
Abstract
Aim
In the pediatric diabetes clinic, patients with type 1 diabetes mellitus (T1D) account for more than 90% of cases, while monogenic forms represent about 6%. Many monogenic diabetes subtypes may respond to therapies other than insulin and have chronic diabetes complication prognosis that is different from T1D. With the aim of providing a better diagnostic pipeline and a tailored care for patients with monogenic diabetes, we set up a monogenic diabetes clinic (MDC).
Methods
In the first 3 years of activity 97 patients with non-autoimmune forms of hyperglycemia were referred to MDC. Genetic testing was requested for 80 patients and 68 genetic reports were available for review.
Results
In 58 subjects hyperglycemia was discovered beyond 1 year of age (Group 1) and in 10 before 1 year of age (Group 2). Genetic variants considered causative of hyperglycemia were identified in 25 and 6 patients of Group 1 and 2, respectively, with a pick up rate of 43.1% (25/58) for Group 1 and 60% (6/10) for Group 2 (global pick-up rate: 45.5%; 31/68). When we considered probands of Group 1 with a parental history of hyperglycemia, 58.3% (21/36) had a positive genetic test for GCK or HNF1A genes, while pick-up rate was 18.1% (4/22) in patients with mute family history for diabetes. Specific treatments for each condition were administered in most cases.
Conclusion
We conclude that MDC may contribute to provide a better diabetes care in the pediatric setting.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Polygenic, autoimmune type 1 diabetes mellitus (T1DM) is the main cause of pediatric diabetes, but a pathogenic variant in a single gene can be identified in a sizeable number of patients referred to the pediatric diabetes clinic. The latter group of patients is affected by Mendelian forms of diabetes (i.e. autosomal dominant, autosomal recessive and X-linked) defects collectively known as "monogenic diabetes" mellitus (MDM) [1,2,3,4,5].
Genes involved in MDM have now surpassed the number of 50 and new genes are discovered at an amazing pace. The list includes "common", non-syndromic forms as well as rare, syndromic subtypes. There are two other genetic forms of diabetes not strictly under the MDM definition but considered part of this group of diseases: chromosome 6 aberrations and mutations of mitochondrial DNA. Chromosome 6 defects include uniparental paternal unidisomy, microduplications and methylation defects (collectively known as 6q24), all causing transient neonatal diabetes mellitus (TNDM), whereas mutations in mitochondrial DNA, such as the recurrent m.3243G > A, cause the maternally inherited diabetes and deafness (MIDD) [6, 7].
With the advent of novel DNA sequencing techniques (next generation sequencing; NGS), the simultaneous screening of coding sequences of all MDM genes has become possible. The screening can then be completed with additional methods that identify medium-large genetic deletions and methylation defects. As a result, many patients suspected of MDM can be genetically diagnosed and may benefit from tailored therapies [1, 6, 8, 9], expanding the aim and scope of pediatric diabetes clinics. Still, the amount of data produced by NGS may prove somehow overwhelming and a strict collaboration between geneticists and diabetologists with expertise in genetics may improve the pick-up rate of cases with robust genetic diagnosis. This in turn allows the appropriateness of customized therapies.
A Monogenic Diabetes Clinic (MDC) within the Diabetology and Growth Disorders Unit of Bambino Gesù Children’s Hospital was started by F.B. and N.R. on January 2019. The idea behind MDC was to convey patients suspected to have a monogenic form of diabetes or with an established genetic diagnosis of monogenic diabetes to a clinic exclusively designated for the diagnosis and care of this subtype of diabetes. The expected advantages of this organization are: 1) the implementation of a standardized pathway toward genetic testing, 2) to ease the revision of complex cases, 3) to administer standardized therapies for monogenic diabetes subtypes, 4) to gather rare cases of monogenic diabetes to the end of acquiring new knowledge on specific subtypes and 5) the identification of MDM in overweight or obese patients, easily diagnosed with type 2 diabetes mellitus (T2D) of youth [10, 11].
In this paper, we report the results of the first 3 years of activity of MDC.
