
Abstract
Background
Vaniprevir (MK-7009) is a hepatitis C virus (HCV) non-structural 3/4a protease inhibitor which significantly increases virologic response rates in HCV genotype (GT) 1-infected patients when added to peginterferon and ribavirin (PR).
Methods
This was a phase II, multicenter, double-blind, randomized, dose-ranging study in Japanese patients with HCV GT1 infection and previous relapse. Patients received twice daily vaniprevir 100, 300, or 600 mg, or placebo plus PR for 4 weeks then PR alone for 2 weeks. Further treatment with PR was continued up to a maximum of 72 weeks. The primary endpoint was rapid virologic response (RVR; undetectable HCV RNA at treatment week 4).
Results
Ninety patients completed 4 weeks of vaniprevir/placebo plus PR. Rates of RVR were significantly higher with vaniprevir compared with placebo (86, 95, and 76 % in the vaniprevir 100-, 300-, and 600-mg arms versus 20 % with control; p<0.001 for all comparisons). Rates of SVR, an exploratory analysis, in the vaniprevir 100-, 300-, 600-mg, and control arms were 95, 100, 100, and 72 %, respectively. No patient had virologic breakthrough or non-response while receiving vaniprevir. There were no serious adverse events (AEs) or discontinuations due to an AE during vaniprevir treatment. Diarrhea and nausea were more common with vaniprevir 600 mg than control or lower vaniprevir doses.
Conclusion
The addition of vaniprevir to PR was associated with an increase in RVR and SVR. Combined with a generally safe and well-tolerated profile, these data supported the further evaluation of vaniprevir in Japanese patients with HCV GT1 infection (#NCT00880763).
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
There are approximately 2 million patients with the hepatitis C virus (HCV) infection in Japan [1]. HCV infection is the leading cause of hepatocellular carcinoma in Japan, causing more than 30,000 deaths each year. Peginterferon plus ribavirin dual therapy remains the cornerstone of treatment for HCV infection, although the direct-acting antiviral agents, telaprevir and simeprevir, are also available in Japan for use as part of triple therapy regimens for selected patients [2]. Triple therapy with telaprevir, peginterferon alfa-2b, and ribavirin has shown improved response rate with room for further improvement, but adverse events (AEs) also increased, including severe anemia and serious skin rashes [2]. Triple therapy with simeprevir, peginterferon, and ribavirin has also recently become available for clinical use in Japan, further improving treatment options for patients with HCV infection in Japan [3]. Additional targeted therapies for the treatment of HCV infection are being developed with the aim of further decreasing HCV-related morbidity and mortality among Japanese patients.
Vaniprevir (MK-7009) is a macrocyclic inhibitor of the HCV non-structural (NS) 3/4a protease [4]. It exhibits potent inhibitory activity in in vitro models of HCV replication and induces a rapid decline in viral load in animal models of HCV infection [5, 6]. In a phase Ib monotherapy study in patients with chronic genotype (GT)1 HCV infection, vaniprevir demonstrated potent antiviral activity, good plasma exposure, and was generally well tolerated [7]. In two phase II clinical trials conducted in non-Japanese patients, addition of vaniprevir to peginterferon alfa-2a and ribavirin significantly improved on-treatment virologic response and sustained virologic response (SVR) in patients with HCV GT1 infection who were either naive to previous therapy or had failed previous treatment with peginterferon and ribavirin [8–10]. In 1 of 2 studies, vaniprevir was administered with peginterferon alfa-2a and ribavirin for 24 or 48 weeks [9, 10]. Alternatively, Manns and colleagues reported the use of vaniprevir (300 or 600 mg twice daily [bid], or 600 or 800 mg once daily [qd]) plus peginterferon alfa-2a and ribavirin for 4 weeks followed by open-label peginterferon alfa-2a and ribavirin for an additional 44 weeks in treatment-naive patients with chronic hepatitis C [8]. In this latter study, the rate of rapid virologic response (RVR; undetectable HCV RNA at treatment week 4), the primary endpoint of the study, was significantly higher in patients receiving vaniprevir than in those receiving placebo. Data from this study suggest that vaniprevir is a potent HCV protease inhibitor with a predictable resistance profile and favorable safety profile that is suitable for once a day or twice a day administration [8].
The aim of the present dose-ranging study was to assess the efficacy and safety of several doses of vaniprevir in combination with peginterferon alfa-2a and ribavirin in Japanese patients with HCV GT1 infection who had relapsed following previous treatment with peginterferon and ribavirin dual therapy to determine the clinical dose of vaniprevir appropriate for use in Japanese patients for further evaluation of vaniprevir in phase III studies.
