred in an orthologous group with CorciTPS089, EgranTPS093, and EglobTPS116 (Fig. 6, O2), and the TPS-e has seen a slight expansion in E. globulus and E. grandis relative to C. citriodora and M. alternifolia. The TPS-f subfamily was also more expanded in the Eucalyptus species compared to the other two Myrtaceae. Only one orthologous pair was observed between E. grandis and E. globulus (Fig. 6, O3), which was unexpected based on previous observations for this subfamily (Külheim et al. 2015). Butler et al. (2018) also reported their TPS-f subfamily results to slightly differ from the phylogeny presented by Külheim et al. (2015) for eucalypts. Low bootstrap supports for some of the clades might have contributed to these differences.
Identification of specific terpene synthases
Based on the phylogenetic tree construction, only two M. alternifolia TPS gene products were identified as potential TPS-b2 proteins (Fig. 5). As shown in Table 2, one of these proteins, MaltTPS043, showed high sequence similarity to a tea tree TPS that was initially annotated as potential monoterpene synthase (GenBank accession AY279379.1; Shelton et al. (2004b)) but was later shown to share conserved amino acids with isoprene synthases (Sharkey et al. 2005). The phylogenetic tree also showed MaltTPS043 to be an orthologue of the functionally characterised isoprene synthase of E. globulus (Fig. 5, O2) (Sharkey et al. 2013). To investigate whether the potential isoprene synthase contained known conserved amino acids, the sequences of the two TPS-b2 proteins MaltTPS042 and MaltTPS043 were aligned to the kudzu isoprene synthase (Sharkey et al. 2005). MaltTPS043 contained the conserved residues Phe-343, Gly-453, and Phe-493, but had a Thr at position Cys-496 (numbering is based on the kudzu amino acid positions). These observations coincide with the findings by Sharkey et al. (2005) and indicate that MaltTPS043 could encode an isoprene synthase. MaltTPS042, on the other hand, only shared the Gly-453 residue with the kudzu isoprene synthase. Calvert et al. (2018) also predicted two TPS-b2 genes to be present in the tea tree genome and considered the second TPS-b2 gene product to be a potential ocimene synthase.
The TPS-b1 family, which is responsible for the production of monoterpenoids, is of special interest considering that tea tree oil gets its medicinal properties from terpinen-4-ol and other monoterpenoids that have to meet specified proportions in commercial essential oils (ISO 4730: 2017 Essential oil of Melaleuca, terpinen-4-ol type (Tea Tree oil)). Terpinen-4-ol is not a direct product of a TPS, but is generated from non-enzymatic conversion of the monoterpene sabinene hydrate (Cornwell et al. 1999). Padovan et al. (2017a) have analysed transcriptomes from different M. alternifolia chemotypes and functionally characterised one terpinolene synthase (MaTPS-Tln), one sabinene hydrate synthase (MaTPS-SaH), and two 1,8-cineole synthase genes (MaTPS-CinA and MaTPS-CinB). Furthermore, they observed a strong correlation between the expression of MaltTPS-SaH, MaTPS-Tln, and MaTPS-CinA, and the terpenoids terpinen-4-ol, terpinolene, and 1,8-cineole, respectively. These findings were consistent with the suggestion by Keszei et al. (2010b) that as few as three enzymes might be responsible for the creation of chemotypes in tea tree. However, they also observed that terpenoid profiles correlated with the expression of other, as yet uncharacterised putative TPS, indicating that more than the 3 functionally characterised TPS could be involved in the formation of chemotypes in M. alternifolia.
By creating multiple sequence alignments of the newly predicted TPS to sequences obtained from Padovan et al. (2017a), we were able to identify four TPS with high sequence similarity to conserved amino acids of MaTPS-Tln, MaTPS-SaH, and MaTPS-Cin (Table 2). All four TPS that putatively account for chemotype grouping in tea tree are located in tandem within a region of around 87 kb on one scaffold. This scaffold, scf7180000030749, has a total length of 731,646 bp and contains genes for 13 TPS proteins (MaltTPS019–MaltTPS031), as well as TPS pseudogenes (Online Resource 8). MaltTPS020 had 98% amino acid identity with sequences conserved in MaTPS-Tln, MaltTPS021 had 100% similarity with MaTPS-SaH amino acids, and MaltTPS023 and MaltTPS025 were 99% and 100%, identical with regions characteristic of MaTPS-Cin, respectively. Among the TPS encoded on scf7180000030749, MaltTPS019 and MaltTPS026 also had high similarities to the SaH and Cin synthases (Table 2). This was in concordance with the previous findings supposing that one more TPS might have a similar product profile to MaTPS-Tln, one more TPS was likely to produce 1,8-cineole and related compounds, and another TPS was similar to MaTPS-SaH (Padovan et al. 2017a). Although these enzymes would have to be functionally characterised in order to get a definitive answer to this theory, the phylogenetic results of our analysis give weight to this hypothesis. MaltTPS019 was placed on a sister branch to the sabinene hydrate synthase MaltTPS021, MaltTPS026 was closely related to the two 1,8-cineole synthases, and two TPS (MaltTPS008 and MaltTPS056) were similar to the putative terpinolene synthase (Fig. 1).
Discussion
TPS occur in tandem arrays in Melaleuca alternifolia
This manual annotation of a scaffold-scale assembly of the tea tree genome revealed that genes and pseudogenes from the same TPS subfamily with high sequence similarities were frequently located in close proximity on the same scaffold. This pronounced clustering of TPS genes into tandem arrays in tea tree paralleled the tandem clusters found in E. grandis and C. citriodora (Külheim et al. 2015; Butler et al. 2018), and supported the conclusion that duplication through unequal crossing over, and subsequent sub- or neo-functionalisation was a key mechanism underpinning the evolution of TPS in M. alternifolia. The mechanism of tandem duplication is believed to contribute to the adaptive diversification of genes involved in stress responses, such as the TPS, as they are more likely to be retained following duplication due to adaptive pressures (Hanada et al. 2008).
A large cluster of 13 TPS-b genes on scf7180000030749 served as an exemplar for the investigation of tandem duplication of TPS genes in tea tree with a maximum of two non-TPS genes interspersed among TPS gene pairs of MaltTPS019-MaltTPS028. Moreover, four TPS genes, including the putative terpinolene, sabinene hydrate, and one 1,8-cineole synthase (MaltTPS020-MaltTPS023), occurred in a single contiguous tandem array (Online Resource 8). Taken together with the high sequence similarity between the sabinene hydrate synthase and the two 1,8-cineole synthases, as well as their grouping on one clade of the phylogenetic tree (Fig. 1), this finding supported a hypothesis that the sabinene hydrate synthase evolved by gene duplication and subsequent neo-functionalisation of a more ancestral 1,8-cineole synthase (Keszei et al. 2010c; Padovan et al. 2017a).