Materials and methods
Patients
Patients with diabetes, impaired fasting glucose (IFG) or impaired glucose tolerance who tested negative for four types 1 diabetes (T1DM)-related autoantibodies (GADA, IA-2A, IAA, ZnT8) were considered eligible for genetic testing. Individuals with at least two independent fasting plasma glucose samples ≥ 100–125 mg/dl (5.6–6.9 mmol/L) were classified as IFG. Most, but not all IFG subjects underwent an oral glucose tolerance test (OGTT) to identify cases with diabetes or impaired glucose tolerance (IGT). A diagnosis of diabetes was established with two independent fasting plasma glucose samples ≥ 126 mg/dl (7.0 mmol/L) or one fasting plasma glucose sample > 126 mg/dl and a HbA1c value ≥ 6.5% (48 mmol/mol) or a value ≥ 200 at 120ʹ of OGTT or a random value ≥ 200 mg/dl. Age at onset of diabetes > 25 of the proband was not considered a criterium of exclusion for genetic testing if family history was indicative of autosomal dominant inheritance (3 consecutive generations). Presentation/accidental discovery of IFG/diabetes was in most cases between 0 and 18 years of age, with a few patients diagnosed beyond the age of 25. Parental history of IFG status or diabetes was obtained or actively established by requesting fasting plasma glucose analysis of parents. History of IFG/diabetes in both proband’s parents was not a criterium of exclusion. Lean patients with high fasting insulinemia (> 22 μU/ml) and acanthosis nigricans with or without fasting and/or post-load hyperglycemia were clinically classified as type A severe insulin resistance (SIR) and screened for insulin receptor (INSR) variants.
We were also consulted to give our opinion on previously identified monogenic diabetes genes defect/variant in two cases with no dysglycemia/diabetes. One patient was investigated for a mild intellectual impairment, while genetic test was requested for the other because of HbA1c repeatedly at the upper limit of reference range and a family history of type 2 diabetes.
Most patients were sent to MDC by physicians belonging to the Diabetology and Growth Disorders Unit of Bambino Gesù Children’s Hospital, but some patients were referred from hospitals outside Rome and Lazio region. A few patients self-referred to the clinic by word-of-mouth. Eight patients with non-autoimmune neonatal diabetes mellitus (NDM) from other centers were directly referred to F.B.
T1DM autoantibodies
Autoantibodies were tested in the Clinical Laboratory Unit with ELISA commercial kits.
Genetic screening
Clinical exome sequencing (CES, including 8245 genes) was performed on genomic DNA by using the Twist Custom Panel kit (Twist Bioscience, San Francisco, CA, USA) according to the manufacturer’s protocol on a NovaSeq6000 platform (Illumina, San Diego, CA, USA).
Coding sequences and intron/exon boundaries of the following genes were filtered out for analysis ("virtual panel"): ABCC8, APPL1, CISD2, CNOT1, DNAJC3, DCAF17, KCNJ11, GCK, EIF2S2, EIF2AK3, INS, INSR, GATA4, GATA6, GLIS3, HNF1A, HNF4A, HNF1B, IER3IP1, NEUROD1, NEUROG3, GLIS3, PDX1, RFX6, MNX1, NKX2-2, PAX6, PCBD1, PTF1A, SLC2A2, SLC19A2, SLC29A3, EIF2S3, WFS1, ZFP57 [5].
The reads were aligned to human genome build GRCh37/UCSC hg19. Variant calling was performed with Dragen Germline Enrichment application of BaseSpace (Illumina, San Diego, CA, USA) while variant annotation and phenotype-based prioritization of candidate genes were carried out through the Geneyx Analysis software (Geneyx Genomex). A minimum depth coverage of 30X was considered suitable for analysis, but most genes had a coverage of 100X. Exome sequencing data filtering was performed to identify protein-altering, putative rare recessive homozygous, compound heterozygous, and pathogenic or likely pathogenic heterozygous variants with an allele frequency < 1%. Variants were classified based on the guidelines of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology (ACMG/AMP) [12]. Putative causative variants were analyzed by Sanger sequencing following a standard protocol (BigDye Terminator v3.1 Cycle Sequencing Kit, Life Technologies) to confirm the next-generation sequencing (NGS) results in probands, and, if possible, were investigated in the parents to check the inheritance status.
Patients with neonatal diabetes mellitus who resulted negative to the CES were analyzed for 6q24 aberrations by MS-MLPA analysis (ME033-A1, MrC Holland, Amsterdam, The Netherlands) that detects also methylation defects (6). Recently, two genes have been described as a novel causes of permanent neonatal diabetes: ONECUT1 and ZNF808 [13, 14]. These genes were examined in patients who were negative for standard screening in addition to INS promoter and INS intron 2 [5].
