
Abstract
Purpose
This study aims to compare the efficacy and safety of oral palonosetron with intravenous (IV) palonosetron for the prevention of cisplatin-related chemotherapy-induced nausea and vomiting (CINV).
Methods
A multinational, randomized, double-blind study enrolling adult chemotherapy-naive patients with malignant solid tumors scheduled to receive cisplatin-based highly emetogenic chemotherapy (HEC). Patients received oral palonosetron (0.50 mg) or IV palonosetron (0.25 mg), each with oral dexamethasone. The primary objective was to demonstrate non-inferiority in terms of patients with a complete response (CR, no emesis/no rescue medication) within the acute phase (0–24 h after chemotherapy administration).
Results
Of the 743 patients randomized, 739 received study medications and 738 were included in the full analysis set. The CR rate in the acute phase was high for both groups (oral 89.4 %; IV 86.2 %). As this difference in proportions (stratum-adjusted Cochran-Mantel-Haenszel method) was 3.21 % (99 % confidence interval (CI) −2.74 to 9.17 %), non-inferiority was demonstrated (since the lower limit of the 99 % CI was closer to zero than the predefined margin of 15 %). Treatment-emergent adverse events (TEAEs) related to the study drug were rare (oral 3.2 %; IV 6.5 %). No TEAEs related to study drug leading to discontinuation were reported.
Conclusion
Non-inferiority of oral versus IV palonosetron was demonstrated. The CR rate in the acute phase was >86 % in both patient groups. The safety profiles were comparable.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Cisplatin-based chemotherapy regimens are a cornerstone of curative cancer treatment for non-seminomatous germ cell tumors and a standard treatment for most advanced lung and some gastrointestinal cancers; however, chemotherapy-induced nausea and vomiting (CINV) is a major side effect [1, 2]. Currently, neurokinin 1 (NK1) and 5-hydroxytryptamine 3 (5-HT3) receptor antagonists (RAs), used in combination with steroids such as dexamethasone, are recommended to control CINV related to cisplatin-based highly emetogenic chemotherapy (HEC) [3].
Palonosetron (Aloxi®, Oncit®, Paloxi®) is a potent and selective second-generation 5-HT3 RA which has demonstrated efficacy in the management of CINV when administered both intravenously (IV) and orally [4]. Its prolonged duration of action offers significant advantages over the other 5-HT3 RAs for the prevention of CINV in patients with cancer receiving both moderately and HEC regimens [5–9]. Palonosetron has also been shown to be effective in managing CINV associated with a variety of chemotherapeutic agents across a range of indications [10–19].
Clinical studies of CINV prevention with oral and IV palonosetron doses ranging from 0.1 to 90 μg/kg have been performed. The clinical development of IV palonosetron for the prevention of CINV comprised three efficacy studies, two in moderately emetogenic CINV [20, 21] and one in highly emetogenic CINV [22], which evaluated the efficacy and safety of two palonosetron doses, 0.25 and 0.75 mg, against doses of approved comparators (ondansetron and dolasetron). The studies reported that 0.25 mg IV palonosetron was the most effective for the prevention of both moderately and highly emetogenic CINV over the study period (0–120 h).
The safety and efficacy of oral palonosetron was based on the results of a double-blind, randomized study, which evaluated the efficacy of single oral doses of 0.25, 0.50, and 0.75 mg palonosetron, versus 0.25 mg IV palonosetron, with/without the concomitant administration of dexamethasone, for the prevention of moderately emetogenic CINV. The study reported that 0.50 mg was the lowest effective oral dose of palonosetron for the prevention of moderately emetogenic CINV [4].
In this multicenter, randomized, double-blind study, we investigated the efficacy and safety of 0.50 mg oral palonosetron, compared with 0.25 mg IV palonosetron, for the prevention of CINV induced by cisplatin-based HEC in patients with malignant solid tumors.
The objective of this study was to demonstrate the efficacy of 0.50 mg oral palonosetron for the prevention of highly emetogenic CINV. Evidence of efficacy would confirm the contribution of the 0.50 mg oral palonosetron component to a netupitant/palonosetron fixed-dose combination product, developed for the prevention of nausea and vomiting induced by HEC, and the suitability of 0.50 mg oral palonosetron as an active comparator in other CINV clinical studies conducted with fixed-dose combination products.