The clustering of pseudo-TPS with putatively functional TPS genes further supported tandem duplication as a mechanism of TPS evolution. Together with the 13 TPS-b genes on scf7180000030749, six additional TPS pseudogenes were located to the same scaffold, suggesting that this region of the genome is a hot spot for non-homologous recombination. While some duplicated genes can obtain new functions through sub- or neo-functionalisation, most functionally redundant genes will be lost if they are not subject to selection (Lynch and Conery 2000). In this process of pseudogenisation, one copy of the gene usually accumulates mutations and becomes functionless, eventually accumulating too many mutations to be identifiable (Zhang 2003).
Divergent adaptive histories may account for TPS subfamily differences in eucalypts and Melaleuca
All angiosperm TPS subfamilies are represented in M. alternifolia but variations in the size of certain subfamilies relative to the other Myrtaceae were observed. The largest differences were evident in subfamilies that produce secondary metabolites, and thus are likely to be subject to adaptive pressures. For example eucalypts had around twice as many TPS-a (sesquiterpenoid) genes (Table 1) compared to M. alternifolia. Even if TPS-a pseudogenes are considered (Online Resource 3, Online Resource 7 a), this subgroup in tea tree is unlikely to have had the same significance historically as it had in extant eucalypts. Many sesquiterpenoids are important signalling molecules mediating plant—insect or plant—pathogen interactions by attracting pollinators (Pichersky and Gershenzon 2002), acting as phytoalexins (Vögeli and Chappell 1988) or defence against insect herbivores (Yuan et al. 2008).
Eucalypts also had a higher number of TPS-b2 than M. alternifolia (Table 1). Enzymes of this subfamily catalyse the formation of the hemiterpene isoprene and the acyclic monoterpenes β-ocimene and myrcene (Sharkey et al. 2013), molecules shown to have roles in biotic and abiotic stress responses (Fäldt et al. 2003; Velikova and Loreto 2005). Eucalyptus grandis, for example possesses a total of nine TPS-b2, including a large cluster of eight TPS-b2 genes located within 107 kb on pseudo-chromosome 11 of its genome (Külheim et al. 2015; Calvert et al. 2018). While all the TPS-b2 genes in tea tree were also located together on a single scaffold separated by around 21.5 kb, there were only two full-length candidates (MaltTPS042 and MaltTPS043), a putative isoprene and ocimene synthase for this subfamily. This suggested that M. alternifolia may not have experienced the same biotic and abiotic adaptive pressures to expand its TPS-b2 subfamily as the eucalypts.
Biotic factors
Tandem duplication and adaptation to biotic stresses such as herbivory are likely to have been drivers of TPS subfamily expansion in both lineages of these long-lived trees, but the degree of expansion may have been a consequence of differences in the guilds of insects, pathogens, and mammals utilising each lineage. Evidence of coevolution in the hosts and their dependants has been found in both lineages. A number of components of both eucalyptus oil and tea tree oil exhibit antibacterial and antifungal properties (Carson et al. 2006; Hendry et al. 2009). Furthermore, eucalypts and tea tree both support a myriad of insects that feed on or otherwise utilise the foliage for shelter, or pollinate the flowers. Members of the Psyllidae and Chrysomelidae (e.g. Paropsisterna tigrina) utilise tea tree as host (Campbell and Maddox 1999). Some have been shown to have developed mechanisms to metabolise its terpenoids (Southwell et al. 1995), and exhibit selective herbivory corresponding with tea tree chemotypes, suggesting long-term associations (Bustos-Segura et al. 2015). Similar evidence of coevolution has been found among insect herbivores of some eucalypts (Edwards et al. 1993; Stone and Bacon 1994). Mammal browsers of eucalypts, such as the marsupial koalas (Phascolarctos cinereus) and common ringtail possums (Pseudocheirus peregrinus), also show feeding behaviours influenced by eucalypt leaf chemistry (Lawler et al. 2000; Moore et al. 2005), and adaptations to digest terpenoids (McLean and Foley 1997; Pass et al. 2002). Similar evidence of utilisation by mammals has not been shown for tea tree, and this may be a point of difference between the lineages.
Abiotic factors
Other differences in the historic environments in which the two lineages evolved likely also contributed to the divergence in TPS gene family compositions. The Myrtaceae originated during the late Cretaceous (Thornhill et al. 2015), and according to phylogenetic studies by Crisp et al. (2011), the most recent common ancestor between Melaleuceae and Eucalypteae was most likely a rainforest tree occurring in wet environments. The eucalypt progenitor is thought to have diverged from the Melaleucae lineage around 68 Ma and emerged from its rainforest environment to inhabit drier habitats. The ancestral eucalypts, therefore, evolved in drier ecosystems with frequent fires (Hill et al. 1999), with present-day species showing constellations of features associated with adaptation to resource-limiting conditions (Keith 1997).
Plants adapted to low-resource environments are thought to be slower growing than resource-rich species, as they place more emphasis on defending precious photosynthetic organs and toughing out challenging conditions by relying on energy storage (e.g. lignotubers and root tubers) (Coley et al. 1985; Chapin et al. 1993). Resource-rich species, on the other hand, are thought to be adapted to exploit moisture and fertile conditions for rapid growth required to outcompete their neighbours, and tend to invest less energy in defence. A richer terpenoid chemistry that provides protection from predation as well as resilience to abiotic stress for eucalypts therefore is consistent with a greater prominence of aridity and nutrient limitations in their evolutionary history relative to Melaleuca.
Such an emphasis on adaptation to aridity may have been particularly important for TPS-b2 expansions. Eucalyptus spp., for instance, are known to emit exceptionally high amounts of isoprene (Benjamin et al. 1996; He et al. 2000), especially under higher temperatures (Guidolotti et al. 2019), which increases a plant’s tolerance to heat and water stress. Even though the exact mechanisms still require further investigation, isoprene emissions have been shown to confer thermotolerance by protecting leaves against high temperatures (Velikova and Loreto 2005), and protect plants against damage from reactive oxygen species (Velikova et al. 2004). Another TPS-b2 product, β-ocimene, could also play a role in mitigating abiotic stresses. For example Faralli et al. (2020) reported that transgenic Arabidopsis plants that constitutively emit ocimene and were subject to soil water stress displayed non-conservative behaviour with late stomata closures. They proposed that this might be a beneficial trait for plants in dry environments with limited resources, enabling them to take up water and nutrients under adverse conditions, thus gaining an advantage over competing species.