Methods
The study was conducted in accordance with principles of Good Clinical Practice, approved by the appropriate institutional review boards and regulatory agencies, and is registered with ClinicalTrials.gov (#NCT00880763).
Patients
Japanese patients with HCV infection and relapse following previous treatment with peginterferon and ribavirin dual therapy were enrolled. Key inclusion and exclusion criteria included: age 20–64 years; chronic, compensated, GT1 HCV infection; and HCV RNA levels ≥5.0 log IU/mL at screening. Patients with cirrhosis were excluded: all patients were required to have fibrosis scores of F0–F2 within 3 years of screening, or F3 within 1 year of screening. Patients with retinopathy, including retinal hemorrhage, at screening were also excluded. All patients were required to meet protocol-defined laboratory criteria within 75 days prior to first dose.
Study design
This was a phase II, multicenter, double-blind, randomized, placebo-controlled, and dose-ranging study. Patients were randomized to receive vaniprevir 100, 300, 600 mg bid, or matched placebo in combination with peginterferon alfa-2a (180 µg/week) and ribavirin (600–1,000 mg/day, according to body weight) per product label for an initial 4 week treatment period, and thereafter they received peginterferon alfa-2a plus ribavirin alone for 2 additional weeks. The initial 4 week treatment period with vaniprevir and peginterferon alfa-2a plus ribavirin was used to align with the primary efficacy endpoint of RVR (the proportion of patients with undetectable HCV RNA at treatment week 4). The additional 2 weeks of treatment with peginterferon alfa-2a and ribavirin after the completion of vaniprevir dosing was implemented to enable continued monitoring of vaniprevir safety after the completion of triple therapy and to facilitate uninterrupted continued treatment with peginterferon and ribavirin, in line with current treatment recommendations [2]. Further treatment with peginterferon alfa-2a plus ribavirin was then administered at the discretion of each investigator, up to a maximum total treatment duration of 72 weeks, while also taking into consideration recommendations from Japanese treatment guidelines. Treatment blinding was achieved through the centralized preparation of identical capsules. Dose reduction and interruption of peginterferon alfa-2a and ribavirin were permitted in accordance with product labels.
Within the total treatment population, a pharmacokinetic (PK) cohort was identified to provide blood samples for PK analyses. Specific sites with the ability to perform an intensive PK sampling scheme were identified, and patients screened at these sites were offered the option to participate in the intensive PK cohort. The computer-generated randomized allocation schedule was prepared by the study sponsor, independent of the study team, and implemented by a third-party vendor. Randomization was stratified according to participation in the PK cohort with the aim of enrolling 4–8 patients in the PK cohort per treatment arm.
Endpoints
The study consisted of 2 parts, an initial 6 week phase (4 weeks of treatment with vaniprevir/placebo plus PR and a subsequent 2 week post-vaniprevir treatment follow-up period) followed by a second treatment phase up to 72 weeks in total duration. Primary efficacy and safety assessments were evaluated based on the treatment period from baseline to treatment week 6. HCV RNA measurements, evaluation of viral resistance, and assessments of serious adverse events were based on the entire study period including treatment period and follow-up period.
The primary efficacy endpoint was RVR, defined as the proportion of patients with undetectable HCV RNA at treatment week 4. Secondary virologic endpoints included the proportion of patients achieving a ≥2-log10 decrease and a ≥3-log10 decrease in viral load from baseline to week 4, and the mean log10 decrease in viral load from baseline to week 4. Analysis of SVR, defined as the proportion of patients with undetectable HCV RNA 24 weeks after completion of treatment, was not defined in the protocol; however, this endpoint was also calculated in an exploratory manner. Patients in the per protocol population for the primary analysis with an HCV RNA measurement at each time point were included in the exploratory SVR analysis.
Resistance-associated variants were to be analyzed at baseline for all patients, and at virologic failure in patients with breakthrough viremia or those with a null response while receiving vaniprevir/placebo through week 4. Breakthrough viremia was defined as a >1-log10 increase from nadir HCV RNA at 2 consecutive HCV RNA measurements, or HCV RNA >100 IU/mL at 2 consecutive visits after becoming undetectable. Null response was defined as ≤1-log decrease in HCV RNA through week 4. The persistence of resistance variants in patients who had taken vaniprevir was also assessed.
