
Abstract
Purpose
A phase II, open-label, dose-finding, randomized study was performed to evaluate the recommended dose of pegfilgrastim in Japanese breast cancer patients.
Methods
Patients received 1.8, 3.6, or 6.0 mg of pegfilgrastim once per chemotherapy cycle for up to 6 cycles. Patients received docetaxel, doxorubicin, and cyclophosphamide (TAC) therapy followed by pegfilgrastim on the next day.
Results
Pegfilgrastim was administered to 87 women with stage II/III invasive breast carcinoma. The duration of grade 4 neutropenia in the first cycle, the primary endpoint, was 2.2 ± 0.9 days, 1.5 ± 0.9 days, and 1.4 ± 0.7 days in the 1.8, 3.6, and 6.0 mg groups, respectively. This finding indicated that pegfilgrastim efficacy peaked at 3.6 mg. Pegfilgrastim doses up to 6.0 mg were considered safe.
Conclusion
A 3.6-mg pegfilgrastim dose may be safe and effective for Japanese patients. A confirmatory study is required to establish safety and efficacy at this dose for intensive anti-cancer chemotherapy.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Febrile neutropenia (FN) is a clinical condition characterized by the development of fever in association with neutropenia. The risk of FN increases with the duration of neutropenia [1, 2]. Infection, often manifested by FN, may become serious and potentially life threatening. Therefore, to prevent FN, chemotherapy doses are often reduced or dose intervals are extended. However, reduced relative dose intensity may reduce the efficacy of chemotherapy regimens, leading to decreased survival rates, particularly in curative settings [3–7]. For these reasons, granulocyte-colony stimulating factor (G-CSF) is used to reduce the risk of chemotherapy-associated FN. G-CSF may improve therapeutic outcomes by helping anticancer drugs to be administered at planned dosages and intervals. Guidelines for the recommended use of G-CSF based on FN risk have been established by the American Society of Clinical Oncology, National Comprehensive Cancer Network, and European Organization for Research and Treatment of Cancer [8–10]. Prophylactic use of G-CSF is based on the intensity of chemotherapy regimens and patient-specific risks and is recommended for patients with a 20 % or greater risk of FN.
Pegfilgrastim is a drug with a polyethylene glycol (PEG) molecule, approximate molecular weight of 20,000, covalently bound to the N-terminal residue of G-CSF (filgrastim). Pegfilgrastim is produced by recombinant DNA technology and expressed in Escherichia coli. PEG-modified protein is known to decrease kidney clearance, inhibit protease-mediated hydrolysis, and prolong serum half-life. The serum half-life of pegfilgrastim has been reported to be 2 to 3 days, which is longer than the approximately 3-h half-life of filgrastim.
A placebo-controlled study in patients with early-stage breast cancer showed that the incidence rates of FN were 17 and 1 % in the placebo and pegfilgrastim groups, respectively [11]. Therefore, prophylactic pegfilgrastim can remarkably reduce the incidence of FN. Furthermore, pegfilgrastim has been shown to reduce the incidence of FN-related hospitalizations and the use of intravenous antibiotics to treat infection. In the curative setting, highly myelosuppressive chemotherapy regimens may be required to treat patients with early-stage breast cancer. TAC, dose-dense doxorubicin plus cyclophosphamide followed by paclitaxel, and TC (docetaxel plus cyclophosphamide) regimens are widely used in the clinical setting and have been shown to result in favorable clinical outcomes.
While pegfilgrastim has been widely studied in the United States and European populations, it has not been evaluated in the Japanese population. Therefore, a phase II dose-finding study of pegfilgrastim was performed in Japanese patients with early-stage breast cancer. To evaluate the efficacy of pegfilgrastim in this population, TAC therapy, a highly myelosuppressive chemotherapy that is used in the adjuvant treatment of early stage breast cancer, was selected. The primary endpoint of this study was the duration of severe (grade 4) neutropenia in the first cycle (DSN).