Patients and methods
Study design
This was a phase III, multicenter (60 centers), multinational, randomized, double-blind, double-dummy, parallel group-stratified study to assess the efficacy and safety of 0.50 mg oral palonosetron in comparison with 0.25 mg IV palonosetron in patients with solid tumors receiving cisplatin-based HEC (EudraCT number: 2010-022223-29). The stratification criteria were gender (male and female) and region (USA, Latin America, Europe, Asia, and the Commonwealth of Independent States).
The trial protocol and documentation were reviewed and approved by the relevant institutional review boards, ethics committees, and health authorities. The trial was conducted in accordance with the International Conference on Harmonization Guidelines for Good Clinical Practice and applicable local regulations as well as the Declaration of Helsinki (1964, amended in 1975, 1983, 1989, 1996, and 2000). All patients provided written informed consent.
Inclusion criteria
Chemotherapy-naive male and female patients receiving a cisplatin-based HEC regimen, aged ≥18 years with histologically or cytologically confirmed malignant solid tumors and an Eastern Cooperative Oncology Group (ECOG) performance status of 0–2, were enrolled in this study. Additional inclusion criteria included adequate hepatic, renal, and hematological function.
Exclusion criteria
Patients could not receive moderately or HEC from days 2–5 following cisplatin administration, radiation therapy to the abdomen or the pelvis within 1 week prior to day 1 or between days 1 and 5, and any medication with known or potential antiemetic efficacy within 24 h prior to day 1. Additional exclusion criteria included symptomatic primary or metastatic CNS malignancy and grade ≥1 vomiting, retching, and mild nausea (as defined by the National Cancer Institute) within 24 h prior to day 1.
Patient treatment
All patients were scheduled to receive their first course of cisplatin-based cytotoxic chemotherapy administered as a single IV dose of ≥70 mg/m2 over 1–4 h on day 1, alone or in combination with other chemotherapeutic agents.
Patients were randomized to one of the following treatment groups: oral palonosetron 0.50 mg or IV palonosetron 0.25 mg. Both groups received oral dexamethasone 20 mg on day 1, followed by dexamethasone (8 mg) twice daily (bid) on days 2–4.
The use of rescue medication for the treatment of nausea/vomiting was allowed after the start of the chemotherapy administration; however, this was considered as a treatment failure. Metoclopramide tablets were provided, and the investigator was authorized to use an alternative rescue treatment at their discretion. However, the use of 5-HT3 RAs was discouraged and the use of palonosetron was not permitted.
Endpoints
The primary study objective was to demonstrate the non-inferiority of single-dose oral palonosetron 0.50 mg versus single-dose IV palonosetron 0.25 mg. The primary efficacy endpoint was the proportion of patients with a complete response (CR, defined as no emesis and no rescue medication) within 24 h (acute phase) of the start of HEC administration on day 1.
Secondary study objectives were to assess the efficacy of single-dose 0.50 mg oral palonosetron versus single-dose 0.25 mg IV palonosetron during the delayed and overall phases and to evaluate the safety and tolerability of 0.50 mg oral palonosetron versus IV palonosetron in preventing CINV. Secondary efficacy endpoints included the CR rate for the delayed (25–120 h) and overall (0–120 h) phases, the severity of nausea, the percentage of patients with complete protection (CP, defined as no vomiting, no rescue therapy, and no nausea), the percentage of patients without emesis, and the percentage of patients without rescue medication for the delayed and overall phases. Time to treatment failure (TTF), defined as the time to the first emetic episode or the administration of rescue medication, was also investigated. The impact of palonosetron on QoL using the modified Functional Living Index-Emesis (FLIE) questionnaire was also investigated [23]. Time-related efficacy assessments commenced at the time of cisplatin administration (time zero). Efficacy parameters were evaluated in the acute, delayed, and overall phases. Safety assessments included physical examination, vital signs, 12-lead electrocardiogram (ECG), laboratory analysis (hematology, blood chemistry, urinalysis), and adverse events (AEs).
AEs were coded using the Medical Dictionary for Regulatory Activities (MedDRA), version 14.0. The incidence of treatment-emergent adverse events (TEAEs) in each treatment group was assessed. Clinical laboratory data were summarized using frequency tables for values within/outside reference ranges, and shift tables were used to evaluate changes in clinical laboratory data versus baseline.