In contrast with eucalypts, where the most parsimonious explanation for the significant evolution of terpene diversity has been one of the overall expansions, the history of the Melaleuceae lineage may have been more complex and multi-directional, with the progenitors of Melaleuceae keeping stronger ties to the wetter environments of their ancestral origins, yet nonetheless experiencing periods of aridity (Crisp et al. 2011). Barlow (1988) hypothesised that the ancestral melaleucas and eucalypts evolved in distinct habitats, with Melaleuca spp. growing in wet areas, and eucalypts in drier environments with the regular occurrence of fires. Subsequent evolution and diversification, especially during the cyclic aridity of the Quaternary, led to the ecological coexistence that we observe in some members of these genera today. More recently, Thornhill et al. (2015) concluded that the last common ancestor of Melaleuceae and its sister tribe Osbornieae must have been a wetland plant, based on the fact that Osbornia grows in mangrove forests, and Melaleuca spp. often occur in swampy habitats. A history for the ancestors of M. alternifolia with periods of aridity but overall more benign, resource-rich habitats than eucalypts, is consistent with an overall less expanded TPS gene family relative to the eucalypt lineage, but one nonetheless that may carry signatures of past expansion and contraction in a legacy of pseudogenes.
Outlook
The terpene synthase gene family is of special interest to the tea tree industry because of its role in essential oil production. Six chemotypes have been identified in M. alternifolia based on the composition of three main terpenoids (Homer et al. 2000). Of these, one chemotype is dominated by terpinen-4-ol, one by 1,8-cineole, and one by terpinolene, whereas the other three non-cardinal types have intermediate levels of each of the three major constituents in different combinations. Only one chemotype, however, produces the commercially valuable high terpinen-4-ol oil (Homer et al. 2000). Chemotypes have been shown to be highly heritable (Shelton et al. 2002) and segregate within bi-parental crosses. The capacity to fix populations for a chemotype within three breeding cycles suggests major gene effects underly these categorical phenotypes. Our finding that the TPS genes putatively controlling terpinolene, sabinene hydrate, and 1,8-cineole synthesis are located in close proximity (within 87 kb) on one scaffold provides a possible explanation for the apparent categorical inheritance of these chemical attributes if the region behaves as a single locus. The availability of an improved draft genome sequence for tea tree (Voelker et al. 2021a) and the TPS annotation reported here are valuable resources for the tea tree breeding program.
Taken all together, this new information could be applied in molecular breeding to identify polymorphisms in the TPS genes that relate to variation in terpenoid profiles, and to investigate the genetic foundation of the six different oil chemotypes in tea tree. Similar approaches have already been used to associate genetic variants with traits connected to terpenoid concentration or oil yield in Eucalyptus sp. (Padovan et al. 2017b; Kainer et al. 2019). Finally, the release of a chromosome-scale genome assembly for M. alternifolia (Zheng et al. 2022) during the preparation of this work will provide a further opportunity to explore larger scale syntenic blocks shared by eucalypts and Melaleuca in relation to the TPS gene organisation among Myrtaceae in the future.
Data availability statement
A more detailed description of the methods and computational commands has been summarised on protocol.io (Voelker 2023). The M. alternifolia genome sequence and accompanying raw-read data are available under NCBI BioProject PRJNA702189 (GenBank assembly accession: GCA_019926035.1). The Fgenesh++ gene prediction and other related data are deposited in the GigaScience Database (Voelker et al. 2021b). All supplementary text and figures mentioned in the article are available in the supplementary material (Online Resource 1 – 8), and other supplementary files contain further information about the annotated genes or proteins, e.g. GFF3 and FASTA files (see Information on Electronic Supplementary Material). DNA sequences for annotated TPS genes can be retrieved with the GFF3 file in Online Resource 10 using the genome assembly in the above-mentioned NCBI BioProject.
References
Baker GR, Doran J, Williams ER, Olesen TD (2010) Improved tea tree varieties for a competitive market. RIRDC Research Paper Publication No. 10/188. RIRDC, Barton
Barlow BA (1988) Patterns of differentiation in tropical species of Melaleuca L. (Myrtaceae). In: Kitching RL (ed) The ecology of Australia’s wet tropics: Proceedings of a symposium held at the University of Queensland. Surrey Beatty & Sons for the Ecological Society of Australia, New South Wales
Benjamin MT, Sudol M, Bloch L, Winer AM (1996) Low-emitting urban forests: a taxonomic methodology for assigning isoprene and monoterpene emission rates. Atmos Environm 30:1437–1452. https://doi.org/10.1016/1352-2310(95)00439-4
Beran F, Rahfeld P, Luck K, Nagel R, Vogel H, Wielsch N, Irmisch S, Ramasamy S, Gershenzon J, Heckel DG, Köllner TG (2016) Novel family of terpene synthases evolved fromtrans-isoprenyl diphosphate synthases in a flea beetle. Proc Natl Acad Sci USA 113:2922–2927. https://doi.org/10.1073/pnas.1523468113
Block AK, Vaughan MM, Schmelz EA, Christensen SA (2019) Biosynthesis and function of terpenoid defense compounds in maize (Zea mays). Planta 249:21–30. https://doi.org/10.1007/s00425-018-2999-2
Bohlmann J, Crock J, Jetter R, Croteau R (1998) Terpenoid-based defenses in conifers: cDNA cloning, characterization, and functional expression of wound-inducible (E)-α-bisabolene synthase from grand fir (Abies grandis). Proc Natl Acad Sci USA 95:6756–6761. https://doi.org/10.1073/pnas.95.12.6756
