
Abstract
We have greatly increased our understanding of the genetics of inflammatory bowel disease (IBD) in the last decade; however, migrant studies highlight the importance of environment in disease risk. The possibility that IBD is an infection has been debated since the first description of Crohn’s disease. Mycobacterium avium paratuberculosis was the first organism to be suggested as an IBD pathogen, and it has been argued that it fulfils Koch’s postulates and could be designated the cause of Crohn’s disease. Other organisms have been postulated as possible IBD pathogens, including various Helicobacter species, one of which has been identified in primate colitis; others are widely used in animal models of IBD. Adherent-invasive Escherichia coli appear specific to ileal Crohn’s disease and have been shown to induce the release of TNF-α, a key cytokine in IBD inflammation. The aim of this article is to give a concise overview of the infections postulated as being relevant to the onset of IBD. We will also briefly cover the immunology underpinning IBD, in addition to reviewing current knowledge regarding other microorganisms that are associated with modifying the risk of developing IBD. It may be that infectious organisms have an orchestrator role in the development of dysbiosis and subsequently IBD.
Similar content being viewed by others


Introduction
“I can only regret that the etiology of the condition remains in obscurity, but I trust that ere long further consideration will clear up the difficulty.”
Thomas Kennedy Dalziel on chronic interstitial enteritis (now Crohn’s disease) in 1913 [1].
We have come a long way since Kennedy Dalziel’s original description of what has since become known as Crohn’s disease [2], but the possibility of an infectious aetiology for the disease remains after 96 years of increasingly complex research into the condition and its clinical bedfellow ulcerative colitis (UC). Dalziel himself likened the condition to tuberculous disease and was the first author to suggest a microbial aetiology by comparing Crohn’s with Johne’s disease of cattle, an association that continues to be quoted and which we will consider later [3].
The areas with the highest incidence and prevalence of inflammatory bowel disease (IBD) are those traditionally classed as Western countries, namely those in Northern Europe and North America [4]. It is clear from migrant studies that regional, ethnic and racial differences are less important to disease aetiology than lifestyle and environmental influences, as individuals moving from low-occurrence areas to high-occurrence areas demonstrate an increased risk of disease [4]. An increase in the incidence of IBD has also been described in previously low-incidence, non-Western countries such as Japan [5]. The root cause of this increase remains unclear, but increasing “Westernisation” and infectious agents may be of importance. The hygiene hypothesis [6] originally suggested that an increasingly clean environment could result in allergic disease aetiology; however, it is now being linked to a broader array of immunological disorders [7]. It may be that this facet of Western culture, alongside poor dietary habits (which often prevail in the West), may play a part in IBD pathogenesis. What is not clear is whether these factors, along with genetic susceptibility, lead directly to IBD or whether they allow the conditions needed for pathogens to thrive. In this article we will discuss the various organisms postulated as infectious agents in IBD.
Immunology
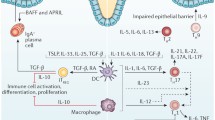
The immunology of IBD at a basic level can be thought of as an imbalance between two populations of T cells: pro-inflammatory effector T (TEff) cells and regulatory T (TReg) cells (see Fig. 1).

T cell balance between intestinal inflammation and health. IBD immunology centres on an imbalance between pro-inflammatory effector T cells typified by TH17 and regulatory T cells (TReg). The differentiation of TH17 is dependent on IL-23, and this cell lineage is responsible for the key pro-inflammatory cytokine IL-17. The development of regulatory T cells is controlled by FOXP3, and the anti-inflammatory cytokines released include IL-10 and IL-35 along with TGF-β, which can be pro-inflammatory or anti-inflammatory
The most commonly described TEff cells in IBD are T-helper 1 (TH1) and 17 (TH17) in Crohn’s disease and T-helper 2 (TH2) in UC [8]. The differentiation of TH17 cells is dependent on the key IL-23 pathway [9, 10]. Interestingly, variations in the IL-23 receptor gene have been shown to be protective against ileal Crohn’s disease [11]. TH17 is largely responsible for the production of IL-17, an important pro-inflammatory cytokine which is increased in IBD [12]. In contrast, the development of TReg cells is thought to be controlled by transcription factor forkhead box P3 (FOXP3) [13], and these cells in turn release the anti-inflammatory cytokines TGF-β, IL-10 and IL-35. TGF-β plays either a pro-inflammatory or an anti-inflammatory role depending on the level of tissue inflammation present [14], promoting either FOXP3-mediated development of TReg cells in a non-inflamed environment or the differentiation of TH17 cells in an inflamed environment. The role of intestinal bacteria in this balance has recently been brought into sharp focus by the seminal work of Mazmanian et al. [15], which has shown the influence of a single microbial molecule on intestinal inflammatory balance. Polysaccharide A (PSA), produced by Bacteroides fragilis, suppresses the production of IL-17 by an as-yet unidentified route which has been shown to be dependent on IL-10-producing T cells. Interestingly, this study made use of Helicobacter hepaticus, which is a benign commensal in wild-type mice but can initiate disease as a pathogen in immunocompromised mice [16]. This mechanism may parallel human IBD, with those individuals now described as being genetically susceptible to IBD in fact being susceptible to pathogens which would be harmless in others [17, 18]. PSA has been termed a “symbiosis factor”, and it is likely the first of many such factors to be discovered. Developments in this field are likely to expand our understanding of the mechanisms of probiotic efficacy and the immunological mechanisms of the role that dysbiosis has in IBD.
The role of pattern-recognising signal proteins in IBD was brought to the fore with the discovery on chromosome 16 of the NOD2/CARD15 (nucleotide-binding oligomerization domain 2/caspase activation recruitment domain 15) gene [19]. This heralded the beginning of an unprecedented era of genetic discovery and improved the understanding of the immunobiology of mucosal defences in the gastrointestinal tract and the defective immune response underpinning IBD. A number of IBD susceptibility loci have since been identified, which are reviewed in detail elsewhere [17, 18]. What has become clear, however, is the role that the innate immune system plays in sensing and responding to bacteria within the gut lumen in order to maintain health in the gastrointestinal tract. The large surface area of the gastrointestinal tract and the sheer number of live microbes in close contact with the epithelial surface mean that there is a constant balance between maintaining and sustaining symbiotic species on one hand and recognising and responding to pathogens on the other. An imbalance in the proportions of “protective” and “harmful” bacteria has been termed “dysbiosis”, and this is thought to be central to IBD pathogenesis [20–22]. NOD2/CARD15 represents an intracellular protein expressed in Paneth cells at the base of intestinal crypts [23] which is involved in sensing muramyl dipeptide (MDP), a key component of the cell walls of both Gram-positive and Gram-negative bacteria [24]. Loss of NOD2/CARD15 function is thought to lead to an altered innate immune response to bacterial cells and resultant inappropriate activation of the NF-κB pathway and uncontrolled inflammation [25]. Nod-like receptors (NLRs) and Toll-like receptors (TLRs) are both involved in bacterial sensing in the mucosal (innate) immune system, and mutations have been shown in both groups in IBD [26–29] featuring variously NODs 1 and 2, TLRs 1, 2, 4, 5, 6 and 9 and NF-κB, a downstream transcription factor within NOD/TLR signalling pathways.