Materials and methods
Patients
This study was conducted in accordance with the Declaration of Helsinki. The institutional review boards of the participating centers approved the protocol. Informed consent was obtained from all patients before beginning any study-related procedures.
Women who met the following inclusion criteria were eligible for study entry: aged 20–64 years of age; pathohistological and clinical diagnosis of stage II or III primary invasive breast carcinoma; Eastern Cooperative Oncology Group performance status ≤2; chemotherapy naïve; plan to receive full-dose adjuvant or neoadjuvant TAC therapy; absolute neutrophil count (ANC) ≥ 2 × 109/L; platelet count ≥100 × 109/L; hemoglobin concentration ≥10 g/dL; aspartate aminotransferase and alanine aminotransferase levels ≤2.5 times the upper limit of the normal range in each institute; total bilirubin content ≤1.5 times the upper limit of the normal range in each institute; creatinine level ≤1.5 mg/dL; left ventricular ejection fraction ≥50 %; and negative hepatitis B (hepatitis B surface antigen and hepatitis B core antibody) test result within 3 months of enrollment. Patients with a history of radiation therapy within 4 weeks of enrollment, history of bone marrow or stem cell transplantation, or comorbid malignancies other than breast cancer were excluded from the study.
Study design
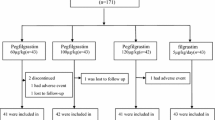
This was a multicenter, randomized, open-label, dose-finding study to evaluate the safety and efficacy of a single injection of pegfilgrastim per chemotherapy cycle in Japanese patients. Patients were randomized via a dynamic allocation method and were assigned in a 1:1:1 ratio to each pegfilgrastim cohort (1.8, 3.6, and 6.0 mg) after stratification according to chemotherapy status (neoadjuvant or adjuvant) and investigational site (Fig. 1).

Study schema Superscript letter a indicates that pegfilgrastim (KRN125) was administered subcutaneously (SC) 24 h after docetaxel administration on day 2 of each cycle. Superscript letter b indicates that chemotherapy cycle was defined as the interval from the day of TAC (docetaxel, doxorubicin, and cyclophosphamide) initiation to the day before the next TAC course. The maximum duration of each chemotherapy cycle was 42 days
Chemotherapy treatment
On day 1 of each chemotherapy cycle, patients received an intravenous infusion of doxorubicin (50 mg/m2), followed by intravenous infusions of cyclophosphamide (500 mg/m2) and docetaxel (75 mg/m2). Chemotherapy was repeated every 3 weeks for up to 6 cycles.
Full-dose chemotherapy was administered on day 1 of the next cycle to patients who met the following criteria: ANC ≥1.5 × 109/L, platelet count ≥100 × 109/L, hemoglobin concentration ≥8 g/dL, aspartate aminotransferase and alanine aminotransferase levels ≤2.5 times the upper limit of the normal range in each institute, total bilirubin content ≤1.5 times the upper limit of the normal range in each institute, and creatinine level ≤1.5 mg/dL. In addition, any adverse events, including peripheral neuropathy, edema, constipation, and weight loss, must have been cured completely or reduced to below grade 2. Any non-hematologic adverse events other than hair loss, peripheral neuropathy, edema, weight loss, and constipation must have been cured completely or reduced to grade 1. After adverse event recovery to the appropriate level, chemotherapy was resumed at the full dose administered in the previous cycle. Patients whose chemotherapy was delayed for more than 3 weeks were eliminated from the study.
If any grade 3 or 4 critical adverse event, including FN, prolonged neutropenia, thrombocytopenia, abdominal pain, diarrhea, nausea, vomiting, or nerve disorder, occurred in the previous cycle of TAC therapy, and dose reduction was deemed necessary by the investigator, the dosage of each anticancer drug was reduced 20 % in the next cycle. Dose reduction was allowed only once.
Supportive treatment consisted of 20 mg of dexamethasone administered intravenously on day 1 and 4 mg of dexamethasone administered orally twice daily on days 2 and 3. Prophylactic antibiotics were prohibited in the first cycle of chemotherapy.