Brophy J, Davies NW, Southwell IA, Stiff IA, Williams LR (1989) Gas chromatographic quality control for oil of Melaleuca terpinen-4-ol type (Australian tea tree). J Agric Food Chem 37:1330–1335. https://doi.org/10.1021/jf00089a027
Brophy J, Craven L, Doran J (2013) Melaleucas: their botany, essential oils and uses. Australian Centre for International Agricultural Research ACIAR Monograph No. 156. ACIAR, Canberra
Bustos-Segura C, Külheim C, Foley W (2015) Effects of terpene chemotypes of Melaleuca alternifolia on two specialist leaf beetles and susceptibility to Myrtle rust. J Chem Ecol 41:937–947. https://doi.org/10.1007/s10886-015-0628-0
Bustos-Segura C, Padovan A, Kainer D, Foley WJ, Kulheim C (2017) Transcriptome analysis of terpene chemotypes of Melaleuca alternifolia across different tissues. Pl Cell Environm 40:2406–2425. https://doi.org/10.1111/pce.13048
Butler JB, Vaillancourt RE, Potts BM, Lee DJ, King GJ, Baten A, Shepherd M, Freeman JS (2017) Comparative genomics of Eucalyptus and Corymbia reveals low rates of genome structural rearrangement. BMC Genomics 18:397. https://doi.org/10.1186/s12864-017-3782-7
Butler JB, Freeman JS, Potts BM, Vaillancourt RE, Grattapaglia D, Silva-Junior OB, Simmons BA, Healey AL, Schmutz J, Barry KW, Lee DJ, Henry RJ, King GJ, Baten A, Shepherd M (2018) Annotation of the Corymbia terpene synthase gene family shows broad conservation but dynamic evolution of physical clusters relative to Eucalyptus. Heredity 121:87–104. https://doi.org/10.1038/s41437-018-0058-1
Calvert J, Baten A, Butler J, Barkla B, Shepherd M (2018) Terpene synthase genes in Melaleuca alternifolia: comparative analysis of lineage-specific subfamily variation within Myrtaceae. Pl Syst Evol 304:111–121. https://doi.org/10.1007/s00606-017-1454-3
Calvert J, Baten A, Butler J, Barkla B, Shepherd M (2021) Correction to: terpene synthase genes in Melaleuca alternifolia: comparative analysis of lineage-specific subfamily variation within Myrtaceae. Pl Syst Evol 307:15. https://doi.org/10.1007/s00606-021-01740-9
Campbell AJ, Maddox CDA (1999) Insect pests of tea tree: can plantation pests be managed? In: Lowe R (ed) Southwell I Tea Tree. The Genus Melaleuca. CRC Press, London
Cao R, Zhang Y, Mann FM, Huang C, Mukkamala D, Hudock MP, Mead ME, Prisic S, Wang K, Lin F-Y, Chang T-K, Peters RJ, Oldfield E (2010) Diterpene cyclases and the nature of the isoprene fold. Proteins Struct Funct Bioinform 78:2417–2432. https://doi.org/10.1002/prot.22751
Carson CF, Hammer KA, Riley TV (2006) Melaleuca alternifolia (Tea Tree) oil: a review of antimicrobial and other medicinal properties. Clin Microbiol Rev 19:50–62. https://doi.org/10.1128/cmr.19.1.50-62.2006
Chapin FS, Autumn K, Pugnaire F (1993) Evolution of suites of traits in response to environmental stress. Amer Naturalist 142:S78–S92
Chen F, Tholl D, Bohlmann J, Pichersky E (2011) The family of terpene synthases in plants: a mid-size family of genes for specialized metabolism that is highly diversified throughout the kingdom. Plant J 66:212–229. https://doi.org/10.1111/j.1365-313X.2011.04520.x
Cheng CY, Krishnakumar V, Chan AP, Thibaud-Nissen F, Schobel S, Town CD (2017) Araport11: a complete reannotation of the Arabidopsis thaliana reference genome. Plant J 89:789–804. https://doi.org/10.1111/tpj.13415
Christianson DW (2006) Structural biology and chemistry of the terpenoid cyclases. Chem Rev 106:3412–3442. https://doi.org/10.1021/cr050286w
Coley PD, Bryant JP, Chapin FS (1985) Resource availability and plant antiherbivore defense. Science 230:895–899. https://doi.org/10.1126/science.230.4728.895
Cornwell CP, Leach DN, Wyllie SG (1999) The origin of terpinen-4-ol in the steam distillates of Melaleuca argentea, M. dissitiflora and M. linariifolia. J Essential Oil Res 11:49–53. https://doi.org/10.1080/10412905.1999.9701068
Crisp MD, Burrows GE, Cook LG, Thornhill AH, Bowman DMJS (2011) Flammable biomes dominated by eucalypts originated at the cretaceous-palaeogene boundary. Nat Commun 2:193. https://doi.org/10.1038/ncomms1191
Dai X, Hu Q, Cai Q, Feng K, Ye N, Tuskan GA, Milne R, Chen Y, Wan Z, Wang Z, Luo W, Wang K, Wan D, Wang M, Wang J, Liu J, Yin T (2014) The willow genome and divergent evolution from poplar after the common genome duplication. Cell Res 24:1274–1277. https://doi.org/10.1038/cr.2014.83
Degenhardt J, Köllner TG, Gershenzon J (2009) Monoterpene and sesquiterpene synthases and the origin of terpene skeletal diversity in plants. Phytochemistry 70:1621–1637. https://doi.org/10.1016/j.phytochem.2009.07.030
Doran JC, Baker G, Murtagh GJ, Southwell IA (1996) Breeding and selection of Australian Tea Tree for improved oil yield and quality. RIRDC Research Paper publication No. DAN-87A. RIRDC, Barton
Dudareva N, Pichersky E, Gershenzon J (2004) Biochemistry of plant volatiles. Pl Physiol 135:1893–1902. https://doi.org/10.1104/pp.104.049981
Dunn NA, Unni DR, Diesh C, Munoz-Torres M, Harris NL, Yao E, Rasche H, Holmes IH, Elsik CG, Lewis SE (2019) Apollo: Democratizing genome annotation. PLoS Comp Biol 15:1006790. https://doi.org/10.1371/journal.pcbi.1006790
Edwards PB, Wanjura WJ, Brown WV (1993) Selective herbivory by Christmas beetles in response to intraspecific variation in Eucalyptus terpenoids. Oecologia 95:551–557. https://doi.org/10.1007/BF00317440
Ehrlich PR, Raven PH (1964) Butterflies and plants: a study in coevolution. Evolution 18:586–608. https://doi.org/10.1111/j.1558-5646.1964.tb01674.x
Emms DM, Kelly S (2019) OrthoFinder: phylogenetic orthology inference for comparative genomics. Genome Biol 20:238. https://doi.org/10.1186/s13059-019-1832-y
Fäldt J, Arimura G-i, Gershenzon J, Takabayashi J, Bohlmann J (2003) Functional identification of AtTPS03 as (E)-β-ocimene synthase: a monoterpene synthase catalyzing jasmonate- and wound-induced volatile formation in Arabidopsis thaliana. Planta 216:745–751. https://doi.org/10.1007/s00425-002-0924-0