The discovery of a Crohn’s disease association with the autophagy gene ATG16L1 has led to work in mice demonstrating that loss of this gene’s function results in aberrant IL-1β production in response to bacterial endotoxins [30, 31]. Autophagy is the process whereby cells recycle internal organelles and proteins, and this process is rapidly increased in times of cell stress or starvation [32, 33]. Interestingly, this process is essential to Mycobacterium tuberculosis inhibition in infected macrophages, and so defective ATG16L1 function may lend support to the idea of a Mycobacterial pathogen in Crohn’s disease [34].
Why genetically susceptible individuals can live for years in good health before developing disease or in many cases never develop disease at all is still a mystery. Intestinal microbes, in particular bacteria, are thought to be pivotal to the development of disease [35]. The exact process of transition from health to disease is, however, still unclear. The role of disease triggers in genetically susceptible populations must be understood as the next step in our greater understanding of IBD aetiopathogenesis.
Bacteria
Our understanding of the role that bacteria play within the human gastrointestinal tract and their role in both health and disease is currently undergoing a revolution. The numerical importance of our cohabitants has been clear for some time; they outnumber human cells by a factor of 10:1 [36, 37]. It has also been clear for some time that they are not metabolically inert, having a metabolic activity equating to that of a virtual organ [38]. This relationship is clearly symbiotic, the most commonly cited example of this symbiosis being that of butyrate production, which is dependent on colonic bacteria. Butyrate is the main fuel source for colonocytes, and it is known to stimulate the proliferation and differentiation of epithelial cells in the gut [39, 40]. It is now becoming clear that the symbiosis between human hosts and intestinal microbes extends beyond mere substrate exchange, as the discovery of PSA, described above, clearly demonstrates. The finding of this anti-inflammatory polysaccharide heralds a new era of understanding the role of our cohabitants in the balance between health and disease [15].
With regards to IBD, it has long been recognised that bacteria are essential to the development of the disease [41], and historical animal models of Crohn’s disease have initiated granulomatous change in both mice and rabbits by infiltrating healthy animal tissue with human Crohn’s tissue [42, 43]. The lack of transmissibility of disease by transfer of stool from human IBD patients to the monkey colon has however been cited as demonstrating a lack of evidence for a specific pathogen [44]. However, animal work has eloquently demonstrated the ability of an organism to induce colitis in immunodeficient (IL-10-deficient) but not immunocompetent mice [16]. This observation may be in keeping with our increasing knowledge of the genetic basis for IBD [17, 18], and perhaps our quest for a specific pathogen in IBD has been hindered by a lack of understanding of the host conditions that are required for pathogenesis.
A fascinating paper by Van Kruiningen et al. [45] examined two French families in which Crohn’s disease was highly prevalent (6 of 6 family members in one family in the same home and 7 of 11 children in another). Despite compelling evidence for an infective organism (clustering of disease, similarity of distribution and extent of disease and timespan) and an exhaustive search for evidence of an infectious organism (examining Campylobacter-like organisms, Mycobacteria, Yersinia, Mycoplasma, Coronavirus, Brucella, influenza, toroviruses and pestiviruses), none was identified. Interestingly, both families were noted to be regular consumers of unpasteurised milk, a recognised source of Mycobacterium avium paratuberculosis (MAP), a frequently proposed Crohn’s pathogen and a possible source of bacterial entry [3, 46].
Many bacterial agents have been postulated as infectious agents in IBD. The most important of these are summarised in Table 1.
Mycobacteria
Mycobacterium avium paratuberculosis has been implicated in Crohn’s pathogenesis since Dalziel’s original description of the condition in 1913 [1]. The link has been based largely on the similarity of Crohn’s to the bovine condition Johne’s disease, characterised by granulomatous inflammation of the intestines [3]. There is growing concern regarding the difficulty of removing MAP from the food supply, particularly in milk products, and this has led some European countries to move towards eradicating it [47]. The literature surrounding MAP and Crohn’s disease is vast. There have been conflicting reports regarding MAP DNA positivity from Crohn’s biopsies, but it has been postulated that this may be due to MAP DNA residing intracellularly in the cytoplasm of human cells; hence the possibility of false negativity [47]. Greenstein fluently argues the case for MAP as the causative organism underlying Crohn’s disease with respect to each of Koch’s four postulates [48], including culture [49–51].
Detractors from the hypothesis that MAP is causative state various arguments, chief of which is the observation that anti-TNF-α antibodies are so effective in treating Crohn’s disease and yet they have been shown to cause reactivation of latent tuberculosis [52–55].
Helicobacter
Cotton-top Tamarin colitis (CTT) is essentially the Helicobacter equivalent of MAP’s Johne’s disease as an animal model for IBD; however, whereas Johne’s parallels Crohn’s disease, CTT parallels UC with similar clinical and pathological findings. The suggestion of an infectious aetiology for CTT which is a highly prevalent pancolitis in captive monkeys but rare in the wild dates back to the observations of Johnson et al. [56]. A novel urease-negative Helicobacter species was identified from monkeys with this condition [57]. This organism, previously dubbed “Flexispira taxon”, is now thought to incorporate at least 10 Helicobacter taxa, including Helicobacter bilis, Helicobacter callitrichis and Helicobacter trogontum, along with other species which have not been formally characterised but which are often referred to as Helicobacter rappini or Helicobacter flexispira [58].
Different species of Helicobacter organisms have been used to initiate colitis in rodent models, for example Helicobacter hepaticus and Helicobacter bilis [59, 60]. Interestingly, a reduction in commensal organisms in response to Helicobacter infection has also been shown, and it has since been demonstrated that an IgG immune response towards commensal bacteria predates the onset of colitis after infection with Helicobacter bilis [61–63]. This suggests a potential role for Helicobacter organisms in orchestrating the switch from a healthy microbiota to dysbiosis [64].
Non-pylori Helicobacter organisms have never been isolated from humans with IBD, but the pathogenic potential of these organisms in humans has been made clear by their association with proctocolitis and enteritis in homosexual men [65]. Attempts to demonstrate their presence in IBD have at times given disappointing results, with Bell et al. [66] failing to demonstrate their presence utilising a PCR-only method, and Grehan et al. [67] also failing to demonstrate their presence utilising nested PCR. In 2006, however, Zhang et al. [68] demonstrated a 92% prevalence of Helicobacter organisms in children with IBD against 25% in paediatric controls utilising multiple techniques (PCR, DGGE, FISH). Our own FISH work has supported this finding, demonstrating a non-pylori Helicobacter prevalence of 79% in adult UC against 23% in adult controls [69] and 87% in paediatric UC against 40% in paediatric controls [70]. We have shown a similarly high prevalence of these organisms in paediatric Crohn’s disease at 83% (unpublished data). Work is currently underway to identify to species level which non-pylori Helicobacter are present. Candidate Helicobacter organisms which have been associated with human gastrointestinal illnesses are summarised in Table 2.