Study treatment
On day 2 of each chemotherapy cycle (24 h after chemotherapy completion), patients received a single subcutaneous injection of pegfilgrastim at a fixed dose of 1.8 g, 3.6, or 6.0 mg.
G-CSF treatment was generally prohibited during the study period. However, rescue G-CSF treatment was allowed only when three consecutive days of concurrent fever (≥38 °C) and grade 4 neutropenia occurred in cycle 1 or when three consecutive days of fever accompanied by ≥1 episode of grade 4 neutropenia occurred in subsequent cycles.
Efficacy measurements
Blood samples were obtained for complete blood counts (CBCs). During cycle 1, CBC was measured daily from days 1 to 4, followed by daily measurements until ANC ≥1 × 109/L post-nadir was achieved, and thereafter measured every 2 days up to day 21. During all subsequent cycles, CBC was measured on days 1 to 3, 8, and 15. Axillary body temperature was measured daily and recorded throughout the study.
The primary efficacy endpoint of this study was DSN, which was defined as the number of days in which the patient had an ANC <0.5 × 109/L during cycle 1 of TAC therapy. The secondary efficacy endpoints included ANC profile, ANC nadir, and proportion of patients who developed FN in the first cycle. FN was defined as an axillary body temperature ≥38 °C with concurrent ANC <0.5 × 109/L.
Safety endpoints
The safety endpoints were incidence of adverse events, changes in vital signs, and laboratory tests. Adverse events that occurred after pegfilgrastim administration were documented. Serum was collected at the beginning and end of the study to detect antibodies capable of neutralizing the biological effects of either pegfilgrastim or filgrastim.
Data sets and statistical analysis
The objective of this trial was to estimate the mean DSN in order to determine the recommended dose of pegfilgrastim. Efficacy and safety were assessed based on the per protocol set and safety analysis set, respectively. Standard deviation of DSN in each group was assumed to be 1.4, based on a previous trial [15]. With 22 patients in each group, the mean DSN would be calculated with the standard error of approximately 0.3. Assuming that 10 % of patients would be withdrawn, the planned total sample size was 75 patients (25 patients per dosage group).
A three-contrast test was conducted in the dose response analysis: linear reduction (1, 0, −1), reduction at 6.0 mg (1, 1, −2), and plateau at 3.6 mg (2, −1, −1).
Adverse events were classified according to the Japanese version of the Medical Dictionary for Regulatory Activities preferred terms and were graded by severity according to the National Cancer Institute Common Terminology Criteria for Adverse Events (version 3.0).
All analyses were performed using SAS software (version 9.1.3).
Results
Patients
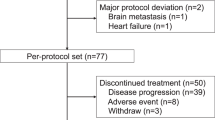
Between November 2009 and April 2010, a total of 90 Japanese patients were randomized to the three pegfilgrastim dosage groups (30 patients per group). A total of 87 patients received pegfilgrastim and were included in the per protocol set and the safety analysis set. Three patients randomized to each group (one each from the 1.8, 3.6, and 6.0 mg groups) were excluded from the analysis of efficacy endpoints because they withdrew from the study before receiving chemotherapy or the study drug. Two of these patients withdrew because of allergic reactions caused by docetaxel. One of these patients could not receive TAC therapy because of health problems and withdrew (Fig. 2).

Patient consort diagram
A total of 13 patients withdrew from the study after receiving the study drug, including 5 (16.7 %) patients in the 1.8-mg group, 5 (16.7 %) patients in the 3.6-mg group, and 3 (10.0 %) patients in the 6.0-mg group. Of these patients, 1 patient in the 1.8-mg group developed drug hypersensitivity (allergic reaction to docetaxel) and discontinued the study. Two patients in the 1.8-mg group, 1 patient in the 3.6-mg group, and 2 patients in the 6.0-mg group were eliminated from the study because of treatment changes. One patient in the 3.6-mg group withdrew from the study because of progression of the primary disease during neoadjuvant treatment. The other patients were eliminated from the study because of informed consent withdrawal. A total of 74 patients completed the trial.