Statistical analysis
The full analysis set (FAS) comprised all patients who were randomized to treatment and received at least one chemotherapy regimen and the study medication. The FAS was used to analyze patient demographic and baseline characteristics and primary and secondary efficacy endpoints. The per-protocol (PP) population comprised all patients included in the FAS who completed the 0–24-h study period with no major protocol violations, i.e., those affecting the primary efficacy endpoint. The PP population was also used for demographic and other baseline characteristics and primary efficacy analysis. The safety population was used for all safety analyses and comprised all patients who received at least one dose of study medication and had at least one safety assessment after treatment administration.
For non-inferiority testing, calculations were based on the two-sided stratum-adjusted Cochran-Mantel-Haenszel (CMH) method for the proportion of patients with CR in the acute phase. The model included gender and region as strata. The non-inferiority of oral palonosetron would be established if the lower limit of the two-sided 99 % confidence intervals (CIs) for the difference between oral palonosetron and IV palonosetron was greater (i.e., closer to zero) than the prespecified non-inferiority margin, which was set at −15 %. For a two-sided test of difference using a type I error of 0.010, a sample size of 322 evaluable patients per treatment group was required to ensure 90 % power (nQuery Advisor, version 6.0, module PTEO1). This was revised up to 370 per treatment group in order to ensure an adequate number of evaluable patients.
For the secondary efficacy endpoints, no test for non-inferiority was performed. The comparison between treatments was performed using a CMH test including gender and region as factors. The results are presented using odds ratios (ORs), two-sided 95 % CIs for the ORs, and p values.
Results
Baseline characteristics
Between 21 June 2011 and 14 November 2012, a total of 743 patients were enrolled and randomized as 1:1 to receive oral and IV palonosetron. Of these, 739 (99.5 %) received study medications and were included in the safety population and 738 were included in the FAS. Of the 743 randomized patients, 33 (4.4 %) discontinued the study after randomization. The main reasons for discontinuation were death which was reported in 6 (1.6 %) patients in the oral palonosetron group and 11 (3.0 %) patients in the IV palonosetron group; withdrawal of consent in 2 (0.5 %) and 3 (0.8 %) patients, respectively; and loss to follow-up in 1 (0.3 %) and 3 (0.8 %) patients, respectively. Deaths observed during the study were generally due to the patient’s underlying cancer and disease progression. No deaths were considered to be due to palonosetron. In total, 710 (95.6 %) patients completed the study.
The majority of patients in the safety population were male (59.3 %) and white (86.7 %) with a mean age of 57.9 years. Overall, nearly half (46.5 %) of all the patients had lung/respiratory tract cancer. Patient baseline characteristics were generally comparable in both treatment arms and are summarized in Table 1.
Efficacy
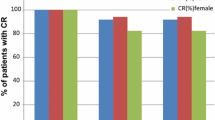
The percentage of patients (FAS) with a CR in the acute phase was 89.4 % in the oral palonosetron group and 86.2 % in the IV palonosetron group (Fig. 1). The difference in the proportions of responders (stratum-adjusted CMH method) in the FAS was 3.21 % (99 % CI −2.74 to 9.17 %) (Table 2). The non-inferiority of oral palonosetron versus IV palonosetron was demonstrated since the lower limit of the two-sided 99 % CI for the difference in proportions was greater than the predefined non-inferiority margin set at −15 %. Consistent results were obtained in the PP population with a difference in proportions between the oral and IV palonosetron groups of 3.77 % (99 % CI −3.22 to 10.76 %). The CR rates were also similar in both the delayed (76.2 vs 74.8 % for oral and IV palonosetron; OR 1.09, 95 % CI 0.77–1.5, p = 0.637) and overall (73.7 vs 70.2 % for oral and IV palonosetron; OR 1.20, 95 % CI 0.87–1.67, p = 0.269) phases (Fig. 1).

Complete response rates during the acute, delayed, and overall phases (full analysis set)
On individual study days, day 1 (0–24 h), day 2 (25–48 h), day 3 (49–72 h), day 4 (73–96 h), and day 5 (97–120 h), the percentage of responders was similar for both the oral palonosetron and IV palonosetron groups. The results were also similar for the cumulative time intervals.