Whether or not Helicobacter organisms have a role in human IBD and understanding what that role is will depend on our culturing these organisms from affected individuals for further work before addressing Koch’s postulates [48] once again.
Escherichia coli
Modern interest in Escherichia coli as a pathogen in IBD began with the dual observations that organisms isolated from patients with Crohn’s disease had greater adherent properties to human cells than those from controls [71], and that previously unrecognised invasive E. coli organisms with adherent properties were present in Crohn’s ileal tissue [72]. These invasive organisms with adherent properties have been termed adherent-invasive E. coli (AIEC) [72]. In addition to being able to invade intestinal epithelial cells, AIEC (strain LF82) can invade human macrophages, survive and replicate extensively for long periods [73]. Such infected macrophages release high levels of tumour necrosis factor (TNF-α) (2.7 times more than cells stimulated by E. coli 01141:B4 lipopolysaccharide) [73]. TNF-α is a key cytokine in intestinal inflammation, and has been recognised as being released in large amounts in Crohn’s disease [74, 75]. In direct contrast to the MAP/anti-TNF-α conundrum, this observation fits nicely with the success of anti-TNF-α antibodies as a treatment in Crohn’s disease [55].
AIEC organisms have been found to be more prevalent in early ileal Crohn’s lesion tissue (36.4%) versus controls (6.2%), but interestingly, these organisms were relatively rarely found in colonic tissue from Crohn’s patients (3.7%) and controls (1.9%), and they were not identified at all in UC colonic tissue, suggesting a specific association with ileal Crohn’s [76]. The number of E. coli organisms identified within the ileum in Crohn’s has recently been significantly correlated with the activity of ileal disease at endoscopy [77]. Similar organisms have also been found to be universally present in granuloma tissue from Boxer dogs with colitis [78], suggesting their ability to initiate pathognomic granulomatous Crohn’s changes in the intestinal mucosa.
Risk attributed to gastroenteritis
Porter et al. [79] recently published a large retrospective case–control study examining the development of IBD in army personnel and its association with prior gastroenteritis. This study reported an increased risk for the development of IBD following an infectious gastroenteritis exposure (odds ratio 1.40, 95% CI 1.19–1.66), with odds ratios for Crohn’s and UC independently being 1.54 (1.17–2.04) and 1.36 (1.08–1.72), respectively. Interestingly, exposure was not based on pathogen identification, and so bacteria, viruses and protozoal infections may all have been included in the cases studied. Whether a small group of specific pathogens accounted for a high risk which was diluted within the large sample or whether any gastroenteritis exposure confers an increased risk is thus unknown. Certainly recent animal work in rats has shown that colitis itself can lead to dysbiosis, so it is possible that a broad range of pathogens can serve to initiate IBD [80]. Porter et al. [79] excluded possible infectious gastroenteritis episodes within 6 months of a formal IBD diagnosis. The reason for this exclusion was to remove false diagnoses of infection, which were in fact first presentations of IBD. The authors themselves acknowledge that this cut-off may have served to under-represent the true association between infectious gastroenteritis and IBD, particularly if the period between exposure to a pathogen and development of IBD is short.
A 15-year follow-up study from Denmark recently examined the risk of developing IBD following infection with either Campylobacter or Salmonella gastroenteritis [81]. The risk for developing IBD following infection was 2.9 (95% CI 2.2–3.9) times higher for the whole 15-year period, with the risk attributable to the first year after exposure being 1.9 (1.4–2.6) times normal. Campylobacter and Salmonella spp. presence conferred similar risks, and the risk of developing Crohn’s versus UC was also broadly similar. This work has mirrored previous findings that are supportive of bacterial gastroenteritis as a risk factor for IBD and UC alone [82–84]. Interestingly, the Campylobacter infections in the Danish study were diagnosed by microscopy and positive oxidase and catalase reactions only. Given the recent finding of higher numbers of non-jejuni Campylobacter species in children with Crohn’s disease by Zhang et al. [85], it would be interesting to examine the Campylobacter organisms isolated in the Danish study to species level by molecular methods. The organism highlighted by Zhang et al. as being the most prevalent was Campylobacter concisus (57% oxidase positive, 0% catalase positive [86]), but other oxidase/catalase-positive organisms such as Campylobacter showae (50% oxidase positive, 100% catalase positive [86]), Campylobacter rectus (100% oxidase positive, 20% catalase positive [86]) and Bacteroides ureolyticus (100% oxidase positive, 20% catalase positive [86]) were also identified from the Crohn’s cohort in the same study [85]. Further exploration of whether infection with specific Campylobacter species predisposes to IBD or whether this finding is generic is warranted.
Fungal, viral and helminthic agents
Fungi have had relatively little exposure as potential pathogens in IBD, despite the longstanding recognition of anti-Saccharomyces cerevisiae antibodies (ASCA) as a diagnostic marker for Crohn’s disease and a common antigen of Saccharomyces cerevisiae cell walls [87–89]. This antigen can also be expressed by Candida albicans [90]. It is clear that fungal species and particularly Candida species form a normal part of the gut microbiota [91]. It is also clear that perturbation of the gastrointestinal microbial community, for example during antibiotic intake, can increase fungal populations [92, 93]. It is not clear, however, whether the organisms themselves are responsible for the diarrhoea associated with antibiotic use [93]. Recent work has shown that Candida albicans is more frequently isolated from Crohn’s patients (44%) and their healthy relatives (38%) than controls (22%) [94]. Another recent study examined the fungal microbiota in IBD versus controls utilising 18S rDNA methods [95]. The study of Ott et al. found that fungal diversity was significantly increased in Crohn’s when compared with both inflamed and non-inflamed controls. UC fungal diversity was also increased, but not to significant levels.
Several viruses have also been implicated as exacerbating agents in IBD. These include chiefly cytomegalovirus (CMV), but recently parvovirus B19, norovirus and Epstein–Barr virus have all been suggested as exacerbating agents [96–98]. CMV appears to act through latent reactivation (reviewed succinctly by Hommes et al. [99]). The paramyxoviruses, and in particular measles, have been extensively debated in the medical literature since the observation that measles virus nucleocapsid protein appeared to be present in intestinal tissue from Crohn’s patients [100]. The link between measles and Crohn’s disease has been broadly disregarded by a body of evidence since; hence it will not be given further discussion within this article. Two excellent reviews which cover this topic have been written by Ghosh et al. [101] and Loftus [4].