Patient baseline characteristics are shown in Table 1. Patient characteristics were not different among the three dosage groups. The median (range) age was 46.0 years (33–63 years), 47.0 years (26–64 years), and 48.0 years (26–64 years); the mean body weight was 52.3, 56.3, and 53.2 kg; and the mean body surface area was 1.50, 1.55, and 1.51 in the 1.8-, 3.6-, and 6.0-mg groups, respectively. A median of 6 cycles of chemotherapy was administered to all dosage groups.
Efficacy
In cycle 1 of TAC therapy, ANC decreased to <0.5 × 109/L in 28 (96.6 %) patients, 25 (86.2 %) patients, and 27 (93.1 %) patients in the 1.8-, 3.6-, and 6.0-mg groups, respectively. DSN (mean ± standard deviation), the primary endpoint, was 2.2 ± 0.9 days, 1.5 ± 0.9 days, and 1.4 ± 0.7 days in the 1.8-, 3.6-, and 6.0-groups, respectively (Fig. 3a). The dose-response analysis, which was conducted based on the three-contrast test, showed a reduction of DSN from 1.8- to 3.6-mg pegfilgrastim followed by a plateau from 3.6 to 6.0 mg (“plateau from 3.6 mg”), with the smallest P value among the three-contrast (P = 0.001). Pegfilgrastim efficacy stratified by chemotherapy setting (adjuvant or neoadjuvant) showed similar dose responses.

a Duration of severe neutropenia (DSN) in the first cycle. b Median absolute neutrophil count (ANC) for cycle 1. SD standard deviation
FN occurred in 2 (6.9 %) patients in the 1.8-mg group, 3 (10.3 %) patients in the 3.6-mg group, and 1 (3.4 %) patient in the 6.0-mg group with no statistically significant difference between the groups (Table 2). In cycle 1 of TAC therapy, 1 patient in the 3.6-mg group received G-CSF on days 7 and 8 as rescue treatments.
Changes in the mean ANC in cycle 1 of TAC therapy are shown in Fig. 3b. The mean ANC peaked on day 3 of study drug administration in all groups, and nadir occurred on day 7 in the 1.8-mg group and day 6 in the 3.6- and 6.0-mg groups. ANC almost reached the baseline values after day 11 in all groups.
The ANC at nadir (mean ± standard deviation) was 0.184 ± 0.212 × 109/L in the 1.8-mg group, 0.240 ± 0.208 × 109/L in the 3.6-mg group, and 0.255 ± 0.287 × 109/L in the 6.0-mg group (Table 2).
Safety
All 87 patients experienced adverse events. Non-hematologic adverse events that occurred in ≥50 % of patients in all groups were alopecia, nausea, malaise, constipation, dysgeusia, decreased appetite, vomiting, and diarrhea (Online Resource 1). Most of these events were considered to be associated with TAC therapy.
No adverse events resulted in death. Serious non-fatal adverse events occurred in four patients in the 1.8-mg group (upper respiratory tract inflammation, cystitis, pneumonia, and radius fracture) and 1 patient in the 6.0-mg group (decreased appetite and malaise).
Bone pain and back pain are common adverse events associated with G-CSF. Bone pain occurred in 1 (3.4 %) patient in each group. Throughout the course of chemotherapy, back pain was observed in 3 (10.3 %) patients in the 1.8-mg group, 6 (20.7 %) patients in the 3.6-mg group, and 8 (27.6 %) patients in the 6.0-mg group. Because these adverse events were grade 2 or lower in severity and could be controlled without treatment or with non-steroidal anti-inflammatory drugs, there were no safety concerns in any of the groups.