Faralli M, Li M, Varotto C (2020) Shoot characterization of isoprene and ocimene-emitting transgenic arabidopsis plants under contrasting environmental conditions. Plants 9:477. https://doi.org/10.3390/plants9040477
Fürstenberg-Hägg J, Zagrobelny M, Bak S (2013) Plant defense against insect herbivores. Int J Molec Sci 14:10242–10297
Grattapaglia D, Vaillancourt RE, Shepherd M, Thumma BR, Foley W, Külheim C, Potts BM, Myburg AA (2012) Progress in Myrtaceae genetics and genomics: eucalyptus as the pivotal genus. Tree Genet Genom 8:463–508. https://doi.org/10.1007/s11295-012-0491-x
Guidolotti G, Pallozzi E, Gavrichkova O, Scartazza A, Mattioni M, Loreto F, Calfapietra C (2019) Emission of constitutive isoprene, induced monoterpenes, and other volatiles under high temperatures in Eucalyptus camaldulensis: a 13C labelling study. Pl Cell Environm 42:1929–1938. https://doi.org/10.1111/pce.13521
Guindon S, Dufayard J-F, Lefort V, Anisimova M, Hordijk W, Gascuel O (2010) New algorithms and methods to estimate maximum-likelihood phylogenies: assessing the performance of PhyML 3.0. Syst Biol 59:307–321. https://doi.org/10.1093/sysbio/syq010
Gutbrod K, Romer J, Dörmann P (2019) Phytol metabolism in plants. Prog Lipid Res 74:1–17. https://doi.org/10.1016/j.plipres.2019.01.002
Hackl T, Ankenbrand MJ, (2022) gggenomes: a grammar of graphics for comparative genomics, R package version 0.9.5.9000. Available at: https://github.com/thackl/gggenomes
Hanada K, Zou C, Lehti-Shiu MD, Shinozaki K, Shiu SH (2008) Importance of lineage-specific expansion of plant tandem duplicates in the adaptive response to environmental stimuli. Pl Physiol 148:993–1003. https://doi.org/10.1104/pp.108.122457
He C, Murray F, Lyons T (2000) Monoterpene and isoprene emissions from 15 Eucalyptus species in Australia. Atmos Environm 34:645–655. https://doi.org/10.1016/S1352-2310(99)00219-8
Healey AL, Shepherd M, King GJ, Butler JB, Freeman JS, Lee DJ, Potts BM, Silva-Junior OB, Baten A, Jenkins J, Shu S, Lovell JT, Sreedasyam A, Grimwood J, Furtado A, Grattapaglia D, Barry KW, Hundley H, Simmons BA, Schmutz J, Vaillancourt RE, Henry RJ (2021) Pests, diseases, and aridity have shaped the genome of Corymbia citriodora. Commun Biol 4:537. https://doi.org/10.1038/s42003-021-02009-0
Hendry ER, Worthington T, Conway BR, Lambert PA (2009) Antimicrobial efficacy of eucalyptus oil and 1,8-cineole alone and in combination with chlorhexidine digluconate against microorganisms grown in planktonic and biofilm cultures. J Antimicrob Chemother 64:1219–1225. https://doi.org/10.1093/jac/dkp362
Hill RS, Beer YK, Hill KE, Maciunas E, Tarran MA, Wainman CC (2016) Evolution of the eucalypts; an interpretation from the macrofossil record. Austral J Bot 64:600–608. https://doi.org/10.1071/BT16117
Hill RS, Truswell EM, McLoughlin S, Dettman ME (1999) The evolution of the Australian flora: fossil evidence. In: Orchard AE (ed) Flora of Australia Introduction, vol. 1. ABRS/CSIRO, Melbourne, pp 251–320
Homer LE, Leach DN, Lea D, Slade Lee L, Henry RJ, Baverstock PR (2000) Natural variation in the essential oil content of Melaleuca alternifolia Cheel (Myrtaceae). Biochem Syst Ecol 28:367–382. https://doi.org/10.1016/S0305-1978(99)00071-X
Jaillon O, Aury J-M, Noel B, Policriti A, Clepet C, Casagrande A, Choisne N, Aubourg S, Vitulo N, Jubin C, Vezzi A, Legeai F, Hugueney P, Dasilva C, Horner D, Mica E, Jublot D, Poulain J, Bruyère C, Billault A, Segurens B, Gouyvenoux M, Ugarte E, Cattonaro F, Anthouard V, Vico V, Del Fabbro C, Alaux M, Di Gaspero G, Dumas V, Felice N, Paillard S, Juman I, Moroldo M, Scalabrin S, Canaguier A, Le Clainche I, Malacrida G, Durand E, Pesole G, Laucou V, Chatelet P, Merdinoglu D, Delledonne M, Pezzotti M, Lecharny A, Scarpelli C, Artiguenave F, Pè ME, Valle G, Morgante M, Caboche M, Adam-Blondon A-F, Weissenbach J, Quétier F, Wincker P (The French-Italian Public Consortium for Grapevine Genome Characterization) (2007) The grapevine genome sequence suggests ancestral hexaploidization in major angiosperm phyla. Nature 449:463–467. https://doi.org/10.1038/nature06148
Jiang S-Y, Jin J, Sarojam R, Ramachandran S (2019) A Comprehensive survey on the terpene synthase gene family provides new insight into its evolutionary patterns. Genome Biol Evol 11:2078–2098. https://doi.org/10.1093/gbe/evz142
Jones P, Binns D, Chang H-Y, Fraser M, Li W, McAnulla C, McWilliam H, Maslen J, Mitchell A, Nuka G, Pesseat S, Quinn AF, Sangrador-Vegas A, Scheremetjew M, Yong S-Y, Lopez R, Hunter S (2014) InterProScan 5: genome-scale protein function classification. Bioinformatics 30:1236–1240. https://doi.org/10.1093/bioinformatics/btu031
Kainer D, Padovan A, Degenhardt J, Krause S, Mondal P, Foley WJ, Külheim C (2019) High marker density GWAS provides novel insights into the genomic architecture of terpene oil yield in Eucalyptus. New Phytol 223:1489–1504. https://doi.org/10.1111/nph.15887
Keane S, Ryan MF (1999) Purification, characterisation, and inhibition by monoterpenes of acetylcholinesterase from the waxmoth, Galleria mellonella (L.). Insect Biochem Molec Biol 29:1097–1104. https://doi.org/10.1016/S0965-1748(99)00088-0
Keeling CI, Dullat HK, Yuen M, Ralph SG, Jancsik S, Bohlmann J (2010) Identification and functional characterization of monofunctional ent-copalyl diphosphate and ent-kaurene synthases in white spruce reveal different patterns for diterpene synthase evolution for primary and secondary metabolism in gymnosperms. Pl Physiol 152:1197–1208. https://doi.org/10.1104/pp.109.151456
Keith H (1997) Nutrient cycling in eucalypt ecosystems. In: Williams J, Woinarski J (eds) Eucalypt ecology: individuals to ecosystems. Cambridge University Press, Cambridge, pp 197–226
Keszei A, Brubaker CL, Carter R, Köllner T, Degenhardt J, Foley WJ (2010a) Functional and evolutionary relationships between terpene synthases from Australian Myrtaceae. Phytochemistry 71:844–852. https://doi.org/10.1016/j.phytochem.2010.03.013