There was no significant difference in the percentage of patients with no emesis in the delayed (78.9 % oral palonosetron, 77.5 % IV palonosetron; OR 1.09, 95 % CI 0.77–1.56, p = 0.625) and overall (75.6 % oral palonosetron, 73.2 % IV palonosetron; OR 1.15, 95 % CI 0.82–1.60, p = 0.430) phases.
The percentage of patients with no nausea in the acute phase was 4.6 % higher in the oral palonosetron group (80.2 vs 75.6 % in the IV group; p = 0.129), while in the delayed and overall phases, patients with no nausea in the oral and IV palonosetron groups were respectively 57.5 vs 53.4 % (p = 0.276) and 51.8 and 47.4 % (p = 0.241). No statistical differences were evidenced between palonosetron oral and IV groups on nausea data in all the acute delayed and overall study phases.
There was no significant difference between patients with no rescue medication in the acute (94.6 % oral palonosetron, 93.2 % IV palonosetron; OR 1.28, 95 % CI 0.69–2.36, p = 0.433), delayed (85.6 % oral palonosetron, 82.9 % IV palonosetron group; OR 1.27, 95 % CI 0.84–1.92, p = 0.258), and overall (84.3 % oral palonosetron, 80.8 % IV palonosetron; OR 1.32, 95 % CI 0.89–1.96, p = 0.174) phases.
Safety
No abnormal clinical test results were reported, and the analysis of ECG data indicated that any changes were at comparable interims of frequency for both treatment groups.
TEAEs were reported for approximately half of all patients in each treatment group (48.6 and 51.8 % for the oral palonosetron and IV palonosetron, respectively) (Table 3). However, TEAEs which were related to the study drug were rare (3.2 and 6.5 % for oral palonosetron and IV palonosetron, respectively) and no study drug-related TEAEs leading to discontinuation were reported (Table 3). The most frequently reported TEAEs assessed as being related to the study drugs were gastrointestinal disorders (1.6 and 3 % in the oral palonosetron group and IV palonosetron group, respectively), nervous system disorders (0.5 and 1.6 %, respectively), constipation (1.4 and 2.4 %, respectively), and headache (0.5 and 1.6 %, respectively). The most frequently reported serious TEAEs were neutropenia (1.4 and 2.4 % in the oral palonosetron group and IV palonosetron group, respectively), anemia (0.3 and 1.6 % in the oral palonosetron group and IV palonosetron group, respectively), thrombocytopenia (0.5 and 1.4 % in the oral palonosetron group and IV palonosetron group, respectively), and febrile neutropenia (0.8 and 1.1 % in the oral palonosetron group and IV palonosetron group, respectively).
Study drug-related TEAEs qualified as serious occurred in two (0.5 %) patients in the oral palonosetron group (Table 4): abdominal pain and constipation (one patient) and diarrhea and asthenia (one patient). For both patients, the events were resolved. No patients in the IV palonosetron group had serious TEAEs assessed as being related to the study drugs.
The death of 7 (1.9 %) patients in the oral palonosetron group and 12 (3.3 %) in the IV palonosetron group was classified as TEAEs. None of these deaths were assessed as being related to the study drugs or dexamethasone.
Discussion
The efficacy of palonosetron in controlling CINV in patients with cancer receiving treatment with a variety of chemotherapy regimens has been demonstrated previously [10–19].
In this study, we reported that in the acute phase, the majority of patients (89.4 % in the oral palonosetron group and 86.2 % in the IV palonosetron group) achieved a CR. The non-inferiority of oral palonosetron compared with IV palonosetron was thus demonstrated, as assessed by the primary efficacy endpoint (CR in the acute phase) in both the FAS and PP populations. All the planned sensitivity analyses supported the conclusion of non-inferiority of oral palonosetron compared with IV palonosetron. The two treatment groups also showed comparable CR in the delayed and overall phases, and the results of the secondary efficacy endpoints in all three phases were similar. Thus, the results of the secondary efficacy endpoints also support the demonstration of non-inferiority.