Hematologic adverse events
Grade 3 or 4 neutropenia occurred in 29 (100 %) patients in the 1.8-mg group, 29 (100 %) patients in the 3.6-mg group, and 28 (96.6 %) patients in the 6.0-mg group in all TAC cycles. Grade 3 or 4 thrombocytopenia occurred in 4 (13.8 %) patients in the 3.6-mg group and 4 (13.8 %) patients in the 6.0-mg group in all TAC cycles. Grade 3 or 4 anemia occurred in 1 (3.4 %) patient in the 1.8-mg group and 1 (3.4 %) patient in the 6.0-mg group in all cycles (Table 3).
Antibody formation
Serum antibodies to pegfilgrastim or filgrastim were tested using the BIAcore assay for all patients who received the study drug; antibodies were not detected in any patients.
Discussion
DSN, the primary endpoint of this study, has been shown to be well correlated with the incidence of FN [1, 2]. The European Medicines Agency (formerly the European Medicines Evaluation Agency) guidelines describe DSN as an acceptable surrogate endpoint for the exploratory and confirmatory trials of G-CSF [12]. According to these guidelines, DSN was set as the primary endpoint of this study. In phase III studies conducted in the USA, Canada, European countries, and Australia, pegfilgrastim administered at a 100 μg/kg dose or 6-mg fixed dose regardless of body weight had a similar effect on DSN [13, 14]. Based on these findings, pegfilgrastim at a fixed dose of 6 mg is widely used in the world.
The safety of pegfilgrastim at doses of 30, 60, and 100 μg/kg was evaluated in a clinical pharmacological study in Japanese lung cancer patients [15]. The study results demonstrated the safety and tolerability of a single subcutaneous pegfilgrastim injection up to 100 μg/kg. In the present study, three fixed pegfilgrastim doses of 1.8, 3.6, or 6.0 mg were used to determine the recommended pegfilgrastim dose in Japanese patients with breast cancer. DSN was 2.2 ± 0.9 days, 1.5 ± 0.9 days, and 1.4 ± 0.7 days in the 1.8-, 3.6-, and 6.0-mg groups, respectively. Analysis of the dose-response relationship between pegfilgrastim dose and DSN revealed a significant “linear reduction” (P = 0.005), significant “plateau at 3.6 mg” (P = 0.001), and non-significant reduction at 6.0 mg (P = 0.092).
A dose-finding study of pegfilgrastim conducted in the USA also showed a linear shortening of DSN in response to pegfilgrastim (30, 60, or 100 μg/kg) injection in breast cancer patients undergoing doxorubicin/docetaxel chemotherapy [16]. In a phase I trial in Japan, lung cancer patients undergoing chemotherapy received a single injection of pegfilgrastim if they experienced grade 4 neutropenia in the previous cycle [15]. In another phase I trial in lung cancer patients conducted in the USA, single-dose pegfilgrastim or daily filgrastim was administered 14 days prior to initial chemotherapy dosing and again after chemotherapy [17]. Both of these clinical trials revealed a non-linear trend in pharmacokinetic parameters, such as C max and area under the concentration curve. However, differences in race, study design, and chemotherapy regimens may have contributed to the discrepancy in pegfilgrastim dose responses in these phase II trials. The approved dose of filgrastim for chemotherapy-induced neutropenia in Japan is one fourth to one half of the dose approved in other foreign countries. Compared with the approved pegfilgrastim dose of 6 mg in Europe and the USA, lower pegfilgrastim doses provided adequate efficacy in Japanese patients undergoing chemotherapy.
Although different regimens were used, the DSN response to 3.6-mg pegfilgrastim in Japanese patients in our study was similar to the DSN response to 6.0-mg pegfilgrastim in phase III trials conducted in the USA, Canada, European countries, and Australia (1.5 ± 0.9 days vs. 1.8 ± 1.4 days) [13, 14]. Considering the difference in regimens and patient populations, as well as the fact that the previous trials were conducted approximately a decade ago, it is difficult to make cross-trial comparisons among these groups.