The report was fully explained and commented to the proband or to the proband’s guardians by F.B. and N.R. Reports regarding NDM cases were also discussed with caring physicians.
F.B. consulted with members of the Monogenic Diabetes Variant Curation Expert Panel (MDEP) [15] of Clinical Genome Resource [16] when novel variants in rare genes were identified.
Results
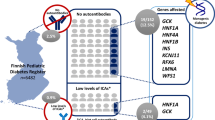
A total of 97 patients were referred to the MDC during the 3-year (Jan/2019-Dec/2021) period. Four were from extra-European countries. Seventeen subjects did not fit inclusion criteria of MDM and no genetic test was requested. Blood sample for DNA extraction was not available for 12 patients at the time of writing. Therefore we were able to evaluate 68 genetic reports (Fig. 1).

Steps from patient referral to MDC to final genetic diagnosis
Fiftysix incident cases were referred to investigate the origin of IFG, IGT or diabetes with onset beyond 1 year of age; in addition, we were requested to evaluate two normoglycemic cases carrying a MDG variant (Group 1, 58 cases) (Fig. 1). Among patients of this group, three lean females with fasting hyperinsulinism, hirsutism, acanthosis nigricans and (two cases) post-load glucose derangement were clinically classified as type A SIR. Only four patients of Group 1 were diagnosed with IFG, IGT or diabetes beyond 18 years of age. Ten patients (8 incident cases plus 2 past cases with permanent neonatal diabetes mellitus of unknown genetic cause) had diabetes onset within 1 year of age and were classified as neonatal diabetes mellitus (NDM; Group 2).
Among cases of Group 1, a pathogenic (P) or likely pathogenic (LP) variant of GCK, HNF1A and HNF1B was identified in 17, 4 and 1 patients, respectively (Table 1, Online resource 1, right panel). In a patient with liver adenomatosis carrying the HNF1A/Arg171Ter an additional somatic mutation has been also identified in the hepatic lesion.
Four cases with a GCK variant (cases 4, 7, 8 and 17) and 1 with HNF1A/Arg171Ter were overweight. Three patients carrying the GCK/Ter466GlyextTer144 (case 9), the HNF1A/Ser247Cysfster96 variant (case 18) and the HNF1B exon 1–9 deletion (case 22) were obese. Interestingly, fasting C-peptide value of two latter patients was normal/high.
All patients with GCK or HNF1A heterozygous variants had a parent with IFG or diabetes who carried the same variant (Table 1). We considered causative two GCK variants of uncertain significance (VUS) (cases 6 and 9) because both proband and affected parent had a clinical phenotype (mild fasting, non-progressive hyperglycemia) consistent with GCK haploinsufficiency. Heterozygous pathogenic or likely pathogenic variants in the INSR were identified in 2 out of three patients with Type A SIR (Table 1, Online resource 1, right panel).
A homozygous variant of the SLC2A2 gene (Fanconi-Bickel syndrome) was found in one patient (Case 25, Table 1). This case showed impaired glucose tolerance at the age of 2 years and diabetes at the age of 15 (oral glucose tolerance test: plasma glucose 262 mg/dl). Massive glycosuria was detected when the patient was 6 years old.
We did not formulate a final diagnosis in six cases belonging to Group 1 with a VUS variant (cases 33–36, 40) (Table 2 and Online resource 1, right panel). In addition, a GCK likely pathogenic variant was identified in a patient with no family history of diabetes who presented with diabetic ketoacidosis; in this case, the variant has not been considered sufficient to determine the clinical presentation (case 32; Table 2).
As part of MDC activity, we were consulted about two patients carrying a MDG variant, but with no defects of glucose metabolism at the time of writing (Table 2). The first one carries a heterozygous, frameshift PDX1 likely pathogenic (LP) variant with premature termination codon (case 38), while the other has a large deletion of chromosome 17q12 encompassing HNF1B (case 39); of note, case 39 did not show any kidney malformation at ultrasound. For these two cases and for case 40 (INSR variant) we decided on a strict follow-up (every 6 months) in order to promptly diagnose any future derangement of glucose metabolism.