The relationship between helminths and the human intestinal tract, and in particular the link between a lack of helminth infection and IBD, is a fascinating one which has recently been the subject of numerous excellent reviews [7, 102, 103]. The first proposal that lack of exposure to helminths predisposed to development of IBD—the so-called “IBD hygiene hypothesis”—was made in 2000 by Elliott et al., who suggested that the rearing of children in increasingly hygienic environments is detrimental to immune development, with a resultant predisposition to immunological diseases such as IBD [104].
Helminths as therapy have been trialled in both Crohn’s disease and UC [105, 106]. The Crohn’s study used an open-label approach in addition to standard treatment; 79.3% of patients responded and 72.4% entered remission. The UC study was a randomised controlled trial against placebo as an adjunct to standard treatment. This trial demonstrated a statistically significant improvement in disease activity in intention-to-treat analysis between the helminth group (43.3% improved) and placebo (16.7% improved, p = 0.04). Both of these trials utilised the pig whipworm Trichuris suis. The hookworm Necator americanus has also been successfully trialled as a potential therapy in a small group of patients with Crohn’s disease [107]. The basis for the beneficial effect of helminths in IBD is through their complex immunological relationship with their host organism. This is summarised in detail by Weinstock and Elliott [103] but involves their direct action on epithelial cells, helminth secretion of potentially anti-inflammatory substances such as TGF-β, and possibly also helminth factors which alter the luminal microbiota of the host.
Clues from diet
The pre-morbid state in Crohn’s disease includes a diet which is more likely to contain more refined sugar, less fibre, and less raw fruit and vegetables [108]. The refined sugar finding has been supported by a case–control study from Sweden demonstrating a positive link between sucrose intake and Crohn’s as well as protective effects of wholegrain bread, muesli and coffee and a surprisingly high risk (RR 3.4, 95% CI 1.3–9.3 for an intake of more than twice per week) associated with fast foods [109]. Vegetable intake and coffee were protective for UC, and fast foods conferred a high risk (3.9, 95% CI 1.4–10.6 for an intake of more than twice per week). A reduced intake of fruit and vegetables in IBD cases against assorted controls has also been shown in a multinational study recruiting in North American, Northern Europe and the Mediterranean [110]. The study of Thornton et al. [108] suggested that “dietary influences may alter the milieu of the intestinal lumen or modify the intestinal flora and so promote the growth of an infective agent or its invasion of the gut wall”. It may well be that the pre-morbid diet provides either specific or sufficient distal intestinal substrates which allow the invasion of a pathological microbe or the establishment of dysbiosis.
The mechanism by which exclusive enteral nutrition (EEN) is efficacious in Crohn’s disease remains a mystery, and does not appear to be due to prebiotic properties of the diet [111]. What remains clear, however, is that this is a therapy as efficacious as steroids for inducing remission in IBD [112]. Different hypotheses have been suggested for its mechanism of action [113], including prebiotic effects, nutritional and micronutrient repletion, removal of dietary antigens (bowel rest), and a reduction in dietary fat leading to a reduction in inflammatory mediator synthesis. Bowel rest has been rejected as a viable hypothesis because of the efficacy of whole protein enteral nutrition [114]. Gerasimidis et al. [111] have shown that despite achieving clinical remission, analysis of stools demonstrated what were thought to be detrimental changes. These changes are presumed to be due to a change to an unhealthy colonic milieu. Butyrate production was seen to decrease in this study. This reduction was deemed due to a lack of indigestible carbohydrates in the diet. A comparison of colonic mucosal bacterial populations before and after enteral nutrition may add greater understanding to the confusing conundrum of how an effective therapy can be detrimental to metabolic indicators of colonic bacteria. This may lend valuable insight into the mechanism of action of EEN, and this may lead to the modification of what can be a burdensome therapy. The fact that the remission induced by enteral nutrition is not long-lasting, with 60–70% of patients relapsing within 12 months of cessation of the therapy, suggests a modulation of bacterial activity rather than any permanent alteration to bacterial cohabitants [115]. This could be due to a reduction in the bacterial substrates available within the colon during EEN, which then return with restoration of a normal diet.
Discussion
The microscopic ecosystem that exists in the human colon and its symbiotic relationship with human health and disease has become the focus of much research interest [116]. It may well be that the unexplained rises in immune-mediated disorders (atopy, allergies, rheumatoid arthritis, IBD, multiple sclerosis, juvenile-onset diabetes mellitus, etc.) are due to our societal evolution away from our symbiotic cohabitants, potentially allowing more pathogenic organisms to take their place.
Many organisms have been studied as possible pathogens, but it seems unlikely that IBD acts as a traditional infection. It appears more likely that infectious agents facilitate a change from the status quo that is sufficient to allow the development of IBD. Thus, in order to understand IBD aetiology, we must study patients at the onset of disease in order to identify the specific trigger events (including infection) which lead to the development of IBD. It may be that certain trigger events can be identified which lead to the development of dysbiosis in genetically susceptible individuals and ultimately to IBD (Fig. 2). If this is the case, it may then be possible to identify these genetically susceptible individuals, perhaps during newborn screening, and then to intervene and prevent the trigger events that initiate disease. This may be achieved, for example, by immunisation for specific pathogens, dietary manipulation or probiotics from infancy, but this could ultimately prevent the development of disease. Clearly this route is not as financially profitable as developing new chronic therapies for advanced disease, but the potential impact of intervention is much larger.

Potential pathological cascade in inflammatory bowel disease. An underlying genetic susceptibility leads to the development of bacterial population shifts within the gastrointestinal tract to the detriment of the host (dysbiosis) after a trigger event. This leads to chronic intestinal inflammation and inflammatory bowel disease
Conclusion
As we increase our knowledge of the aetiology of IBD, we must take into account the differences in genetic susceptibility between Crohn’s disease and UC, explain the variable disease onset and presentations within each disease, and critically explain the mucosal population changes seen which lead to “dysbiosis” [20–22]. It may be that by considering Crohn’s and UC together we do both conditions a disservice, but they are naturally grouped together and each forms an intriguing control for the other. The quest for specific pathogens goes on, but until a microorganism isolated from a human with IBD can be shown to initiate the disease state in an animal model, we will be unable to state causation, and—as has been the case thus far—most studies are at best circumstantial. The key question relating to specific pathogens which needs an answer is: does the presence of these organisms initiate disease, or does the initiation of disease cause conditions suitable for the proliferation of these organisms?
References
Dalziel TK. Chronic interstitial enteritis. BMJ. 1913;2:1068–70.
Crohn BB, Ginsberg L, Oppenheimer GD. Regional ileitis: a clinical and pathological entity. JAMA. 1932;99:1323–9.
Pierce ES. Where are all the Mycobacterium avium subspecies paratuberculosis in patients with Crohn’s disease? PLoS Pathog. 2009;5(3):e1000234.
Loftus EV. Clinical epidemiology of inflammatory bowel disease: incidence, prevalence, and environmental influences. Gastroenterology. 2004;126(6):1504–17.