Pharmacokinetic analyses were also performed to evaluate the following vaniprevir parameters: area under the plasma concentration versus time curve from the time of dosing to 12 h post-dose (AUC0–12 h), maximum plasma drug concentration (C max), trough plasma drug concentration(C trough; i.e., concentration at approximately 12 h post-dose), time to reach C max (T max), and apparent terminal half-life (t ½). For PK analyses, blood sampling points were set at pre-dosing on day 1, week 1, and week 4, and anytime during the visits at weeks 5 and 6 in the all subjects population. In addition, blood samples for PK analyses were taken at 0.5, 1, 1.5, 2, 3, 4, 8, and 12 h post-dose on day 1 and week 4, and at 24 h post-dose on week 4 in the PK cohort. At each time point, 4 mL blood was placed into a vacuumized blood collection tube with EDTA-2 K, and plasma was immediately obtained by centrifugation at 0–10 °C and 1,200 relative centrifugal force (× g) for 15 min.
Safety evaluations included AEs, laboratory values, physical examinations, 12-lead electrocardiogram, and vital sign assessments.
Assays
Serum HCV RNA concentrations
Serum HCV RNA levels were measured using Roche COBAS® TaqMan® HCV Auto assay. The limit of quantification (LoQ) was 1.2 log IU/mL (15 IU/mL) and the limit of detection (LoD) was <1.2 log IU/mL, but with no specific value.
Vaniprevir plasma concentrations
Plasma samples underwent liquid–liquid extraction with methyl t-butyl ether in a 96-well format. The organic supernatant was subsequently evaporated under a nitrogen stream at approximately 40 °C, and the remaining residue was reconstituted in 200 μL 50:50 acetonitrile/water, v/v. The resulting samples were assessed using a validated liquid chromatography/tandem mass spectrometry assay with positive ion electrospray (range 1–1,000 ng/mL). Pharmacokinetic analyses of vaniprevir plasma concentrations were performed using WinNonlin (Certara, ver. 5.2.1).
Resistance testing
Blood samples were obtained for analysis of vaniprevir resistance-associated variants at baseline for all patients and at virologic failure in patients with breakthrough viremia or those with a null response while receiving vaniprevir through week 4. Samples were also collected after completion of vaniprevir treatment to assess the persistence of NS3/4A resistance mutations in patients who have taken vaniprevir. Viral resistance testing utilized population sequence analysis of the HCV NS3/4A region. Due to the sensitivity of this assay, resistance analysis was performed only on samples with HCV RNA >1,000 IU/mL.
Statistics
Target randomization was approximately 80 patients in this study. With evaluable data expected from approximately 18 patients in each treatment arm, this study had 91 % power to declare that vaniprevir is superior to placebo, assuming true RVR rates of 60 % in the vaniprevir plus PR, and 10 % in the placebo plus PR at week 4.
Primary efficacy analyses were based on the per protocol population which excluded patients with important deviations from the protocol that might have substantially affected the results of the primary efficacy analysis. For the primary endpoint, between treatment arm differences were assessed using 95 % confidence intervals (95 % CI) and associated p-values calculated using the Miettinen and Nurminen method [11]. A closed testing procedure with a fixed sequence of tests was used to adjust for multiplicity of multiple dose levels. For the secondary efficacy endpoints, 95 % CIs of between treatment arm differences were calculated using the Miettinen and Nurminen method [11]. A longitudinal data analysis model proposed by Liang and Zeger [12] was used to compare vaniprevir with placebo with respect to the mean log10 decrease in viral load. Missing data were excluded from analysis for each time point. SVR was also summarized in an exploratory manner.
Summary statistics (geometric mean and 95 % CI) for PK parameters were presented by treatment arm and by day. Median (range) was also provided for T max, and harmonic mean (pseudo standard deviation) was also provided for t ½.
Safety analysis was based on the all patients as a treated (APaT) population. AEs (specific terms as well as system organ class terms) were summarized by treatment arm. Rashes that were categorized as serious AEs and anemia were pre-specified as events of interest. The p values and 95 % CIs for between-treatment differences were calculated using the Miettinen and Nurminen method [11]. Summary statistics for baseline, on-treatment, and change from baseline values were provided in laboratory parameters, 12-lead electrocardiogram, and vital signs.
Results
Patient disposition and baseline characteristics
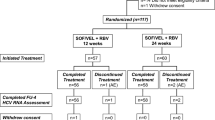
This study was performed at 38 study sites in Japan between May 2009 and February 2012. In total, 118 patients provided informed consent, of whom 90 were randomized and completed 4 weeks of treatment with vaniprevir/placebo plus PR and 2 weeks of follow-up [control arm (placebo plus PR), n = 22; vaniprevir 100-mg bid arm (vaniprevir 100 mg bid plus PR), n = 23; vaniprevir 300-mg bid arm (vaniprevir 300 mg bid plus PR), n = 22; and vaniprevir 600-mg bid arm (vaniprevir 600 mg bid plus PR), n = 23] (Fig. 1). Seven patients were excluded from the per protocol population due to various reasons such as insufficient dosing of study medications.