In the present study, all patients experienced adverse events; however, most adverse events were associated with cancer chemotherapy. Pegfilgrastim was well tolerated up to 6.0 mg.
Based on our results, 3.6 mg of pegfilgrastim may be a safe and effective dose. However, because this study was conducted as an exploratory dose-finding study in Japanese patients, a confirmatory study is required.
This study was conducted in breast cancer patients receiving TAC therapy. The BCIRG001 study compared the efficacy and safety of TAC therapy and FAC (fluorouracil/doxorubicin/cyclophosphamide) therapy as adjuvant chemotherapy in 1047 node-negative breast cancer patients. The BCIRG001 study showed that 5-year progression-free survival and overall survival were significantly longer in the TAC therapy group than in the FAC therapy group, and this significant difference was maintained at 10 years [18, 19]. At the time 237 subjects were enrolled, the incidence of FN was significantly higher in the TAC therapy group than in the FAC therapy group (24.7 vs. 2.5 %; P < 0.001) [20]. Although the use of prophylactic antibiotics was initially mandated in the TAC therapy group, the prophylactic use of G-CSF was not allowed. Therefore, the study protocol was revised to specify the prophylactic use of G-CSF in the TAC therapy group. Prophylactic G-CSF decreased the incidence of FN in the TAC therapy group from 24.7 to 6.5 %. Based on the results of the BCIRG001 study, the National Comprehensive Cancer Network guidelines indicate the use of filgrastim support in all cycles of TAC therapy.
Because G-CSF is not currently indicated for the primary prophylaxis of FN induced by breast cancer chemotherapies in Japan, intensive chemotherapy regimens are rarely used in Japan. If breast cancer is treated with intensive chemotherapy regimens, such as TAC, which is commonly used in Europe and the USA, a pre-planned dose reduction from the standard dose is needed. Conversely, according to various reports, dose reduction in cancer chemotherapy attenuates therapeutic effects [3–7]. It is important to administer cancer chemotherapy at effective doses, as maintenance of relative dose intensity may affect clinical outcomes, particularly in the adjuvant setting.
In our study, TAC therapy with prophylactic pegfilgrastim support was conducted safely, even in the outpatient setting. The proportion of patients who developed FN was 6.9 % in all pegfilgrastim dosage groups. Prophylactic pegfilgrastim at a fixed dose of 3.6 mg is expected to allow cancer chemotherapy to be administered at the same dose intensity for Japanese breast cancer patients as that used for patients in Western countries.
References
Bodey GP, Buckley M, Sathe YS, Freireich EJ (1966) Quantitative relationships between circulating leukocytes and infection in patients with acute leukemia. Ann Intern Med 64:328–340
Blackwell S, Crawford J (1994) Filgrastim (r-metHuG-CSF) in the chemotherapy setting. In: Morstyn G, Dexter TM (eds) Filgrastim (r-metHuG-CSF) in Clinical Practice. Marcel Dekker, Inc., New York, pp 103–116
Bosly A, Bron D, Van Hoof A, De Bock R, Berneman Z, Ferrant A, Kaufman L, Dauwe M, Verhoef G (2008) Achievement of optimal average relative dose intensity and correlation with survival in diffuse large B-cell lymphoma patients treated with CHOP. Ann Hematol 87:277–283
Chirivella I, Bermejo B, Insa A, Pérez-Fidalgo A, Magro A, Rosello S, García-Garre E, Martín P, Bosch A, Lluch A (2009) Optimal delivery of anthracycline-based chemotherapy in the adjuvant setting improves outcome of breast cancer patients. Breast Cancer Res Treat 114:479–484