Keszei A, Hassan Y, Foley WJ (2010b) A Biochemical interpretation of terpene chemotypes in Melaleuca alternifolia. J Chem Ecol 36:652–661. https://doi.org/10.1007/s10886-010-9798-y
Keszei A, Webb H, Kulheim C, Foley W (2010c) Genetic tools for improving tea tree oils. RIRDC Publication No. 10/189. RIRDC, Barton
Külheim C, Padovan A, Hefer C, Krause ST, Köllner TG, Myburg AA, Degenhardt J, Foley WJ (2015) The Eucalyptus terpene synthase gene family. BMC Genomics 16:450. https://doi.org/10.1186/s12864-015-1598-x
Larkman T (2020) Communication of research findings to tea tree oil industry levy payers. AgriFutures, Wagga Wagga, pp 20–135
Lawler IR, Foley WJ, Eschler BM (2000) Foliar concentration of a single toxin creates habitat patchiness for a marsupial folivore. Ecology 81:1327–1338. https://doi.org/10.1890/0012-9658(2000)081[1327:FCOAST]2.0.CO;2
Lee DW, Kim JK, Lee S, Choi S, Kim S, Hwang I (2008) Arabidopsis nuclear-encoded plastid transit peptides contain multiple sequence subgroups with distinctive chloroplast-targeting sequence motifs. Pl Cell 20:1603–1622. https://doi.org/10.1105/tpc.108.060541
Letunic I, Bork P (2021) Interactive Tree Of Life (iTOL) v5: an online tool for phylogenetic tree display and annotation. Nucleic Acids Res 49:W293–W296. https://doi.org/10.1093/nar/gkab301
Lomsadze A, Burns PD, Borodovsky M (2014) Integration of mapped RNA-Seq reads into automatic training of eukaryotic gene finding algorithm. Nucleic Acids Res 42:e119–e119. https://doi.org/10.1093/nar/gku557
Lynch M, Conery JS (2000) The evolutionary fate and consequences of duplicate genes. Science 290:1151–1155. https://doi.org/10.1126/science.290.5494.1151
Martin DM, Aubourg S, Schouwey MB, Daviet L, Schalk M, Toub O, Lund ST, Bohlmann J (2010) Functional annotation, genome organization and phylogeny of the grapevine (Vitis vinifera) terpene synthase gene family based on genome assembly, FLcDNA cloning, and enzyme assays. BMC Pl Biol 10:226. https://doi.org/10.1186/1471-2229-10-226
McLean S, Foley WJ (1997) Metabolism of Eucalyptus terpenes by herbivorous marsupials. Drug Metab Rev 29:213–218. https://doi.org/10.3109/03602539709037582
Moore BD, Foley WJ, Wallis IR, Cowling A, Handasyde KA (2005) Eucalyptus foliar chemistry explains selective feeding by koalas. Biol Lett 1:64–67. https://doi.org/10.1098/rsbl.2004.0255
Moore BD, Andrew RL, Külheim C, Foley WJ (2014) Explaining intraspecific diversity in plant secondary metabolites in an ecological context. New Phytol 201:733–750. https://doi.org/10.1111/nph.12526
Myburg AA, Grattapaglia D, Tuskan GA, Hellsten U, Hayes RD, Grimwood J, Jenkins J, Lindquist E, Tice H, Bauer D, Goodstein DM, Dubchak I, Poliakov A, Mizrachi E, Kullan ARK, Hussey SG, Pinard D, Van Der Merwe K, Singh P, Van Jaarsveld I, Silva-Junior OB, Togawa RC, Pappas MR, Faria DA, Sansaloni CP, Petroli CD, Yang X, Ranjan P, Tschaplinski TJ, Ye C-Y, Li T, Sterck L, Vanneste K, Murat F, Soler M, Clemente HS, Saidi N, Cassan-Wang H, Dunand C, Hefer CA, Bornberg-Bauer E, Kersting AR, Vining K, Amarasinghe V, Ranik M, Naithani S, Elser J, Boyd AE, Liston A, Spatafora JW, Dharmwardhana P, Raja R, Sullivan C, Romanel E, Alves-Ferreira M, Külheim C, Foley W, Carocha V, Paiva J, Kudrna D, Brommonschenkel SH, Pasquali G, Byrne M, Rigault P, Tibbits J, Spokevicius A, Jones RC, Steane DA, Vaillancourt RE, Potts BM, Joubert F, Barry K, Pappas GJ, Strauss SH, Jaiswal P, Grima-Pettenati J, Salse J, Van De Peer Y, Rokhsar DS, Schmutz J (2014) The genome of Eucalyptus grandis. Nature 510:356–362. https://doi.org/10.1038/nature13308
Okonechnikov K, Golosova O, Fursov M (2012) Unipro UGENE: a unified bioinformatics toolkit. Bioinformatics 28:1166–1167. https://doi.org/10.1093/bioinformatics/bts091
Padovan A, Keszei A, Külheim C, Foley WJ (2014) The evolution of foliar terpene diversity in Myrtaceae. Phytochem Rev 13:695–716. https://doi.org/10.1007/s11101-013-9331-3
Padovan A, Keszei A, Hassan Y, Krause ST, Köllner TG, Degenhardt J, Gershenzon J, Külheim C, Foley WJ (2017a) Four terpene synthases contribute to the generation of chemotypes in tea tree (Melaleuca alternifolia). BMC Pl Biol 17:160. https://doi.org/10.1186/s12870-017-1107-2
Padovan A, Webb H, Mazanec R, Grayling P, Bartle J, Foley WJ, Külheim C (2017b) Association genetics of essential oil traits in Eucalyptus loxophleba: explaining variation in oil yield. Molec Breed 37:73. https://doi.org/10.1007/s11032-017-0667-z
Pass GJ, McLean S, Stupans I, Davies NW (2002) Microsomal metabolism and enyzme kinetics of the terpene p-cymene in the common brushtail possum (Trichosurus vulpecula), koala (Phascolarctos cinereus) and rat. Xenobiotica 32:383–397. https://doi.org/10.1080/00498250210124138
Pichersky E, Gershenzon J (2002) The formation and function of plant volatiles: perfumes for pollinator attraction and defense. Curr Opin Pl Biol 5:237–243. https://doi.org/10.1016/S1369-5266(02)00251-0
R Core Team, (2022) R: A language and environment for statistical computing. R Foundation for Statistical Computing, Vienna. Available at: https://www.R-project.org/
Ryan MF, Byrne O (1988) Plant-insect coevolution and inhibition of acetylcholinesterase. J Chem Ecol 14:1965–1975. https://doi.org/10.1007/bf01013489
Schmidt-Dannert C (2015) Biosynthesis of terpenoid natural products in fungi. In: Schrader J, Bohlmann J (eds) Biotechnology of isoprenoids. Springer International Publishing, Cham, pp 19–61. https://doi.org/10.1007/10_2014_283
Schwab W (2003) Metabolome diversity: too few genes, too many metabolites? Phytochemistry 62:837–849. https://doi.org/10.1016/s0031-9422(02)00723-9
Schwacke R, Ponce-Soto GY, Krause K, Bolger AM, Arsova B, Hallab A, Gruden K, Stitt M, Bolger ME, Usadel B (2019) MapMan4: a refined protein classification and annotation framework applicable to multi-omics data analysis. Molec Pl 12:879–892. https://doi.org/10.1016/j.molp.2019.01.003