The safety of IV palonosetron has been demonstrated in several phase III clinical trials [20–22, 24–26]. The present study further supports these findings as the safety profiles of oral palonosetron and IV palonosetron were comparable and raised no new concerns.
The use of orally administered antiemetics for prophylaxis of nausea and emesis in patients receiving HEC treatment, in particular, for the control of nausea and vomiting on the first day of cisplatin treatment has been questioned [27]. This trial clearly demonstrated the non-inferiority of oral palonosetron versus IV palonosetron administration when both are used with dexamethasone. The use of oral palonosetron may be beneficial as it would allow a more convenient prophylaxis which is proven to be effective in controlling CINV caused by HEC. This may be of particular help for patients who receive treatment in an outpatient setting.
The investigators are aware that the study did not include an NK1 RA, as recommended by guidelines. However, the following of such guidelines in the clinical setting remains low and it is still important to evaluate the contribution of individual agents to CINV control [28]. In conclusion, this study demonstrated the non-inferiority of oral palonosetron versus IV palonosetron, in combination with dexamethasone, in the prevention and control of CINV in cancer patients receiving cisplatin-based HEC.
References
Griffin AM, Butow PN, Coates AS, Childs AM, Ellis PM, Dunn SM, Tattersall MH (1996) On the receiving end. V: patient perceptions of the side effects of cancer chemotherapy in 1993. Ann Oncol 7(2):189–195
Laszlo J, Lucas VS Jr (1981) Emesis as a critical problem in chemotherapy. N Engl J Med 305(16):948–949. doi:10.1056/NEJM198110153051609
Roila F, Herrstedt J, Aapro M, Gralla RJ, Einhorn LH, Ballatori E, Bria E, Clark-Snow RA, Espersen BT, Feyer P, Grunberg SM, Hesketh PJ, Jordan K, Kris MG, Maranzano E, Molassiotis A, Morrow G, Olver I, Rapoport BL, Rittenberg C, Saito M, Tonato M, Warr D, Group EMGW (2010) Guideline update for MASCC and ESMO in the prevention of chemotherapy- and radiotherapy-induced nausea and vomiting: results of the Perugia consensus conference. Ann Oncol 21(Suppl 5):v232–v243. doi:10.1093/annonc/mdq194
Boccia R, Grunberg S, Franco-Gonzales E, Rubenstein E, Voisin D (2013) Efficacy of oral palonosetron compared to intravenous palonosetron for the prevention of chemotherapy-induced nausea and vomiting associated with moderately emetogenic chemotherapy: a phase 3 trial. Support Care Cancer 21(5):1453–1460. doi:10.1007/s00520-012-1691-5
Navari RM (2010) Palonosetron for the prevention of chemotherapy-induced nausea and vomiting in patients with cancer. Future Oncol 6(7):1073–1084. doi:10.2217/fon.10.74
Rojas C, Slusher BS (2012) Pharmacological mechanisms of 5-HT(3) and tachykinin NK(1) receptor antagonism to prevent chemotherapy-induced nausea and vomiting. Eur J Pharmacol 684(1–3):1–7. doi:10.1016/j.ejphar.2012.01.046
Navari RM (2003) Pathogenesis-based treatment of chemotherapy-induced nausea and vomiting—two new agents. J Support Oncol 1(2):89–103
Rojas C, Li Y, Zhang J, Stathis M, Alt J, Thomas AG, Cantoreggi S, Sebastiani S, Pietra C, Slusher BS (2010) The antiemetic 5-HT3 receptor antagonist palonosetron inhibits substance P-mediated responses in vitro and in vivo. J Pharmacol Exp Ther 335(2):362–368. doi:10.1124/jpet.110.166181
Rojas C, Stathis M, Thomas AG, Massuda EB, Alt J, Zhang J, Rubenstein E, Sebastiani S, Cantoreggi S, Snyder SH, Slusher B (2008) Palonosetron exhibits unique molecular interactions with the 5-HT3 receptor. Anesth Analg 107(2):469–478. doi:10.1213/ane.0b013e318172fa74