Twenty-five patients belonging to Group 1 were negative to genetic testing. Twelve had a parent with dysglycemia, (Table 3), while 13 were sporadic cases (Table 4). We formulated a diagnosis of T2D in two patients (cases 52 and 58) (Tables 3 and 4) who had normal, not declining c-peptide at onset of hyperglycemia and at follow-up. Other 2 (cases 64, 65) had a single diagnostic OGTT, leaving the T2D diagnosis dubious (Table 4). For cases 49, 51 (Table 3), 57, 60–63 (Table 4) our temptative diagnosis was autoantibody-negative T1D; however, we can not exclude an inherited (Table 3) or spontaneous (Table 4) small HNF1A/HNF4A deletion or pathogenic variant(s) in regulatory regions of these genes.
Six patients of Group 2 carried a pathogenic or likely pathogenic variant in KCNJ11 (3 variants), ABCC8 (1 variant) and PDX1 (biallelic variants); a patient with transient neonatal diabetes mellitus had 6q24 methylation defects (Table 1; Online resource 1, left panel).
In a patient with TNDM the VUS ABCC8/Ser53Cys was identified (Table 2); the mother, carrying the variant, showed normal plasma glucose values at OGTT. We thus considered ABCC8/Ser53Cys not causative. This patient was also negative for KCNJ11, 6q24 and SLC2A2.
In three non-syndromic NDM cases (one incident, two past patients) we did not find any variant in the MDG of the panel or in the new NDM genes ONECUT1 and ZNF808. Among these 3, two presented with diabetes beyond six months and before 1 year (cases 66, 68) and were classified as early-onset antibody-negative T1D (Table 4).
Therapeutic decisions consequent to genetic diagnosis are reported in Supplemental Table 1.
Discussion
Next-generation sequencing has dramatically improved our capability of identifying even rare genetic causes of monogenic diabetes. Overall, genetic testing resulted positive and conclusive in 45.5% of cases (31/68). For patients of Group 1, the positive genetic testing rate was 43.1% (25/58 probands investigated) an acceptable percentage if compared to that obtained by the exceptionally meticulous services offered in the UK [3]. Among 36 patients of Group 1 with a parental history of hyperglycemia, 21 (Cases 1–21; Table 1) carried a heterozygous causative variant in GCK or HNF1A, with a pick-up rate of 58.3% (21/36) in this subgroup.
Among 22 cases with unknown or no parental history of hyperglycemia of Group 1, 3 carried a dominant variant and 1 a homozygous, recessive variant (Table 1, cases 22–25). All these cases have been investigated on the basis of specific clinical features: hyperinsulinism, hirsutism and acanthosis in lean females (cases 23 and 24), renal cysts (case 22), and tubular nephropathy (case 25). In one case with SIR, the INSR pathogenic variant arose spontaneously, while for the other proband parental DNA was not available for analysis. Therefore, among the subgroup lacking parental history, pick up rate was 18.1% (4/22). Recently, biallelic variants of WFS1 have been identified in patients lacking syndromic features of Wolfram disease [17]. Moreover, biallelic WFS1 pathogenic variants either syndromic or not, are quite frequent in pediatric patients born to consanguineous parents, where autosomal recessive mutations represent more than 40% [18]. However, no WFS1 variant was identified in this series. In contrast, a SLC2A2 homozygous variant was found in a single patient, setting the frequency of recessive mutations of Group 1 to 1.7% (1/58) or 4.5% (1/22) when considering the subgroup of patients with mute family history of diabetes. Though based on a relatively small number of patients, it seems reasonable to conclude that genetic testing in individuals with onset of hyperglycemia beyond 1 year of age and without parental history of hyperglycemia should be mainly reserved for cases with extrapancreatic features and/or consanguineous parents [19]. Interestingly, but not surprisingly, autosomal dominant -negative mutations of INSR are not found in populations with high consanguinity rate [18] but can be identified in probands lacking parental history of hyperglycemia (our two cases) on the basis of extrapancreatic features [20].