Wildiers H, Reiser M (2011) Relative dose intensity of chemotherapy and its impact on outcomes in patients with early breast cancer or aggressive lymphoma. Crit Rev Oncol Hematol 77:221–240
Kuderer NM, Dale DC, Crawford J, Lyman GH (2007) Impact of primary prophylaxis with granulocyte colony-stimulating factor on febrile neutropenia and mortality in adult cancer patients receiving chemotherapy: a systematic review. J Clin Oncol 25:3158–3167
Lyman GH, Dale DC, Friedberg J, Crawford J, Fisher RI (2004) Incidence and predictors of low chemotherapy dose-intensity in aggressive non-Hodgkin’s lymphoma: a nationwide study. J Clin Oncol 22:4302–4311
Smith TJ, Khatcheressian J, Lyman GH, Ozer H, Armitage JO, Balducci L, Bennett CL, Cantor SB, Crawford J, Cross SJ, Demetri G, Desch CE, Pizzo PA, Schiffer CA, Schwartzberg L, Somerfield MR, Somlo G, Wade JC, Wade JL, Winn RJ, Wozniak AJ, Wolff AC (2006) 2006 update of recommendations for the use of white blood cell growth factors: an evidence-based clinical practice guideline. J Clin Oncol 24:3187–3205
National Comprehensive Cancer Network (2014) Clinical practice guidelines in oncology: myeloid growth factors, version 1. http://www.nccn.org/professionals/physician_gls/pdf/myeloid_growth.pdf. Accessed 6 Mar 2014
Aapro MS, Bohlius J, Cameron DA, Dal Lago L, Donnelly JP, Kearney N, Lyman GH, Pettengell R, Tjan-Heijnen VC, Walewski J, Weber DC, Zielinski C, European Organisation for Research and Treatment of Cancer (2011) 2010 update of EORTC guidelines for the use of granulocyte-colony stimulating factor to reduce the incidence of chemotherapy-induced febrile neutropenia in adult patients with lymphoproliferative disorders and solid tumours. Eur J Cancer 47:8–32
Vogel CL, Wojtukiewicz MZ, Carroll RR, Tjulandin SA, Barajas-Figueroa LJ, Wiens BL, Neumann TA, Schwartzberg LS (2005) First and subsequent cycle use of pegfilgrastim prevents febrile neutropenia in patients with breast cancer: a multicenter, double-blind, placebo-controlled phase III study. J Clin Oncol 23:1178–1184
Committee for medical products for human use (CHMP); Europe medicines agency (2007) Guideline on clinical trials with haematopoietic growth factors for the prophylaxis of infection following myelosuppressive or myeloablative therapy. http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2009/12/WC500017737.pdf. Accessed 10 Apr 2013
Holmes FA, O’Shaughnessy JA, Vukelja S, Jones SE, Shogan J, Savin M, Glaspy J, Moore M, Meza L, Wiznitzer I, Neumann TA, Hill LR, Liang BC (2002) Blinded, randomized, multicenter study to evaluate single administration pegfilgrastim once per cycle versus daily filgrastim as an adjunct to chemotherapy in patients with high-risk stage II or stage III/IV breast cancer. J Clin Oncol 20:727–731
Green MD, Koelbl H, Baselga J, Galid A, Guillem V, Gascon P, Siena S, Lalisang RI, Samonigg H, Clemens MR, Zani V, Liang BC, Renwick J, Piccart MJ, International Pegfilgrastim 749 Study Group (2003) A randomized double-blind multicenter phase III study of fixed-dose single-administration pegfilgrastim versus daily filgrastim in patients receiving myelosuppressive chemotherapy. Ann Oncol 14:29–35
Yamamoto N, Sekine I, Nakagawa K, Takada M, Fukuoka M, Tanigawara Y, Saijo N (2009) A pharmacokinetic and dose escalation study of pegfilgrastim (KRN125) in lung cancer patients with chemotherapy-induced neutropenia. Jpn J Clin Oncol 39:425–430