Aogi K, Sakai H, Yoshizawa H, Masuda N, Katakami N, Yanagita Y, Inoue K, Kuranami M, Mizutani M (2012) A phase III open-label study to assess safety and efficacy of palonosetron for preventing chemotherapy-induced nausea and vomiting (CINV) in repeated cycles of emetogenic chemotherapy. Support Care Cancer 20(7):1507–1514. doi:10.1007/s00520-011-1239-0
Feinberg B, Gilmore J, Haislip S, Jackson J, Jain G, Balu S, Buchner D (2012) Impact of initiating antiemetic prophylaxis with palonosetron versus ondansetron on risk of uncontrolled chemotherapy-induced nausea and vomiting in patients with lung cancer receiving multi-day chemotherapy. Support Care Cancer 20(3):615–623. doi:10.1007/s00520-011-1140-x
Longo F, Mansueto G, Lapadula V, Stumbo L, Del Bene G, Adua D, De Filippis L, Bonizzoni E, Quadrini S (2012) Combination of aprepitant, palonosetron and dexamethasone as antiemetic prophylaxis in lung cancer patients receiving multiple cycles of cisplatin-based chemotherapy. Int J Clin Pract 66(8):753–757. doi:10.1111/j.1742-1241.2012.02969.x
Aapro M, Fabi A, Nole F, Medici M, Steger G, Bachmann C, Roncoroni S, Roila F (2010) Double-blind, randomised, controlled study of the efficacy and tolerability of palonosetron plus dexamethasone for 1 day with or without dexamethasone on days 2 and 3 in the prevention of nausea and vomiting induced by moderately emetogenic chemotherapy. Ann Oncol 21(5):1083–1088. doi:10.1093/annonc/mdp584
Brugnatelli S, Gattoni E, Grasso D, Rossetti F, Perrone T, Danova M (2011) Single-dose palonosetron and dexamethasone in preventing nausea and vomiting induced by moderately emetogenic chemotherapy in breast and colorectal cancer patients. Tumori 97(3):362–366. doi:10.1700/912.10035
Celio L, Denaro A, Agustoni F, Bajetta E (2012) Palonosetron plus 1-day dexamethasone for the prevention of nausea and vomiting due to moderately emetogenic chemotherapy: effect of established risk factors on treatment outcome in a phase III trial. J Support Oncol 10(2):65–71. doi:10.1016/j.suponc.2011.06.007
Celio L, Frustaci S, Denaro A, Buonadonna A, Ardizzoia A, Piazza E, Fabi A, Capobianco AM, Isa L, Cavanna L, Bertolini A, Bichisao E, Bajetta E (2011) Palonosetron in combination with 1-day versus 3-day dexamethasone for prevention of nausea and vomiting following moderately emetogenic chemotherapy: a randomized, multicenter, phase III trial. Support Care Cancer 19(8):1217–1225. doi:10.1007/s00520-010-0941-7
Hesketh PJ, Sanz-Altamira P (2012) Aprepitant, dexamethasone, and palonosetron in the prevention of doxorubicin/cyclophosphamide-induced nausea and vomiting. Support Care Cancer 20(3):653–656. doi:10.1007/s00520-011-1312-8
Noor R, Bedikian AY, Mahoney S, Bassett R Jr, Kim K, Papadopoulos N, Hwu WJ, Hwu P, Homsi J (2012) Comparison of two dosing schedules of palonosetron for the prevention of nausea and vomiting due to interleukin-2-based biochemotherapy. Support Care Cancer 20(10):2583–2588. doi:10.1007/s00520-011-1359-6
Choi BS, Borsaru GP, Ballinari G, Voisin D, Di Renzo N (2013) Multicenter phase IV study of palonosetron in the prevention of chemotherapy-induced nausea and vomiting (CINV) in patients with non-Hodgkin lymphomas undergoing repeated cycles of moderately emetogenic chemotherapy. Leuk Lymphoma. doi:10.3109/10428194.2013.813498
Gralla R, Lichinitser M, Van Der Vegt S, Sleeboom H, Mezger J, Peschel C, Tonini G, Labianca R, Macciocchi A, Aapro M (2003) Palonosetron improves prevention of chemotherapy-induced nausea and vomiting following moderately emetogenic chemotherapy: results of a double-blind randomized phase III trial comparing single doses of palonosetron with ondansetron. Ann Oncol 14(10):1570–1577. doi:10.1093/annonc/mdg417