Follow-up was recommended for cases 38, 39 and 40 who are currently normoglycemic (Table 2). Case 38 bears a 17q12 deletion that includes HNF1B; this abnormality arises spontaneously in 70% of cases and has been associated with high frequency of diabetes (63%) with onset in adulthood [21]. Our case showed abnormally high glucose levels at 30ʹ of OGTT, indicating a poor first-phase insulin secretion, a finding that is in keeping with those of Ng et al. that indicate a marked insulin deficiency in HNF1B patients [22]. Case 39 underwent genetic testing because of strong family history of diabetes from the paternal side (father deceased). She carries a likely pathogenic PDX1 variant (Ala228GlyfsTer33) which is similar to PDX1/Pro63ArgfsTer60, the only PDX1 variant linked to diabetes with well demonstrated dominant-negative effect [23]. INSR VUS detected in case 40 may concur with fasting hyperinsulinemia but without functional data, it is not possible to opt for a dominant-negative effect.
It is well established that NDM is quite rare (about 1:100,000 live births) in populations with low consanguinity rate [24]. Nine NDM patients out of 10 in the present study were not syndromic and in 6 a causative variant was identified, including a patient with pancreas hypoplasia linked to biallelic PDX1 variants (case 31). Two new NDM genes [13, 14] were additionally screened in the remaining four with no success. Of interest, we found a heterozygous, LP missense variant of ONECUT1 in another individual diagnosed with PNDM 18 years ago and not included in the present study [25].
A genetic diagnosis may guide therapeutic changes (Supplemental Table 1) such as switch from insulin to sulfonylureas (SU) in patients with NDM due to KCNJ11 or ABCC8 (ATP-sensitive potassium channel genes, KATP) variants [8, 9]. We attempted the transfer to glibenclamide (the most used SU in neonatal diabetes) all NDM patients with KATP variants and succeeded in 3, while one patient was resistant even at high glibenclamide dosage [26]. Metformin was introduced after genetic diagnosis in one patient with INSR variant (case 24). Individuals with type A insulin resistance may show severe hyperglycemia over time [27]; however, we do not know whether the early use of metformin in case 23 may prevent the onset of full-blown diabetes later in life. Recently, new therapies aimed at handling hyperinsulinemia and diabetes seen in patients with congenital SIR due to INSR mutations and in type A SIR have been proposed [28, 29].
An attempt to transfer to SU HNF1A patients treated with insulin is common practice [1]. However, the patient with liver transplant (case 21) recently stopped insulin, while case 19 continued insulin because she became pregnant. Metformin was confirmed by the caring physician in an obese patient after HNF1A diagnosis (case 18), while the obese (BMI 30.8) patient carryng the HNF1B deletion (case 22) is on diet. Obesity "complicating" MODY [10, 11] is becoming a frequent issue in Italian patients. Intriguingly, also the affected parent of HNF1A case was obese, making clinical diagnosis even more complex.
Among limitations of this work, there is the very small size of our cohort. Nevertheless, results are in line with those obtained by others and positive diagnostic rate was acceptable in cases with parental history of hyperglycemia. The second limitation is that we did not analyze genes causing lipodystrophies and the frequent mitochondrial mutation causing maternally inherited diabetes and deafness (MIDD), m.3243A > G. As for m.3243A > G, while this mutation is a relatively common cause of hyperglycemia in data sets of adult-onset diabetes [3, 7], it seems to be very rare in the pediatric setting [18].
Conclusions
In conclusion, during the first 3 years of activity MDC seemed to fulfill the objectives that were set at its start, especially points 2, 3 and 5 described in the introduction. A slight change in strategy for selection of "sporadic" cases with non-autoimmune diabetes, focused on thorough, systematic workup of expancreatic features, will be implemented in future MDC activities.
Data availability
Data obtained with the genetic analysis are not archived publicly.