Patient disposition. AE adverse event, bid twice daily, PR peginterferon alfa-2a and ribavirin
In total, 9 patients discontinued the study after completing the post-vaniprevir 2 week follow-up period (i.e.,>2 weeks after completing vaniprevir/placebo dosing). Four patients withdrew consent, 2 patients were withdrawn at the investigator’s discretion, 2 patients discontinued because of AE(s) (1 patient in the vaniprevir 600-mg bid arm and 1 patient in the control arm), and 1 patient in the control arm was withdrawn because of lack of efficacy (Fig. 1). The median (range) duration of PR therapy in each treatment arm was as follows: vaniprevir 100-mg bid arm, 48.1 weeks (47.7, 72.1 weeks); vaniprevir 300-mg bid arm, 48.1 weeks (6.1, 72.1); vaniprevir 600-mg bid arm, 48.0 weeks (12.0, 72.0); and control arm, 49.4 weeks (29.1, 72.1). In total, 7 patients in the vaniprevir 100-mg bid arm, 5 patients in the vaniprevir 300-mg bid arm, 5 patients in the vaniprevir 600-mg bid arm, and 10 patients in the control arm received >48 weeks of PR therapy.
Patient characteristics were generally similar across treatment arms (Table 1). Most patients had HCV GT1b infection: the proportion of women was slightly higher in the control arm and the proportion of men was slightly higher in the vaniprevir 300-mg bid arm. Approximately 80 % of patients had previously received peginterferon alfa-2b in combination with ribavirin.
Efficacy
Virologic response
Treatment with vaniprevir was associated with significant improvement in virologic response compared with control during the first 4 weeks of therapy. The proportion of patients achieving RVR (the primary endpoint) was significantly higher in each of the vaniprevir treatment arms compared with the control arm (p<0.001 for all comparisons) (Fig. 2). Rates of RVR were 20 % in the control arm compared with 86, 95, and 76 % in the vaniprevir 100-, 300-, and 600-mg bid treatment arms, respectively.

Rapid virologic response. PR peginterferon alfa-2a and ribavirin, RVR rapid virologic response (undetectable HCV RNA at treatment week 4). *p < 0.001 versus PR control
Secondary virologic endpoints also indicated profound virologic suppression associated with vaniprevir therapy. All patients in vaniprevir arms regardless of dose achieved a ≥3-log reduction in HCV RNA from baseline by treatment week 4. By comparison, of the 20 patients in control arm, 19 (95 %) achieved a ≥2-log reduction in HCV RNA from baseline at week 4 and 17 (85 %) achieved a ≥3-log decline. Change from baseline in mean log HCV RNA during the first 4 weeks of treatment was also significantly greater in patients in vaniprevir arms than in the control arm (Fig. 3). The model-based mean treatment differences (vaniprevir minus control) in log HCV RNA change from baseline at treatment week 4 were −1.8 log10 (95 % CI −2.3, −1.2), −1.9 log10 (95 % CI −2.4, −1.3), and −1.6 log10 (95 % CI −2.2, −1.1) in the vaniprevir 100-, 300-, and 600-mg bid arms, respectively.

Mean decline in HCV RNA from baseline to week 6. bid twice daily, PR peginterferon alfa-2a and ribavirin. *Administered with peginterferon alfa-2a and ribavirin
The proportion of patients achieving SVR in the control, vaniprevir 100-, 300- and 600-mg bid arms were 72, 95, 100, and 100 %, respectively (Fig. 4).

Sustained virologic response. PR peginterferon alfa-2a and ribavirin, SVR sustained virologic response (undetectable HCV RNA 24 weeks after completing last dose of treatment)
Resistance-associated variants
Baseline sequences of NS3/4A were obtained from all but 2 patients by population-based sequencing. Virus from 2 patients was found to encode variants known to confer reduced susceptibility to vaniprevir, including the A156T variant (300-mg bid arm) or the D168E variant (600-mg bid arm), respectively. Both patients achieved RVR and SVR.
No patient in the vaniprevir arms met the criteria for breakthrough or non-response. Only 1 patient in the control arm met the criteria for breakthrough with a viral load >1,000 IU/mL during the first 6 weeks of the study. Population sequencing of the HCV NS3/4A RNA was performed with the samples obtained from this patient at baseline and at the virologic failure time points; however, no resistance variant was observed at either time point consistent with placebo plus P/R treatment.