Sell C (2010) Chemistry of essential oils. In: Husnu Can Baser K, Buchbauer G (eds) Handbook of essential oils: science, technology, and applications. CRC Press, Boca Raton. https://doi.org/10.1201/b19393
Sharkey TD, Yeh S, Wiberley AE, Falbel TG, Gong D, Fernandez DE (2005) Evolution of the isoprene biosynthetic pathway in Kudzu. Pl Physiol 137:700–712. https://doi.org/10.1104/pp.104.054445
Sharkey TD, Gray DW, Pell HK, Breneman SR, Topper L (2013) Isoprene synthase genes form a monophyletic clade of acyclic terpene synthases in the Tps-B terpene synthase family. Evolution 67:1026–1040. https://doi.org/10.1111/evo.12013
Shelton D, Aitken K, Doimo L, Leach D, Baverstock P, Henry R (2002) Genetic control of monoterpene composition in the essential oil of Melaleuca alternifolia (Cheel). Theor Appl Genet 105:377–383. https://doi.org/10.1007/s00122-002-0948-7
Shelton D, Leach D, Henry R (2004a) Isopentenyl pyrophosphate isomerases from Melaleuca alternifolia (Cheel) and their role in isoprenoid biosynthesis. J Hortic Sci Biotechnol 79:289–292. https://doi.org/10.1080/14620316.2004.11511762
Shelton D, Zabaras D, Chohan S, Wyllie SG, Baverstock P, Leach D, Henry R (2004b) Isolation and partial characterisation of a putative monoterpene synthase from Melaleuca alternifolia. Pl Physiol Biochem 42:875–882. https://doi.org/10.1016/j.plaphy.2004.10.010
Slowikowski K (2021) ggrepel: Automatically Position Non-Overlapping Text Labels with ‘ggplot2’, R package version 0.9.1. Available at: https://CRAN.R-project.org/package=ggrepel
Solovyev V, Kosarev P, Seledsov I, Vorobyev D (2006) Automatic annotation of eukaryotic genes, pseudogenes and promoters. Genome Biol 7:11–12. https://doi.org/10.1186/gb-2006-7-s1-s10
Southwell IA, Maddox CDA, Zalucki MP (1995) Metabolism of 1,8-cineole in tea tree (Melaleuca alternifolia and M. linariifolia) by pyrgo beetle (Paropsisterna tigrina). J Chem Ecol 21:439–453. https://doi.org/10.1007/BF02036741
Stone C, Bacon PE (1994) Relationships among moisture stress, insect herbivory, foliar cineole content and the growth of river red gum Eucalyptus camaldulensis. J Appl Ecol 31:604–612. https://doi.org/10.2307/2404151
Strachan T, Read A (2011) Model organisms, comparative genomics, and evolution. In: Human molecular genetics, vol. 4. Garland Science/Taylor & Francis Group, New York, pp 297–344
Suyama M, Torrents D, Bork P (2006) PAL2NAL: robust conversion of protein sequence alignments into the corresponding codon alignments. Nucleic Acids Res 34:W609–W612. https://doi.org/10.1093/nar/gkl315
The Galaxy Community (2022) The Galaxy platform for accessible, reproducible and collaborative biomedical analyses: 2022 update. Nucleic Acids Res 50:W345–W351. https://doi.org/10.1093/nar/gkac247
Tholl D (2006) Terpene synthases and the regulation, diversity and biological roles of terpene metabolism. Curr Opin Pl Biol 9:297–304. https://doi.org/10.1016/j.pbi.2006.03.014
Tholl D (2015) Biosynthesis and biological functions of terpenoids in plants. Advances Biochem Engin Biotechnol 148:63–106. https://doi.org/10.1007/10_2014_295
Thornhill AH, Ho SYW, Külheim C, Crisp MD (2015) Interpreting the modern distribution of Myrtaceae using a dated molecular phylogeny. Molec Phylogen Evol 93:29–43. https://doi.org/10.1016/j.ympev.2015.07.007
Trapp SC, Croteau RB (2001) Genomic organization of plant terpene synthases and molecular evolutionary implications. Genetics 158:811–832
Tuskan GA, Difazio S, Jansson S, Bohlmann J, Grigoriev I, Hellsten U, Putnam N, Ralph S, Rombauts S, Salamov A, Schein J, Sterck L, Aerts A, Bhalerao RR, Bhalerao RP, Blaudez D, Boerjan W, Brun A, Brunner A, Busov V, Campbell M, Carlson J, Chalot M, Chapman J, Chen GL, Cooper D, Coutinho PM, Couturier J, Covert S, Cronk Q, Cunningham R, Davis J, Degroeve S, Dejardin A, Depamphilis C, Detter J, Dirks B, Dubchak I, Duplessis S, Ehlting J, Ellis B, Gendler K, Goodstein D, Gribskov M, Grimwood J, Groover A, Gunter L, Hamberger B, Heinze B, Helariutta Y, Henrissat B, Holligan D, Holt R, Huang W, Islam-Faridi N, Jones S, Jones-Rhoades M, Jorgensen R, Joshi C, Kangasjarvi J, Karlsson J, Kelleher C, Kirkpatrick R, Kirst M, Kohler A, Kalluri U, Larimer F, Leebens-Mack J, Leple JC, Locascio P, Lou Y, Lucas S, Martin F, Montanini B, Napoli C, Nelson DR, Nelson C, Nieminen K, Nilsson O, Pereda V, Peter G, Philippe R, Pilate G, Poliakov A, Razumovskaya J, Richardson P, Rinaldi C, Ritland K, Rouze P, Ryaboy D, Schmutz J, Schrader J, Segerman B, Shin H, Siddiqui A, Sterky F, Terry A, Tsai CJ, Uberbacher E, Unneberg P, Vahala J, Wall K, Wessler S, Yang G, Yin T, Douglas C, Marra M, Sandberg G, Van de Peer Y, Rokhsar D (2006) The genome of black cottonwood, Populus trichocarpa (Torr. & Gray). Science 313:1596–1604. https://doi.org/10.1126/science.1128691
Velikova V, Loreto F (2005) On the relationship between isoprene emission and thermotolerance in Phragmites australis leaves exposed to high temperatures and during the recovery from a heat stress. Pl Cell Environm 28:318–327. https://doi.org/10.1111/j.1365-3040.2004.01314.x
Velikova V, Edreva A, Loreto F (2004) Endogenous isoprene protects Phragmites australis leaves against singlet oxygen. Physiol Pl 122:219–225. https://doi.org/10.1111/j.0031-9317.2004.00392.x
Voelker J (2023) Additional information for the manual annotation of the terpene synthase gene family in Melaleuca alternifolia (tea tree). Protocols Io. https://doi.org/10.17504/protocols.io.3byl4bjr2vo5/v1
Voelker J, Shepherd M, Mauleon R (2021a) A high-quality draft genome for Melaleuca alternifolia (tea tree): a new platform for evolutionary genomics of myrtaceous terpene-rich species. GIGAbyte 2021:1–15. https://doi.org/10.46471/gigabyte.28