Change history
05 November 2022
A Correction to this paper has been published: https://doi.org/10.1007/s00592-022-01998-6
References
Delvecchio M, Mozzillo E, Salzano G, Iafusco D, Frontino G, Patera PI et al (2017) Monogenic diabetes accounts for 6.3% of cases referred to 15 Italian pediatric diabetes centers during 2007–2012. J Clin Endocrinol Metab 102(6):1826–34
Johansson BB, Irgens HU, Molnes J, Sztromwasser P, Aukrust I, Juliusson PB et al. (2017) Targeted next-generation sequencing reveals MODY in up to 6.5% of antibody-negative diabetes cases listed in the Norwegian Childhood Diabetes Registry. Diabetologia 60(4):625–635
Pang L, Colclough KC, Shepherd MH, McLean J, Pearson ER, Ellard S et al. (2022) Improvements in awareness and testing have led to a threefold increase over 10 years in the identification of monogenic diabetes in the UK. Diabetes Care 45(3):642–649
De Franco E (2020) From biology to genes and back again: gene discovery for monogenic forms of beta-cell dysfunction in diabetes. J Molec Biol 432(5):1535–1550
Barbetti F, Rapini N, Schiaffini R, Bizzarri C, Cianfarani S (2022) The application of precision medicine to monogenic diabetes. Expert Rev Endocrinol Metab 17(2):111–119
Bonfanti R, Iafusco D, Rabbone I, Diedenhofen G, Bizzarri C, Patera PI et al (2021) Differences between transient neonatal diabetes mellitus subtypes can guide diagnosis and therapy. Eur J Endocrinol 184(4):575–585
Saint-Martin C, Bouvet D, Bastide M, Chantelot CB (2022) Gene panel sequencing of patients with monogenic diabetes brings to light genes typically associated with syndromic presentations. Diabetes 71(3):578–584
Bowman P, Sulen A, Barbetti F, Beltrand J, Svalastoga P, Codner E et al (2018) Effectiveness and safety of long-term treatment with sulfonylureas in patients with neonatal diabetes due to KCNJ11 mutations: an international cohort study. Lancet Diabetes Endocrinol 6(8):637–646
Bowman P, Mathews F, Barbetti F, Shepherd MH, Sanchez J, Piccini B et al (2021) Long-term follow-up of glycemic and neurological outcomes in an international series of patients with sulfonylurea-treated ABCC8 permanent neonatal diabetes. Diabetes Care 44(1):35–42
Kleinberger JW, Copeland KC, Gandica RG, Haymond RW, Levitsky LL, Linder B et al (2018) Monogenic diabetes in overweight and obese youth diagnosed with type 2 diabetes:the TODAY clinical trial. Genet Med 20(6):583–590
Todd JN, Kleinberger JW, Zhang H, Srinivasan S, Tollefsen SE, Levitsky LL et al (2021) Monogenic diabetes in youth with presumed type 2 diabetes:results from the progress in diabetes genetics in youth (ProDiGY) collaboration. Diabetes Care 44(10):2312–2319
Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J et al (2015) Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med 17(5):405–424
Philippi A, Heller S, Costa IG, Senée V, Breunig M, Li Z et al (2021) Mutations and variants of ONECUT1 in diabetes. Nat Med 27(11):1928–1940
De Franco E, Owens NDL, Montaser H, Wakeling MN, Saarimäki-Vire J, Ibrahim H, et al (2021) Primate-specific ZNF808 is essential for pancreatic development in humans. medRxiv 08.23.21262262
Monogenic Diabetes Variant Curation Expert Panel. Available online: https://www.clinical genome.org/affiliation/50016/ (Accessed 29/April/2022)
Clinical Genome Resource (ClinGen). Available online: https://clinicalgen.org (accessed 29/April/2022).
Li M, Wang S, Xu K, Chen Y, Fu Q, Gu Y et al (2020) High prevalence of a monogenic cause in Han Chinese diagnosed with type 1 diabetes, partly driven by nonsyndromic recessive WFS1 mutations. Diabetes 69(1):121–126
Patel KA, Ozbek MN, Yildiz M, Guran T, Kocyigit C, Acar S et al (2022) Systematic genetic testing for recessively inherited monogenic diabetes: a cross-sectional studies in paedaitric diabetes clinics. Diabetologia 65(2):336–342
Brener A, Zeitlin L, Wilnai Y, et al. (2022) Looking for the skeleton in the closet-rare genetic diagnosis in patients with diabetes and skeletal manifestation. Acta Diabetol 59(5):711–719
Grasso V, Colombo C, Favalli V, Galderisi A, Rabbone I, Gombos S et al (2013) Six cases with severe insulin resistance (SIR) associated with mutations of insulin receptor: Is a Bartter-like syndrome a feature of congenital SIR ? Acta Diabetol 50(6):951–957
Roehlen N, Hilger H, Stock F, Gläser B, Guhl J, Schmitt-Graeff A et al (2018) 17q12 deletion syndrome as a rare cause for diabetes mellitus type MODY 5. J Clin Endocrinol Metab 103(10):3601–3610