After the peginterferon alfa-2a and ribavirin dual-therapy treatment period, only 1 patient who previously received vaniprevir experienced relapse. This patient, in the vaniprevir 100-mg bid arm, experienced relapse 24 weeks after completing a 48 week treatment regimen with peginterferon and ribavirin (including the initial 4 week treatment with vaniprevir). No vaniprevir-associated RAVs were detected at treatment failure in this patient.
Pharmacokinetics
Non-compartmental analysis was performed based on intensive bioanalytical data of vaniprevir in the PK cohort (n = 5 in the 100-mg bid arm, n = 4 in the 300-mg bid arm, and n = 5 in the 600-mg bid arm). As a result, vaniprevir was rapidly eliminated from the plasma. Plasma exposures (AUC0–12 h and C max) on day 1 increased non-linearly with dose, with disproportionately greater exposure seen at higher doses (Supplementary Table 1 and Fig. 1). Plasma exposures at steady state at week 4 also indicated non-linearity with dose, similar to day 1 observations. There was a trend towards an increase in T max on day 1 with increasing dose. Median T max values were 1.0 h with vaniprevir 100 mg bid, 2.5 h with 300 mg bid, and 3.0 h with 600 mg bid. This trend remained consistent with multiple dosing, and overall T max values were similar on day 1 and week 4. Elimination half-life (t ½) was similar on day 1 and at week 4. Day 1 t ½ harmonic means were 1.8–5.3 h compared to 1.6–4.1 h at week 4. Accumulation ratios at week 4 were 2.5-fold for AUC0–12 h and 1.6–3.3 fold for C max with the vaniprevir 100- and 300-mg bid doses, but no accumulation was observed in the 600-mg bid treatment arm. Trough concentrations (C12 h) of vaniprevir ranged from 32–305 nM in the 100-, 300-, and 600-mg bid treatment arms (Supplementary Table 2).
After completion of the vaniprevir dosing period, quantifiable drug levels (>1.3 nM) were detected at week 5 in 1 patient from the 600-mg bid arm, and at week 6 in 1 patient from the 300-mg bid arm. These plasma concentrations were extremely low, less than 1 % of the vaniprevir C max. In all other patients, vaniprevir plasma concentrations at week 5 and week 6 were unquantifiable.
Safety
All patients experienced at least 1 AE during the vaniprevir dosing period, all of which were mild to moderate severity except for a severe AE of depressed level of consciousness that resolved within 2 h in 1 patient in the vaniprevir 600-mg bid arm. There were no serious AEs and no patient discontinued treatment due to an AE or serious AE during the vaniprevir treatment period (day 1 to week 4) or the 2 week follow-up period (up to week 6). Adverse events occurring during the first 6 weeks of therapy with an incidence rate >30 % in any treatment arm are shown in Table 2. Gastrointestinal AEs, including diarrhea and nausea, were more common in patients in the vaniprevir 600-mg bid arm than those in the control arm or lower vaniprevir dose arms. Other AEs were consistent with the known safety profile of peginterferon and ribavirin, and did not display a dose-dependent relationship with vaniprevir dosing.
Rash categorized as a serious adverse event and anemia were pre-specified AEs of interest in the protocol. The incidence of anemia was similar in the control arm and vaniprevir treatment arms [14 % (3/22) in control arm, 9 % (2/23) in the vaniprevir 100-mg bid arm, 18 % (4/22) in the vaniprevir 300-mg bid arm, and 13 % (3/23) in the vaniprevir 600-mg bid arm] and no significant difference was observed in each vaniprevir treatment arm compared with the control arm (p = 0.602 in the vaniprevir 100-mg bid arm, p = 0.684 in the vaniprevir 300-mg bid arm, and p = 0.954 in the vaniprevir 600-mg bid arm). No rashes categorized as serious AEs were reported in the control or vaniprevir treatment arms.
Alanine aminotransferase and aspartate aminotransferase levels declined from baseline during the first 6 weeks of treatment with no notable difference between the control and vaniprevir arms (Fig. 5). Early, transient bilirubin elevations were noted in patients in the vaniprevir 600-mg bid arm; however, these elevations returned to baseline with continued therapy. Hemoglobin levels also declined during treatment in all treatment arms, with no notable difference between the control and vaniprevir treatment arms (Fig. 5).

Change from baseline laboratory observations during vaniprevir therapy: a alanine aminotransferase; b aspartate aminotransferase; c bilirubin; d hemoglobin. bid twice daily, PR peginterferon alfa-2a and ribavirin. *Administered with peginterferon alfa-2a and ribavirin
There were no clinically meaningful differences in vital signs or in ECG parameters between treatment arms during the vaniprevir treatment period or the 2 week safety follow-up period.