Holmes FA, Jones SE, O’Shaughnessy J, Vukelja S, George T, Savin M, Richards D, Glaspy J, Meza L, Cohen G, Dhami M, Budman DR, Hackett J, Brassard M, Yang BB, Liang BC (2002) Comparable efficacy and safety profiles of once-per-cycle pegfilgrastim and daily injection filgrastim in chemotherapy-induced neutropenia: a multicenter dose-finding study in women with breast cancer. Ann Oncol 13:903–909
Johnston E, Crawford J, Blackwell S, Bjurstrom T, Lockbaum P, Roskos L, Yang BB, Gardner S, Miller-Messana MA, Shoemaker D, Garst J, Schwab G (2000) Randomized, dose-escalation study of SD/01 compared with daily filgrastim in patients receiving chemotherapy. J Clin Oncol 18:2522–2528
Martin M, Pienkowski T, Mackey J, Pawlicki M, Guastalla JP, Weaver C, Tomiak E, Al-Tweigeri T, Chap L, Juhos E, Guevin R, Howell A, Fornander T, Hainsworth J, Coleman R, Vinholes J, Modiano M, Pinter T, Tang SC, Colwell B, Prady C, Provencher L, Walde D, Rodriguez-Lescure A, Hugh J, Loret C, Rupin M, Blitz S, Jacobs P, Murawsky M, Riva A, Vogel C, Breast Cancer International Research Group 001 Investigators (2005) Adjuvant docetaxel for node-positive breast cancer. N Engl J Med 352:2302–2313
Mackey JR, Martin M, Pienkowski T, Rolski J, Guastalla JP, Sami A, Glaspy J, Juhos E, Wardley A, Fornander T, Hainsworth J, Coleman R, Modiano MR, Vinholes J, Pinter T, Rodríguez-Lescure A, Colwell B, Whitlock P, Provencher L, Laing K, Walde D, Price C, Hugh JC, Childs BH, Bass K, Lindsay MA, Wilson V, Rupin M, Houé V, Vogel C, TRIO/BCIRG 001 Investigators (2013) Adjuvant docetaxel, doxorubicin, and cyclophosphamide in node-positive breast cancer: 10-year follow-up of the phase 3 randomised BCIRG 001 trial. Lancet Oncol 14:72–80
Martín M, Lluch A, Seguí MA, Ruiz A, Ramos M, Adrover E, Rodríguez-Lescure A, Grosse R, Calvo L, Fernandez-Chacón C, Roset M, Antón A, Isla D, del Prado PM, Iglesias L, Zaluski J, Arcusa A, López-Vega JM, Muñoz M, Mel JR (2006) Toxicity and health-related quality of life in breast cancer patients receiving adjuvant docetaxel, doxorubicin, cyclophosphamide (TAC) or 5-fluorouracil, doxorubicin and cyclophosphamide (FAC): impact of adding primary prophylactic granulocyte-colony stimulating factor to the TAC regimen. Ann Oncol 17:1205–1212
Acknowledgments
We would like to thank all of the participating patients and their families and the investigators, research nurses, study coordinators, and operations staff who were involved in this study. Study principal investigators and their medical facilities are listed in Online Resource 2. This study was sponsored by Kyowa Hakko Kirin Co., Ltd. The JAPIC registry number for the study is JapicCTI-090954.
Conflict of interest
YT performed research and received research funding from Kyowa Hakko Kirin. RS is employed by and owns stock in Kyowa Hakko Kirin. YI received payment in relation to the role of the safety review committee. KT received payment in relation to the role of medical advisor. NM and SN performed research and have declared that they have no disclosure and financial support.
Author information
Authors and Affiliations
Corresponding author
Additional information
All the authors contributed equally to the work.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Online Resource 1
(DOC 74 kb)
Online Resource 2
(DOC 66 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and the source are credited.
About this article

Cite this article
Masuda, N., Tokuda, Y., Nakamura, S. et al. Dose response of pegfilgrastim in Japanese breast cancer patients receiving six cycles of docetaxel, doxorubicin, and cyclophosphamide therapy: a randomized controlled trial. Support Care Cancer 23, 2891–2898 (2015). https://doi.org/10.1007/s00520-015-2654-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00520-015-2654-4