Asakura K, Nishiwaki Y, Inoue N, Hibi T, Watanabe M, Takebayashi T. Prevalence of ulcerative colitis and Crohn’s disease in Japan. J Gastroenterol. 2009;44(7):659–65.
Strachan DP. Hay fever, hygiene, and household size. BMJ. 1989;299(6710):1259–60.
Rook GAW. Review series on helminths, immune modulation and the hygiene hypothesis: the broader implications of the hygiene hypothesis. Immunology. 2009;126(1):3–11.
Himmel ME, Hardenberg G, Piccirillo CA, Steiner TS, Levings MK. The role of T-regulatory cells and Toll-like receptors in the pathogenesis of human inflammatory bowel disease. Immunology. 2008;125(2):145–53.
Yen D, Cheung J, Scheerens H, Poulet F, McClanahan T, Mckenzie B, et al. IL-23 is essential for T cell-mediated colitis and promotes inflammation via IL-17 and IL-6. J Clin Invest. 2006;116(5):1310–6.
McGovern D, Powrie F. The IL23 axis plays a key role in the pathogenesis of IBD. Gut. 2007;56(10):1333–6.
Duerr RH, Taylor KD, Brant SR, Rioux JD, Silverberg MS, Daly MJ, et al. A genome-wide association study identifies IL23R as an inflammatory bowel disease gene. Science. 2006;314(5804):1461–3.
Fujino S, Andoh A, Bamba S, Ogawa A, Hata K, Araki Y, et al. Increased expression of interleukin 17 in inflammatory bowel disease. Gut. 2003;52(1):65–70.
Fontenot JD, Rudensky AY. A well adapted regulatory contrivance: regulatory T cell development and the forkhead family transcription factor Foxp3. Nat Immunol. 2005;6(4):331–7.
Kim JM, Rudensky A. The role of the transcription factor Foxp3 in the development of regulatory T cells. Immunol Rev. 2006;212(1):86–98.
Mazmanian SK, Round JL, Kasper DL. A microbial symbiosis factor prevents intestinal inflammatory disease. Nature. 2008;453(7195):620–5.
Kullberg MC, Ward JM, Gorelick PL, Caspar P, Hieny S, Cheever A, et al. Helicobacter hepaticus triggers colitis in specific-pathogen-free interleukin-10 (IL-10)-deficient mice through an IL-12-and gamma interferon-dependent mechanism. Infect Immunol. 1998;66(11):5157–66.
Van Limbergen J, Russell RK, Nimmo ER, Satsangi J. The genetics of inflammatory bowel disease. Am J Gastroenterol. 2007;102(12):2820–31.
Ishihara S, Aziz MM, Yuki T, Kazumori H, Kinoshita Y. Inflammatory bowel disease: review from the aspect of genetics. J Gastroenterol. 2009;44(11):1097–108.
Hugot JP, Laurent-Puig P, Gower-Rousseau C, Olson JM, Lee JC, Beaugerie L, et al. Mapping of a susceptibility locus for Crohn’s disease on chromosome 16. Nature. 1996;379:821–3.
Sartor RB. Intestinal microflora in human and experimental inflammatory bowel disease. Curr Opin Gastroenterol. 2001;17(4):324–30.
Farrell RJ, LaMont JT. Microbial factors in inflammatory bowel disease. Gastroenterol Clin North Am. 2002;31(1):41–62.
Tamboli CP, Neut C, Desreumaux P, Colombel JF. Dysbiosis in inflammatory bowel disease. Gut. 2004;53(1):1–4.
Rosenstiel P, Fantini M, Bräutigam K, Kühbacher T, Waetzig GH, Seegert D, et al. TNF-α and IFN-γ regulate the expression of the NOD2 (CARD15) gene in human intestinal epithelial cells. Gastroenterology. 2003;124(4):1001–9.
van Heel DA, Hunt KA, King K, Ghosh S, Gabe SM, Mathew CG, et al. Detection of muramyl dipeptide-sensing pathway defects in patients with Crohn’s disease. Inflamm Bowel Dis. 2006;12(7):598–605.
Inohara N, Ogura Y, Nuñez G. Nods: a family of cytosolic proteins that regulate the host response to pathogens. Curr Opin Microbiol. 2002;5(1):76–80.
Goyette P, Labbe C, Trinh TT, Xavier RJ. Molecular pathogenesis of inflammatory bowel disease: genotypes, phenotypes and personalized medicine. Ann Med. 2007;39(3):177–99.
Franchimont D, Vermeire S, El Housni H, Pierik M, Van Steen K, Gustot T, et al. Deficient host-bacteria interactions in inflammatory bowel disease? The Toll-like receptor (TLR)-4 Asp299gly polymorphism is associated with Crohn’s disease and ulcerative colitis. Gut. 2004;53(7):987–92.
Török HP, Glas J, Tonenchi L, Bruennler G, Folwaczny M, Folwaczny C. Crohn’s disease is associated with a Toll-like receptor-9 polymorphism. Gastroenterology. 2004;127(1):365–6.
Mizoguchi A, Mizoguchi E. Inflammatory bowel disease, past, present and future: lessons from animal models. J Gastroenterol. 2008;43(1):1–17.
Hampe J, Franke A, Rosenstiel P, Till A, Teuber M, Huse K, et al. A genome-wide association scan of nonsynonymous SNPs identifies a susceptibility variant for Crohn disease in ATG16L1. Nat Genet. 2006;39(2):207–11.
Saitoh T, Fujita N, Jang MH, Uematsu S, Yang BG, Satoh T, et al. Loss of the autophagy protein Atg16L1 enhances endotoxin-induced IL-1b production. Nature. 2008;456:264–8.
Levine B, Deretic V. Unveiling the roles of autophagy in innate and adaptive immunity. Nat Rev Immunol. 2007;7(10):767–77.
Hold GL, El-Omar EM. Genetic aspects of inflammation and cancer. Biochem J. 2008;410(2):225–35.
Gutierrez MG, Master SS, Singh SB, Taylor GA, Colombo MI, Deretic V. Autophagy is a defense mechanism inhibiting BCG and Mycobacterium tuberculosis survival in infected macrophages. Cell. 2004;119(6):753–66.
Xavier RJ, Podolsky DK. Unravelling the pathogenesis of inflammatory bowel disease. Nature. 2007;448:427–34.
Tancrède C. Role of human microflora in health and disease. Eur J Clin Microbiol Infect Dis. 1992;11(11):1012–5.
Bengmark S. Ecological control of the gastrointestinal tract. The role of probiotic flora. Gut. 1998;42(1):2–7.
O’Hara AM, Shanahan F. The gut flora as a forgotten organ. EMBO Rep. 2006;7(7):688–93.
Frankel WL, Zhang W, Singh A, Klurfeld DM, Don S, Sakata T, et al. Mediation of the trophic effects of short-chain fatty acids on the rat jejunum and colon. Gastroenterology. 1994;106(2):375–80.