After the post-vaniprevir treatment follow-up period (>2 weeks after completing vaniprevir dosing); 8 serious AEs were reported in 6 patients. One patient in the control arm with severe acute renal failure and severe disseminated intravascular coagulation died from a severe bacterial infection (this patient was also counted as discontinuing PR due to AEs). These serious AEs were not considered related to vaniprevir/placebo by the investigator. The remaining 5 serious AEs reported in single patients were cataract, hepatic steatosis, diabetes mellitus, inguinal hernia, and infective spondylitis: in all cases, a causal relationship to vaniprevir/placebo was excluded by the investigator. One additional patient in the vaniprevir 600-mg bid arm discontinued PR after the post-vaniprevir treatment follow-up period because of a non-serious AE.
Discussion
This study shows that addition of vaniprevir to peginterferon alfa-2a plus ribavirin in Japanese patients with HCV GT1 infection was associated with a rapid decline in the HCV RNA levels, and at treatment week 4 rates of RVR were significantly increased compared with peginterferon alfa-2a plus ribavirin alone. All AEs but 1 were of mild or moderate severity, and generally similar to the established safety profile of peginterferon plus ribavirin dual therapy. An increase in gastrointestinal AEs was noted in patients receiving vaniprevir 600 mg bid. These efficacy and safety findings are consistent with previous observations from phase II studies of vaniprevir in non-Japanese patients [8–10].
Manns and colleagues have reported the results of a previous phase II study incorporating a similar study design to that used in the present study but with a different patient population [8]. In the study reported by Manns and colleagues, vaniprevir (300 or 600 mg bid, or 600 or 800 mg qd) was dosed with peginterferon alfa-2a and ribavirin for 4 weeks followed by peginterferon alfa-2a plus ribavirin dual therapy for an additional 44 weeks in treatment-naive, non-Japanese patients with chronic hepatitis C [8]. RVR was achieved by 69–83 % of patients receiving peginterferon alfa-2a plus ribavirin and vaniprevir compared with 5.6 % of patients receiving peginterferon alfa-2a plus ribavirin and placebo (control arm). SVR was achieved by 61–84 % of patients in the vaniprevir arms compared with 63 % in the control arm [8]. Thus, both the study by Manns and colleagues [8] and data from the present study indicate that addition of vaniprevir to peginterferon plus ribavirin is associated with a rapid decline in HCV RNA and high rates of RVR in patients with HCV GT1 infection.
Although in the present study an analysis of SVR was not defined in the protocol, this endpoint was assessed in an exploratory fashion, and the data demonstrate high rates of SVR associated with vaniprevir therapy. Careful interpretation of these data is required. The total treatment duration of peginterferon alfa-2a plus ribavirin was not identical in all patients because the protocol allowed a maximum of 72 weeks of treatment at the discretion of the investigator. Furthermore, SVR rates were calculated based on the per protocol population for consistency with the primary analysis population. Despite these limitations, SVR rates in the present study indicate that a 4 week dosing period with vaniprevir coadministered with peginterferon alfa-2a plus ribavirin followed by a maximum total 72 weeks of peginterferon alfa-2a plus ribavirin offers substantial benefit to Japanese patients with HCV infection. The relatively high SVR rate in the control arm may also be attributed to the extended treatment duration.
Japanese treatment guidelines recommend extending peginterferon plus ribavirin therapy from 48–72 weeks for patients in whom HCV RNA becomes undetectable between treatment weeks 13 and 36 [2]. Similarly, elderly patients and those with advanced fibrosis who attain undetectable HCV RNA between weeks 9 and 12 are also candidates for a 72 week treatment duration. Thus, patients with delayed virologic response are potentially faced with a long and arduous peginterferon and ribavirin treatment regimen with a well-described tolerability profile, including the influenza-like symptoms of interferon-based therapy and the hemolytic anemia associated with ribavirin therapy. Addition of a targeted therapy such as vaniprevir to peginterferon and ribavirin has the potential to substantially reduce overall treatment duration by reducing the on-treatment time to first undetectable HCV RNA.