Ng N, Zamuner MM, Siddique N, Kim J, Burke M, Byrne MM (2022) Genotype-phenotype correlations and response to glucose-lowering therapy in subjects with HNF1b associated diabetes. Acta Diabetol 59(1):83–93
Stoffers DA, Stanojevic V, Habener JF (1998) Insulin promoter factor-1 gene mutation linked to early-onset type 2 diabetes mellitus directs expression of a dominant negative isoprotein. J Clin Invest 102(1):232–241
Iafusco D, Massa O, Pasquino B, Colombo C, Iughetti L, Bizzarri C et al (2012) Minimal incidence of Neonatal/Infancy onset diabetes in Italy is 1:90,000 live births. Acta Diabetol 49(5):405–408
Prudente S, Andreozzi F, Mercuri L, Alberico F, Digiamberardino A, Mannino GC et al (2022) Contribution of ONECUT1 variants to different forms of non-autoimmune diabetes mellitus in Italian patients. Acta Diabetol 59(8):113–116
McClenaghan C, Rapini N, De Rose UD, Gao J, Roeglin J, Bizzarri C et al (2022) Sulfonylurea-insensitive permanent neonatal diabetes caused by a severe gain-of-function Tyr330His substitution in Kir6.2. Horm Res Paediatr 95(3):215–223
Musso C, Cochran E, Moran SA, Skarulis MC, Oral EA, Taylor SI, Gorden P (2004) Clinical course of genetic disease of the insulin receptor (Type A and Rabson-Mendenhall syndromes). A 30 year prospective. Medicine 83(4):209–222
Martínez-Montoro JI, Pinzón-Martín JL, Damas-Fuentes M, Fernández-Valero A, Tinahones FJ (2022) Combination therapy with semaglutide and dapagliflozin as an effective approach for the management of type A insulin resistance syndrome: a case report. Front Endocrinol 13:838887
Galderisi A, Tamborlane W, Taylor SI, Attia N, Moretti C, Barbetti F (2022) SGLT2i improve glycemic control in patients with congenital severe insulin resistance. Pediatrics 150:e2021055671
Acknowledgements
We thank all collegues for referring cases to us. MDEP members’ opinion and sharing of data are gratefully acknowledged.
Funding
Open access funding provided by Università degli Studi di Roma Tor Vergata within the CRUI-CARE Agreement. This research received no funding.
Author information
Authors and Affiliations
Contributions
Conceptualization: FB; Methodology: FB, GB, OP, RR, AN, MM; Validation: RR, AN, MM, NR, FB; Investigation: NR, PIP, RS, PC, VP, MCM, AD, SC and FB; Data Curation: NR, FB; Original Draft Preparation: FB; Writing -Review & Editing: SC, RR, FB; Visualization: FB; Supervision: FB All Authors have approved the submitted version of this work.
Corresponding author
Ethics declarations
Conflict of interest
Authors have no financial or non-financial interests directly or indirectly related to the work submitted for publication.
Informed consent
Informed consent to the genetic testing was obtained by all patients involved in the study or by their guardians.
Institutional review board statement
The study was conducted in accordance with the Declaration of Helsinki, and approved by the Institutional Review Board (or Ethics Committee) of Bambino Gesù Children’s Hospital protocol code # RRC-2018–2365812.
Additional information
Managed by Antonio Secchi.
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
The original online version of this article was revised: Corrections under “Genetic screening” and in Table 2 caption updated.
Supplementary Information
Below is the link to the electronic supplementary material.
Supplementary file1
Center pie chart shows incident NDM cases (in orange), 57 cases with clinical diagnosis of MODY plus 2 past cases with NDM (in blue) and cases with clinical diagnosis of type A severe insulin resistance (in grey). Pie chart on the left shows pathogenic, likely pathogenic and variants of uncertain significance (VUS) identified in incident patients with NDM. Pie chart on the right shows MODY cases positive to GCK (in orange), HNF1A (in grey), INSR (in dark blue) or causative variants in other genes (in red). In light blue variants of undetermined effect and in 2 shades of green cases negative to genetic testing. (PDF 37 KB)
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Rapini, N., Patera, P.I., Schiaffini, R. et al. Monogenic diabetes clinic (MDC): 3-year experience. Acta Diabetol 60, 61–70 (2023). https://doi.org/10.1007/s00592-022-01972-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00592-022-01972-2