Pryde SE, Duncan SH, Hold GL, Stewart CS, Flint HJ. The microbiology of butyrate formation in the human colon. FEMS Microbiol Lett. 2002;217(2):133–9.
Sellon RK, Tonkonogy S, Schultz M, Dieleman LA, Grenther W, Balish E, et al. Resident enteric bacteria are necessary for development of spontaneous colitis and immune system activation in interleukin-10-deficient mice. Infect Immunol. 1998;66(11):5224–31.
Mitchell DN, Rees RJ. Agent transmissible from Crohn’s disease tissue. Lancet. 1970;2(7665):168–71.
Cave DR, Mitchell DN, Kane SP, Brooke BN. Further animal evidence of a transmissible agent in Crohn’s disease. Lancet. 1973;2(7838):1120–2.
Victor RG, Kirsner JB, Palmer WL. Failure to induce ulcerative colitis experimentally with filtrates of feces and rectal mucosa. Gastroenterology. 1950;14:398–400.
Van Kruiningen HJ, Colombel JF, Cartun RW, Whitlock RH, Koopmans M, Kangro HO, et al. An in-depth study of Crohn’s disease in two French families. Gastroenterology. 1993;104(2):351–60.
Herthnek D, Nielsen SS, Lindberg A, Bölske G. A robust method for bacterial lysis and DNA purification to be used with real-time PCR for detection of Mycobacterium avium subsp. paratuberculosis in milk. J Microbiol Methods. 2008;75:335–40.
Greenstein RJ. Is Crohn’s disease caused by a mycobacterium? Comparisons with leprosy, tuberculosis, and Johne’s disease. Lancet Infect Dis. 2003;3(8):507–14.
Koch R. Die Aetiologie der Tuberkulose. Mittbeilungen Kaiserlichen Gesundbeitsamte. 1884;2:1–88.
Chiodini RJ, Van Kruiningen HJ, Thayer WR, Merkal RS, Coutu JA. Possible role of mycobacteria in inflammatory bowel disease. Dig Dis Sci. 1984;29(12):1073–9.
Gitnick G, Collins J, Beaman B, Brooks D, Arthur M, Imaeda T, et al. Preliminary report on isolation of mycobacteria from patients with Crohn’s disease. Dig Dis Sci. 1989;34(6):925–32.
McFadden JJ, Butcher PD, Chiodini R, Hermon-Taylor J. Crohn’s disease-isolated mycobacteria are identical to Mycobacterium paratuberculosis, as determined by DNA probes that distinguish between mycobacterial species. J Clin Microbiol. 1987;25(5):796–801.
Sartor RB. Does Mycobacterium avium subspecies paratuberculosis cause Crohn’s disease? Gut. 2005;54(7):896–8.
Knight P, Campbell BJ, Rhodes JM. Host–bacteria interaction in inflammatory bowel disease. Br Med Bull. 2008;88(1):95–113.
Keane J, Gershon S, Wise RP, Mirabile-Levens E, Kasznica J, Schwieterman WD, et al. Tuberculosis associated with infliximab, a tumor necrosis factor α-neutralizing agent. NEJM. 2001;345(15):1098–104.
Hanauer SB, Feagan BG, Lichtenstein GR, Mayer LF, Schreiber S, Colombel JF, et al. Maintenance infliximab for Crohn’s disease: the ACCENT I randomised trial. Lancet. 2002;359(9317):1541–9.
Johnson LD, Ausman LM, Sehgal PK, King NW. A prospective study of the epidemiology of colitis and colon cancer in cotton-top tamarins (Saguinus oedipus). Gastroenterology. 1996;110(1):102–15.
Saunders KE, Shen Z, Dewhirst FE, Paster BJ, Dangler CA, Fox JG. Novel intestinal Helicobacter species isolated from cotton-top tamarins (Saguinus oedipus) with chronic colitis. J Clin Microbiol. 1999;37(1):146–51.
Dewhirst FE, Fox JG, Mendes EN, Paster BJ, Gates CE, Kirkbride CA, et al. ‘Flexispira rappini’ strains represent at least 10 Helicobacter taxa. Int J Syst Evol Microbiol. 2000;50(5):1781–7.
Cahill RJ, Foltz CJ, Fox JG, Dangler CA, Powrie F, Schauer DB. Inflammatory bowel disease: an immunity-mediated condition triggered by bacterial infection with Helicobacter hepaticus. Infect Immunol. 1997;65(8):3126–31.
Shomer NH, Dangler CA, Schrenzel MD, Fox JG. Helicobacter bilis-induced inflammatory bowel disease in scid mice with defined flora. Infect Immunol. 1997;65(11):4858–64.
Kuehl CJ, Wood HD, Marsh TL, Schmidt TM, Young VB. Colonization of the cecal mucosa by Helicobacter hepaticus impacts the diversity of the indigenous microbiota. Infect Immunol. 2005;73(10):6952–61.
Whary MT, Danon SJ, Feng Y, Ge Z, Sundina N, Ng V, et al. Rapid onset of ulcerative typhlocolitis in B6. 129P2-IL10 tm1Cgn (IL-10−/−) mice infected with Helicobacter trogontum is associated with decreased colonization by altered Schaedler’s Flora. Infect Immunol. 2006;74(12):6615–23.
Jergens AE, Wilson-Welder JH, Dorn A, Henderson A, Liu Z, Evans RB, et al. Helicobacter bilis triggers persistent immune reactivity to antigens derived from the commensal bacteria in gnotobiotic C3H/HeN mice. Gut. 2007;56(7):934–40.
Fox JG. Helicobacter bilis: bacterial provocateur orchestrates host immune responses to commensal flora in a model of inflammatory bowel disease. Gut. 2007;56(7):898–900.
Totten PA, Fennell CL, Tenover FC, Wezenberg JM, Perine PL, Stamm WE, et al. Campylobacter cinaedi (sp. nov.) and Campylobacter fennelliae (sp. nov.): two new Campylobacter species associated with enteric disease in homosexual men. J Infect Dis. 1985;151(1):131–9.
Bell SJ, Chisholm SA, Owen RJ, Borriello SP, Kamm MA. Evaluation of Helicobacter species in inflammatory bowel disease. Aliment Pharmacol Ther. 2003;18(5):481–6.
Grehan M, Danon S, Lee A, Daskalopoulos G, Mitchell H. Absence of mucosa-associated colonic helicobacters in an Australian urban population. J Clin Microbiol. 2004;42(2):874–6.
Zhang L, Day A, McKenzie G, Mitchell H. Nongastric Helicobacter species detected in the intestinal tract of children. J Clin Microbiol. 2006;44(6):2276–9.
Thomson JM, Hold G, Berry SH, El-Sakka NE, Mowat NA, Shen Z, et al. Variable detection of entero-hepatic Helicobacter species in colonic mucosal pinch biopsies by different molecular techniques. Gastroenterology. 2008;134(4):A655.