The main safety observations from previous vaniprevir phase II studies that were conducted in non-Japanese patients included an increase in mild-to-moderate gastrointestinal AEs, primarily nausea, vomiting, and diarrhea. This occurred when vaniprevir was added to peginterferon and ribavirin [8–10]. In these studies there was no significant difference in the rates of anemia and rash between the vaniprevir and control arms [8, 9]. Safety findings from the present study are consistent with those previous reports. In the present study, gastrointestinal AEs, including diarrhea and nausea, were more common in patients receiving vaniprevir 600 mg bid than those receiving placebo or lower vaniprevir doses. All gastrointestinal AEs were mild or moderate in intensity and none of them resulted in discontinuation of study treatment during the vaniprevir treatment or 14 day safety follow-up period; thus, vaniprevir-based triple therapy was considered generally well tolerated.
Drug resistance is an important consideration in the use of HCV protease inhibitors [13]. In the present study, the high efficacy demonstrated in vaniprevir-treated patients limits the number of cases of drug resistance provided by this study and restricts evaluation of drug limitations due to resistance-derived failure. Baseline variants at positions A156 and D168 known to confer reduced susceptibility to vaniprevir [14] were observed in 2 patients in vaniprevir treatment arms, with both patients achieving RVR and SVR. It is difficult to draw general conclusions from this, however, due to the limited data set.
PK analyses indicate that, based on a twice daily dose regimen, vaniprevir exposure increases non-linearly with increasing dose, with disproportionately higher exposure achieved at higher doses. Mean trough concentrations of vaniprevir were 32, 86, and 305 nM with the 100-, 300-, and 600-mg bid dosing regimens. These values are several fold above the serum-adjusted in vitro vaniprevir 50 % effective concentration (EC50) of 5.9 nM against the HCV GT1b replicon (Data on file, Merck & Co., Inc.), suggesting that viral replication will remain suppressed at trough plasma concentrations.
In conclusion, the results from the present study indicate that the addition of vaniprevir to peginterferon plus ribavirin is associated with a rapid decline in HCV RNA in Japanese patients with HCV GT1 infection, reducing the time to undetectable HCV RNA for a substantial number of patients. Combined with an acceptable tolerability profile, these data support the further evaluation of vaniprevir in Japanese patients with HCV GT1 infection. Based on the cumulative efficacy, safety, and PK data, vaniprevir at a dose of 300 mg bid was selected for further evaluation in phase III clinical trials.
References
Chung H, Ueda T, Kudo M. Changing trends in hepatitis C infection over the past 50 years in Japan. Intervirology. 2010;53(1):39–43.
Guidelines for the management of hepatitis. C virus infection: first edition, May 2012, the Japan Society of Hepatology. Hepatol Res. 2013;43(1):1–34.
Hayashi N, Seto C, Kato M, et al. Once-daily simeprevir (TMC435) with peginterferon/ribavirin for treatment-naive hepatitis C genotype 1-infected patients in Japan: the DRAGON study. J Gastroenterol. 2014;49(1):138–47.
McCauley JA, McIntyre CJ, Rudd MT, et al. Discovery of vaniprevir (MK-7009), a macrocyclic hepatitis C virus NS3/4a protease inhibitor. J Med Chem. 2010;53(6):2443–63.
Olsen DB, Davies ME, Handt L, et al. Sustained viral response in a hepatitis C virus-infected chimpanzee via a combination of direct-acting antiviral agents. Antimicrob Agents Chemother. 2011;55(2):937–9.
Liverton NJ, Carroll SS, Dimuzio J, et al. MK-7009, a potent and selective inhibitor of hepatitis C virus NS3/4A protease. Antimicrob Agents Chemother. 2010;54(1):305–11.
Lawitz E, Sulkowski M, Jacobson I, et al. Characterization of vaniprevir, a hepatitis C virus NS3/4A protease inhibitor, in patients with HCV genotype 1 infection: safety, antiviral activity, resistance, and pharmacokinetics. Antiviral Res. 2013;99(3):214–20.
Manns MP, Gane E, Rodriguez-Torres M, et al. Vaniprevir with peginterferon alfa-2a and ribavirin in treatment-naive patients with chronic hepatitis C: a randomized phase 2 study. Hepatology. 2012;56(3):884–93.
Lawitz E, Rodriguez-Torres M, Stoehr A, et al. A phase 2b study of Mk-7009 (vaniprevir) in patients with genotype 1 HCV infection who have failed previous pegylated interferon and ribavirin treatment. J Hepatol. 21 Feb 2013.
Rodriguez-Torres M, Stoehr A, Gane EJ, et al. Combination of vaniprevir with peginterferon and ribavirin significantly increases the rate of sustained viral response in treatment-experienced patients with chronic HCV genotype 1 infection and cirrhosis. Clin Gastroenterol Hepatol. 2014;12(6):1029–37.
Miettinen O, Nurminen M. Comparative analysis of two rates. Stat Med. 1985;4(2):213–26.