Voelker J, Shepherd M, Mauleon R (2021b) Supporting data for “A high-quality draft genome for Melaleuca alternifolia (tea tree)—a new platform for evolutionary genomics of myrtaceous terpene-rich species.” GigaSci Database. https://doi.org/10.5524/100917
Vögeli U, Chappell J (1988) Induction of sesquiterpene cyclase and suppression of squalene synthetase activities in plant cell cultures treated with fungal elicitor 1. Pl Physiol 88:1291–1296. https://doi.org/10.1104/pp.88.4.1291
Vranová E, Coman D, Gruissem W (2013) Network analysis of the MVA and MEP pathways for isoprenoid synthesis. Annual Rev Pl Biol 64:665–700. https://doi.org/10.1146/annurev-arplant-050312-120116
Wickham H (2016) ggplot2: Elegant graphics for data analysis. Springer-Verlag, New York
Wilkins D (2020) gggenes: Draw Gene Arrow Maps in ‘ggplot2’, R package version 0.4.1. Available at: https://CRAN.R-project.org/package=gggenes
Wilson PG (2011) Myrtaceae. In: Kubitzki K (ed) The families and genera of vascular plants flowering plants eudicots: Sapindales, Cucurbitales Myrtaceae, vol. X. Springer-Verlag, Heidelberg, pp 212–271
Yamada Y, Kuzuyama T, Komatsu M, Shin-ya K, Omura S, Cane DE, Ikeda H (2015) Terpene synthases are widely distributed in bacteria. Proc Natl Acad Sci USA 112:857–862. https://doi.org/10.1073/pnas.1422108112
Yang Z (2007) PAML 4: Phylogenetic analysis by maximum likelihood. Molec Biol Evol 24:1586–1591. https://doi.org/10.1093/molbev/msm088
Yu SJ (2008) Detoxification mechanisms in insects. In: Capinera JL (ed) Encyclopedia of entomology. Springer, Dordrecht, pp 1187–1201. https://doi.org/10.1007/978-1-4020-6359-6_891
Yu F, Utsumi R (2009) Diversity, regulation, and genetic manipulation of plant mono- and sesquiterpenoid biosynthesis. Cell Molec Life Sci 66:3043–3052. https://doi.org/10.1007/s00018-009-0066-7
Yuan JS, Köllner TG, Wiggins G, Grant J, Degenhardt J, Chen F (2008) Molecular and genomic basis of volatile-mediated indirect defense against insects in rice. Plant J 55:491–503. https://doi.org/10.1111/j.1365-313X.2008.03524.x
Zhang J (2003) Evolution by gene duplication: an update. Trends Ecol Evol 18:292–298. https://doi.org/10.1016/S0169-5347(03)00033-8
Zheng X, Chen S, Lin G, Chen J, Li H, Xiao Y, Chen X, Chen J, Wu Y, Xiao H, He W, Chen Y, Chen D, Zhu J, Xue T (2022) The chromosome-level Melaleuca alternifolia genome provides insights into the molecular mechanisms underlying terpenoids biosynthesis. Industr Crops Prod 189:115819. https://doi.org/10.1016/j.indcrop.2022.115819
Zhou R, Macaya-Sanz D, Rodgers-Melnick E, Carlson CH, Gouker FE, Evans LM, Schmutz J, Jenkins JW, Yan J, Tuskan GA, Smart LB, DiFazio SP (2018) Characterization of a large sex determination region in Salix purpurea L. (Salicaceae). Molec Genet Genomics 293:1437–1452. https://doi.org/10.1007/s00438-018-1473-y
Acknowledgements
The authors would like to thank the Australian BioCommons and Operational Partners for providing access to the Apollo annotation software via the Australian Apollo Service (https://apollo-portal.genome.edu.au/). InterProScan (Jones et al. 2014) for Pfam analyses was run on the public servers of Galaxy Europe (https://usegalaxy.eu/), and the patmatdb search tool for sequence motifs was accessed via Galaxy Australia (https://usegalaxy.org.au/) (The Galaxy Community 2022). The authors would also like to thank the two anonymous reviewers whose valuable feedback helped to further improve this manuscript.
Funding
Open Access funding enabled and organized by CAUL and its Member Institutions. Julia Voelker was supported by a PhD stipend and operating expenses from Southern Cross University and the Australian Tea Tree Industry Association (ATTIA). Ramil Mauleon and Mervyn Shepherd were funded by salaries from Southern Cross University.
Author information
Authors and Affiliations
Contributions
JV, RM, and MS helped in conceptualisation; JV worked on methodology and formal analysis; JV undertook writing—original draft preparation; JV, RM, and MS helped in writing—review and editing; MS worked in funding acquisition; and RM and MS helped in supervision. All authors read and approved the manuscript.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that no competing interests exist.
Additional information
Handling Editor: Mike Thiv.
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Information on Electronic Supplementary Material
Information on Electronic Supplementary Material
Online Resource 1. Biosynthetic pathway information for terpenoid synthesis.
Online Resource 2. Examples of manual TPS-b annotation evidence.
Online Resource 3. Phylogenetic tree for putatively functional Melaleuca alternifolia TPS sequences.
Online Resource 4. Sequence logo representation of the NSE/DTE motif in tea tree TPS.
Online Resource 5. Information on the screen for organelle-targeting signal peptides.
Online Resource 6. Species tree inferred from Orthofinder results.
Online Resource 7. Inter-species phylogenetic trees of TPS subfamilies including tea tree pseudogenes.
Online Resource 8. Visualisation of the large TPS-b tandem cluster organisation.
Online Resource 9. General information about the annotated putatively functional TPS and pseudogenes, including a summary of conserved motifs encoded in each gene.
Online Resource 10. GFF3 file listing the genome coordinates from the manual annotation of putatively functional TPS genes and pseudogenes.
Online Resource 11. GFF3 file for the 15 TPS sequences that were excluded from further analysis due to potential redundancy and assembly errors.
Online Resource 12. FASTA file containing the protein sequences of the 58 analysed full-length TPS.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Voelker, J., Mauleon, R. & Shepherd, M. The terpene synthase genes of Melaleuca alternifolia (tea tree) and comparative gene family analysis among Myrtaceae essential oil crops. Plant Syst Evol 309, 13 (2023). https://doi.org/10.1007/s00606-023-01847-1
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s00606-023-01847-1