Hansen R, El-Sakka NE, Berry SH, Thompson JM, Bisset WM, Mahdi G, et al. Are non-pylori helicobacter organisms associated with paediatric ulcerative colitis? Retrospective observational study. Gastroenterology. 2009;136(5):A671.
Darfeuille-Michaud A, Neut C, Barnich N, Lederman E, Di Martino P, Desreumaux P, et al. Presence of adherent Escherichia coli strains in ileal mucosa of patients with Crohn’s disease. Gastroenterology. 1998;115(6):1405–13.
Boudeau J, Glasser AL, Masseret E, Joly B, Darfeuille-Michaud A. Invasive ability of an Escherichia coli strain isolated from the ileal mucosa of a patient with Crohn’s disease. Infect Immunol. 1999;67(9):4499–509.
Glasser AL, Boudeau J, Barnich N, Perruchot MH, Colombel JF, Darfeuille-Michaud A. Adherent invasive Escherichia coli strains from patients with Crohn’s disease survive and replicate within macrophages without inducing host cell death. Infect Immunol. 2001;69(9):5529–37.
MacDonald TT, Hutchings P, Choy MY, Murch S, Cooke A. Tumour necrosis factor-alpha and interferon-gamma production measured at the single cell level in normal and inflamed human intestine. Clin Exp Immunol. 1990;81(2):301–5.
Murch SH, Braegger CP, Walker-Smith JA, MacDonald TT. Location of tumour necrosis factor alpha by immunohistochemistry in chronic inflammatory bowel disease. Gut. 1993;34(12):1705–9.
Darfeuille-Michaud A, Boudeau J, Bulois P, Neut C, Glasser AL, Barnich N, et al. High prevalence of adherent-invasive Escherichia coli associated with ileal mucosa in Crohn’s disease. Gastroenterology. 2004;127(2):412–21.
Baumgart M, Dogan B, Rishniw M, Weitzman G, Bosworth B, Yantiss R, et al. Culture independent analysis of ileal mucosa reveals a selective increase in invasive Escherichia coli of novel phylogeny relative to depletion of Clostridiales in Crohn’s disease involving the ileum. ISME J. 2007;1(5):403–18.
Simpson KW, Dogan B, Rishniw M, Goldstein RE, Klaessig S, McDonough PL, et al. Adherent and invasive Escherichia coli is associated with granulomatous colitis in Boxer dogs. Infect Immunol. 2006;74(8):4778–92.
Porter CK, Tribble DR, Aliaga PA, Halvorson HA, Riddle MS. Infectious gastroenteritis and risk of developing inflammatory bowel disease. Gastroenterology. 2008;135(3):781–6.
Valcheva R, Slingerland B, Farrant A, Gänzle MG, Dieleman LA. Prebiotics maintain biodiversity of the intestinal microbiota and reduce colitis in HLA-B27 transgenic rats. Gastroenterology. 2009;136(5):A21.
Gradel KO, Nielsen HL, Schønheyder HC, Ejlertsen T, Kristensen B, Nielsen H. Increased short-and long-term risk of inflammatory bowel disease after Salmonella or Campylobacter gastroenteritis. Gastroenterology. 2009;137(2):495–501.
Rodríguez LAG, Ruigómez A, Panés J. Acute gastroenteritis is followed by an increased risk of inflammatory bowel disease. Gastroenterology. 2006;130(6):1588–94.
Helms M, Simonsen J, Molbak K. Foodborne bacterial infection and hospitalization: a registry-based study. Clin Infect Dis. 2006;15,42(4):498–506.
Ternhag A, Törner A, Svensson Å, Ekdahl K, Giesecke J. Short-and long-term effects of bacterial gastrointestinal infections. Emerg Infect Dis. 2008;14(1):143–8.
Zhang L, Man SM, Day AS, Leach ST, Lemberg DA, Dutt S, et al. Detection and isolation of Campylobacter species other than C. jejuni from children with Crohn’s disease. J Clin Microbiol. 2009;47(2):453–5.
On SLW. Taxonomy, phylogeny, and methods for the identification of campylobacter species. In: Ketley JM, editor. Campylobacter: molecular and cellular biology. 1st ed. Norwich: Horizon Bioscience; 2005. p. 13–42.
Barnes RM, Allan S, Taylor-Robinson CH, Finn R, Johnson PM. Serum antibodies reactive with Saccharomyces cerevisiae in inflammatory bowel disease: is IgA antibody a marker for Crohn’s disease? Int Arch Allergy Appl Immunol. 1990;92(1):9–15.
McKenzie H, Main J, Pennington CR, Parratt D. Antibody to selected strains of Saccharomyces cerevisiae (baker’s and brewer’s yeast) and Candida albicans in Crohn’s disease. Gut. 1990;31(5):536–8.
Heelan BT, Allan S, Barnes RMR. Identification of a 200-kDa glycoprotein antigen of Saccharomyces cerevisiae. Immunol Lett. 1991;28(3):181–5.
Standaert–Vitse A, Jouault T, Vandewalle P, Mille C, Seddik M, Sendid B, et al. Candida albicans is an immunogen for anti-Saccharomyces cerevisiae antibody markers of Crohn’s disease. Gastroenterology. 2006;130(6):1764–75.
Bernhardt H, Knoke M. Mycological aspects of gastrointestinal microflora. Scand J Gastroenterol Suppl. 1997;222:102–6.
Mavromanolakis E, Maraki S, Cranidis A, Tselentis Y, Kontoyiannis DP, Samonis G. The impact of norfloxacin, ciprofloxacin and ofloxacin on human gut colonization by Candida albicans. Scand J Infect Dis. 2001;33(6):477–8.
Krause R, Schwab E, Bachhiesl D, Daxbock F, Wenisch C, Krejs GJ, et al. Role of Candida in antibiotic-associated diarrhea. J Infect Dis. 2001;184(8):1065–9.
Standaert-Vitse A, Sendid B, Joossens M, François N, Vandewalle-El Khoury P, Branche J, et al. Candida albicans colonization and ASCA in familial Crohn’s disease. Am J Gastroenterol. 2009;104(7):1745–53.
Ott SJ, Kuhbacher T, Musfeldt M, Rosenstiel P, Hellmig S, Rehman A, et al. Fungi and inflammatory bowel diseases: alterations of composition and diversity. Scand J Gastroenterol. 2008;43(7):831–41.
Pironi L, Bonvicini F, Gionchetti P, D’Errico A, Rizzello F, Corsini C, et al. Parvovirus B19 infection localized in the intestinal mucosa and associated with severe inflammatory bowel disease. J Clin Microbiol. 2009;47(5):1591–5.
Khan RR, Lawson AD, Minnich LL, Martin K, Nasir A, Emmett MK, et al. Gastrointestinal norovirus infection associated with exacerbation of inflammatory bowel disease. JPGN. 2009;48(3):328–33.