Liang K-Y, Zeger SL. Longtitudinal data analysis using generalized linear models. Biometrika. 1986;73(1):13–22.
Halfon P, Locarnini S. Hepatitis C virus resistance to protease inhibitors. J Hepatol. 2011;55(1):192–206.
Barnard RJ, McHale CM, Newhard W, et al. Emergence of resistance-associated variants after failed triple therapy with vaniprevir in treatment-experienced non-cirrhotic patients with hepatitis C-genotype 1 infection: a population and clonal analysis. Virology. 2013;443(2):278–84.
Acknowledgments
We thank all patients, their families, investigators, and their staff in the following 38 study sites for completion of the study (ordered according to the site number assigned in the study): Tetsuo Takehara (Osaka University Hospital); Atsuo Inoue (Osaka General Medical Center); Namiki Izumi (Musashino Red Cross Hospital); Junichi Tazawa (Tsuchiura Kyodo General Hospital); Hideki Hagiwara (Kansai Rosai Hospital); Harumasa Yoshihara (Osaka Rosai Hospital); Masahide Oshita (Osaka Police Hospital); Toshihide Shima (Saiseikai Suita Hospital); Shoichi Takahashi (Hiroshima University Hospital); Keisuke Iizuka (Isesaki municipal hospital); Kiyomi Yasuda (Kiyokawa Hospital); Takashi Kumada (Ogaki Municipal Hospital); Eiji Mita (National Hospital Organization Osaka National Hospital); Kazuyoshi Nagayama (Namegata District General Hospital); Tatsuji Komatsu (National Hospital Organization Yokohama Medical Center); Teruaki Kawanishi (Hokkaido Health Coop Sapporo Ryokuai Hospital); Kunihiko Tsuji (Teine Keijinkai Hospital); Akira Ohashi (Japanese Red Cross Koga Hospital); Makoto Shimizu (International Goodwill Hospital); Satoru Kaneda (National Hospital Organization Chiba Medical Center); Michiyasu Yagura (National Hospital Organization Tokyo National Hospital); Toshifumi Ito (Osaka Koseinenkin Hospital); Ryo Nakata (Japanese Red Cross Medical Center); Yasuyuki Aisaka (Hiroshima Red Cross Hospital & Atomic-bomb Survivors Hospital); Yuji Yoshikawa (Sanraku Hospital); Akihisa Ishikawa (Hitachi General Hospital); Kazuhiro Katayama (Osaka Medical Center for Cancer and Cardiovascular Diseases); Eijiro Hayashi (Kinki Central Hospital of the Mutual Aid Association of Public School Teachers); Seiji Kawazoe (Saga-Ken Medical Centre KOSEIKAN); Yasuharu Imai (Ikeda Municipal Hospital); Tatsuho Sugimoto (Tonami General Hospital); Hiroshi Yatsuhashi (National Hospital Organization Nagasaki Medical Center); Michio Kato (National Hospital Organization Minamiwakayama Medical Center); Yutaka Sasaki (Kumamoto University Hospital); Makoto Nakamuta (National Hospital Organization Kyushu Medical Center); Youichi Morimoto (Kurashiki Central Hospital); Akio Ido (Kagoshima University Hospital); Yoshiaki Inui (Hyogo Prefectural Nishinomiya Hospital). We also thank Go Fujimoto, Richard J.O. Barnard, Luzelena Caro, Takashi Iwasa, Keisuke Nakamura, Akiko Takase, Victoria Enwemadu, and Yoshiyuki Tanaka of Merck and MSD K.K. for their support in data analysis and/or preparation of this manuscript. This study was funded by MSD K.K., a subsidiary of Merck and Co., Inc., Whitehouse Station, NJ, USA. Medical writing and editorial assistance were provided by Tim Ibbotson, PhD, and Bianca B Ruzicka, PhD, of ApotheCom, Yardley, PA. This assistance was funded by Merck and Co., Inc., Whitehouse Station, NJ.
Conflict of interest
Norio Hayashi has no conflict of interest. Niloufar Mobashery is an employee of Merck and has company stock ownership. Namiki Izumi received lecture fees from MSD K.K.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and the source are credited.
About this article

{kind=link}
Cite this article
Hayashi, N., Mobashery, N. & Izumi, N. Vaniprevir plus peginterferon alfa-2a and ribavirin in treatment-experienced Japanese patients with hepatitis C virus genotype 1 infection: a randomized phase II study. J Gastroenterol 50, 238–248 (2015). https://doi.org/10.1007/s00535-014-0979-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00535-014-0979-2