Eisenberg P, Figueroa-Vadillo J, Zamora R, Charu V, Hajdenberg J, Cartmell A, Macciocchi A, Grunberg S (2003) Improved prevention of moderately emetogenic chemotherapy-induced nausea and vomiting with palonosetron, a pharmacologically novel 5-HT3 receptor antagonist: results of a phase III, single-dose trial versus dolasetron. Cancer 98(11):2473–2482. doi:10.1002/cncr.11817
Aapro MS, Grunberg SM, Manikhas GM, Olivares G, Suarez T, Tjulandin SA, Bertoli LF, Yunus F, Morrica B, Lordick F, Macciocchi A (2006) A phase III, double-blind, randomized trial of palonosetron compared with ondansetron in preventing chemotherapy-induced nausea and vomiting following highly emetogenic chemotherapy. Ann Oncol 17(9):1441–1449. doi:10.1093/annonc/mdl137
Martin AR, Pearson JD, Cai B, Elmer M, Horgan K, Lindley C (2003) Assessing the impact of chemotherapy-induced nausea and vomiting on patients’ daily lives: a modified version of the Functional Living Index-Emesis (FLIE) with 5-day recall. Support Care Cancer 11(8):522–527. doi:10.1007/s00520-003-0482-4
Saito M, Aogi K, Sekine I, Yoshizawa H, Yanagita Y, Sakai H, Inoue K, Kitagawa C, Ogura T, Mitsuhashi S (2009) Palonosetron plus dexamethasone versus granisetron plus dexamethasone for prevention of nausea and vomiting during chemotherapy: a double-blind, double-dummy, randomised, comparative phase III trial. Lancet Oncol 10(2):115–124. doi:10.1016/S1470-2045(08)70313-9
Gonullu G, Demircan S, Demirag MK, Erdem D, Yucel I (2012) Electrocardiographic findings of palonosetron in cancer patients. Support Care Cancer 20(7):1435–1439. doi:10.1007/s00520-011-1226-5
Yavas C, Dogan U, Yavas G, Araz M, Ata OY (2012) Acute effect of palonosetron on electrocardiographic parameters in cancer patients: a prospective study. Support Care Cancer 20(10):2343–2347. doi:10.1007/s00520-011-1348-9
Tsavaris NB, Tsaroucha-Noutsou E, Karvounis N, Bacoyannis C, Pagou M, Kosmidis P (1995) Comparison of different schedules of ondansetron (GR 38032F) administration during cisplatin-based chemotherapy: a randomized trial. Chemotherapy 41(1):70–76
Aapro M, Molassiotis A, Dicato M, Pelaez I, Rodriguez-Lescure A, Pastorelli D, Ma L, Burke T, Gu A, Gascon P, Roila F, investigators P (2012) The effect of guideline-consistent antiemetic therapy on chemotherapy-induced nausea and vomiting (CINV): the Pan European Emesis Registry (PEER). Ann Oncol 23(8):1986–1992. doi:10.1093/annonc/mds021
Acknowledgments
The authors would like to thank the patients, the investigators, and the study teams at each of the participating centers. The study was supported by Helsinn Healthcare S.A. Medical writing assistance was provided by Dr. Matthew Young, Cancer Communications and Consultancy Ltd., Knutsford, Cheshire, UK, and funded by Helsinn Healthcare, S.A.
Conflict of interest
As for disclosures in relation to Helsinn Healthcare S.A., MK, TC, VL, RS-A, and AF all received honoraria; G. Rizzi, G. Rossi, and MEB received compensated employment; SMG declares no conflict of interest. The authors declare that they have full control of all primary data, and this can be made available to the journal on request.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial 4.0 International License, which permits any non-commercial use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made.
The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder.
To view a copy of this licence, visit https://creativecommons.org/licenses/by-nc/4.0/.
About this article

Cite this article
Karthaus, M., Tibor, C., Lorusso, V. et al. Efficacy and safety of oral palonosetron compared with IV palonosetron administered with dexamethasone for the prevention of chemotherapy-induced nausea and vomiting (CINV) in patients with solid tumors receiving cisplatin-based highly emetogenic chemotherapy (HEC). Support Care Cancer 23, 2917–2923 (2015). https://doi.org/10.1007/s00520-015-2657-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00520-015-2657-1