Weinberg I, Neuman T, Margalit M, Ayman F, Wolf DG, Ben-Yehuda A. Epstein–Barr virus-related diarrhea or exacerbation of inflammatory bowel disease: a diagnostic dilemma. J Clin Microbiol. 2009;47(5):1588–90.
Hommes DW, Sterringa G, van Deventer SJH, Tytgat GNJ, Weel J. The pathogenicity of cytomegalovirus in inflammatory bowel disease. A systematic review and evidence-based recommendations for future research. Inflamm Bowel Dis. 2004;10(3):245–50.
Wakefield AJ, Pittilo RM, Sim R, Cosby SL, Stephenson JR, Dhillon AP, et al. Evidence of persistent measles virus infection in Crohn’s disease. J Med Virol. 1993;39(4):345–53.
Ghosh S, Armitage E, Wilson DC, Minor PD, Afzal MA. Detection of persistent measles virus infection in Crohn’s disease: current status of experimental work. Gut. 2001;48(6):748–52.
Reddy A, Fried B. An update on the use of helminths to treat Crohn’s and other autoimmunune diseases. Parasitol Res. 2009;104(2):217–21.
Weinstock JV, Elliott DE. Helminths and the IBD hygiene hypothesis. Inflamm Bowel Dis. 2009;15(1):128–33.
Elliott DE, Urban JOEF, Argo CK, Weinstock JV. Does the failure to acquire helminthic parasites predispose to Crohn’s disease? FASEB J. 2000;14(12):1848–55.
Summers RW, Elliott DE, Urban JF, Thompson R, Weinstock JV. Trichuris suis therapy in Crohn’s disease. Gut. 2005;54(1):87–90.
Summers RW, Elliott DE, Urban JF, Thompson RA, Weinstock JV. Trichuris suis therapy for active ulcerative colitis: a randomized controlled trial. Gastroenterology. 2005;128(4):825–32.
Croese J, O’Neil J, Masson J, Cooke S, Melrose W, Pritchard D, et al. A proof of concept study establishing Necator americanus in Crohn’s patients and reservoir donors. Gut. 2006;55(1):136–7.
Thornton JR, Emmett PM, Heaton KW. Diet and Crohn’s disease: characteristics of the pre-illness diet. BMJ. 1979;2(6193):762–4.
Persson PG, Ahlbom A, Hellers G. Diet and inflammatory bowel disease: a case–control study. Epidemiology. 1992;3(1):47–52.
Gilat T, Hacohen D, Lilos P, Langman MJS. Childhood factors in ulcerative colitis and Crohn’s disease: an international cooperative study. Scand J Gastroenterol. 1987;22(8):1009–24.
Gerasimidis K, McGrogan P, Garrick V, Hassan K, Edwards CA. Effect of exclusive enteral nutrition on colonic bacterial activity in paediatric Crohn’s disease. Proc Nutr Soc. 2009;67(OCE8):E425.
Heuschkel RB, Menache CC, Megerian JT, Baird AE. Enteral nutrition and corticosteroids in the treatment of acute Crohn’s disease in children. JPGN. 2000;31(1):8–15.
Griffiths AM. Enteral feeding in inflammatory bowel disease. Curr Opin Clin Nutr Metab Care. 2006;9(3):314–8.
Heuschkel R. Enteral nutrition in Crohn disease: more than just calories. JPGN. 2004;38(3):239–41.
Griffiths AM, Carricato M. Enteral nutrition in inflammatory bowel disease. In: Koletzko B, Cooper P, Makrides M, Garza C, Uauy R, Wang W, editors. Pediatric nutrition in practice. Basel: Karger; 2008. p. 219–23.
Round JL, Mazmanian SK. The gut microbiota shapes intestinal immune responses during health and disease. Nat Rev Immunol. 2009;9:313–23.
Man SM, Zhang L, Day AS, Leach S, Mitchell H. Detection of enterohepatic and gastric Helicobacter species in fecal specimens of children with Crohn’s disease. Helicobacter. 2008;13(4):234–8.
Fox JG, Yan LL, Dewhirst FE, Paster BJ, Shames B, Murphy JC, et al. Helicobacter bilis sp. nov., a novel Helicobacter species isolated from bile, livers, and intestines of aged, inbred mice. J Clin Microbiol. 1995;33(2):445–54.
Fox JG, Dewhirst FE, Shen Z, Feng Y, Taylor NS, Paster BJ, et al. Hepatic Helicobacter species identified in bile and gallbladder tissue from Chileans with chronic cholecystitis. Gastroenterology. 1998;114(4):755–63.
Stanley J, Linton D, Burnens AP, Dewhirst FE, Owen RJ, Porter A, et al. Helicobacter canis sp. nov., a new species from dogs: an integrated study of phenotype and genotype. Microbiology. 1993;139(10):2495–504.
Vandamme P, Harrington CS, Jalava K, On SLW. Misidentifying helicobacters: the Helicobacter cinaedi example. J Clin Microbiol. 2000;38(6):2261–6.
Vandamme P, Falsen E, Rossau R, Hoste B, Segers, Tytgat R, et al. Revision of Campylobacter, Helicobacter, and Wolinella taxonomy: emendation of generic descriptions and proposal of Arcobacter gen. nov. Int J Syst Evol Microbiol. 1991;41(1):88–103.
Fox JG, Dewhirst FE, Tully JG, Paster BJ, Yan L, Taylor NS, et al. Helicobacter hepaticus sp. nov., a microaerophilic bacterium isolated from livers and intestinal mucosal scrapings from mice. J Clin Microbiol. 1994;32(5):1238–45.
Stanley J, Linton D, Burnens AP, Dewhirst FE, On SLW, Porter A, et al. Helicobacter pullorum sp. nov.-genotype and phenotype of a new species isolated from poultry and from human patients with gastroenteritis. Microbiology. 1994;140(12):3441–9.
Fox JG, Chien CC, Dewhirst FE, Paster BJ, Shen Z, Melito PL, et al. Helicobacter canadensis sp. nov. isolated from humans with diarrhea as an example of an emerging pathogen. J Clin Microbiol. 2000;38(7):2546–9.
Tee W, Montgomery J, Dyall-Smith M. Bacteremia caused by a Helicobacter pullorum-like organism. Clin Infect Dis. 2001;33(10):1789–91.
Mendes EN, Queiroz DMM, Dewhirst FE, Paster BJ, Moura SB, Fox JG. Helicobacter trogontum sp. nov., isolated from the rat intestine. Int J Syst Evol Microbiol. 1996;46(4):916–21.
Acknowledgments
The authors wish to acknowledge funding from the Broad Foundation, USA, and the Chief Scientist Office, Scotland. RH is funded by a fellowship from the Chief Scientist Office in Scotland.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Hansen, R., Thomson, J.M., El-Omar, E.M. et al. The role of infection in the aetiology of inflammatory bowel disease. J Gastroenterol 45, 266–276 (2010). https://doi.org/10.1007/s00535-009-0191-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00535-009-0191-y