
Abstract
A variety of factors regulate the efficiency of phosphate absorption in the intestine and phosphate reabsorption in kidney. Apart from the well-known regulators of phosphate homeostasis, namely parathyroid hormone (PTH) and the vitamin D–endocrine system, a number of peptides collectively known as the “phosphatonins” have been recently identified as a result of the study of various diseases associated with hypophosphatemia. These factors, fibroblast growth factor 23 (FGF-23), secreted frizzled-related protein 4 (sFRP-4), fibroblast growth factor 7 (FGF-7) and matrix extracellular phosphoglycoprotein (MEPE), have been shown to play a role in the pathogenesis of various hypophosphatemic and hyperphosphatemic disorders, such as oncogenic osteomalacia, X-linked hypophosphatemic rickets, autosomal dominant hypophosphatemic rickets, autosomal recessive hypophosphatemia and tumoral calcinosis. Whether these factors are true hormones, in the sense that they are regulated by the intake of dietary phosphorus and the needs of the organism for higher or lower amounts of phosphorus, remains to be firmly established in humans. Additionally, new information demonstrates that the intestine “senses” luminal concentrations of phosphate and regulates the excretion of phosphate in the kidney by elaborating novel factors that alter renal phosphate reabsorption.
Similar content being viewed by others
Phosphorus homeostasis
Phosphorus distribution: Phosphorus plays a critical role in many biological processes, including energy metabolism, cellular signaling through the phosphorylation of proteins and other substances, nucleic acid metabolism, membrane integrity, and bone mineralization. In the male human adult, total body phosphorus is between 15 mol and 20 mol (12.0 g/kg), the majority of which (80–90%) is present in bone in the form of hydroxyapatite [1, 2]. The remainder is present in soft tissues, extracellular fluid and erythrocytes. Soft tissue phosphorus is between 0.1% and 0.3% wet weight tissue (∼59 mmol/kg wet weight muscle). In plasma or serum, phosphorus exists as inorganic phosphate, lipid phosphorus and phosphoric ester phosphorus (concentrations, in millimoles, of each being 0.71–1.36, 2.23–3.13 and 0.86–1.45, respectively). Clearly, the plasma compartment contains only a small fraction of total body phosphorus, and changes in concentrations of inorganic phosphate do not necessarily reflect total body stores of phosphorus.
Phosphorus homeostasis and its regulation: The amounts of phosphorus moving across various epithelial tissues and organs are depicted in Fig. 1 [3]. Phosphorus is absorbed in the small intestine, predominantly in the jejunum, by both transcellular and paracellular processes (Fig. 1), the former process being mediated by sodium–phosphate type IIb cotransporters [4]. The paracellular pathway for phosphate/phosphorus absorption in the intestine is dependent, in large part, on the concentration of phosphorus present in the intestinal lumen. Increasing amounts of dietary phosphorus are associated with larger amounts of phosphorus absorption, with little evidence of saturation of the process. After entering the extracellular fluid space and circulation, phosphorus enters various tissues, including bone, as a result, at least in part, of the activity of sodium–phosphate type III cotransporters [4, 5]. Plasma inorganic phosphate is filtered at the glomerulus and is reabsorbed in the proximal tubule, largely via the sodium–phosphate cotransporter type IIa [6].

Phosphorus homeostasis in humans. Reprinted with permission [3]
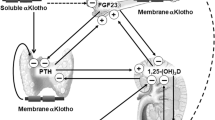
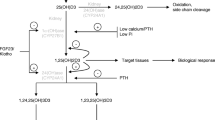
The regulation of phosphorus homeostasis is a complex process that involves the interplay between parathyroid hormone and vitamin D endocrine system (Fig. 2) [3, 7]. Phosphorus balance is primarily determined by processes that regulate the efficiency of intestinal phosphorus absorption and renal phosphorus reabsorption. Recent studies have provided evidence that parathyroid hormone (PTH) and vitamin D are not the sole regulators of inorganic phosphate (Pi) homeostasis and have led to the identification of other factors, such as the phosphatonins that contribute to the maintenance of Pi homeostasis. In addition, dietary Pi intake, dopamine, adrenergic activity and blood pH also influence plasma Pi concentrations (Fig. 3) [8]. Recent findings from our laboratory suggest that unique intestinal factors (“intestinal phosphatonins”), released by increases in intestinal luminal phosphate concentrations, alter the renal reabsorption of phosphate (Figs. 4 and 5) [9]. It is likely that these intestinal phosphatonins mediate the short-term changes in the fractional excretion of phosphate observed after ingestion of a meal, and it is likely that they play a role in the short-term adaptation to changes in dietary phosphate. On the other hand, long-term changes in dietary phosphate may be associated with changes in PTH, 1,25-dihydroxyvitamin D and the phosphatonins (see below).

The interaction between parathyroid hormone and vitamin D–endocrine system in the regulation of phosphorus homeostasis

Factors regulating phosphorus homeostasis in humans (FGF fibroblast growth factor, sFRP-4 secreted frizzled-related protein 4)

Experimental evidence for the presence of intestinal phosphatonins that mediate changes in renal phosphate excretion following increases in luminal phosphate concentrations in the intestine. Sodium phosphate (Na P) or sodium chloride (NaCl) was infused into the duodena of rats, and fractional excretion (FE) of phosphate was measured at short intervals following the infusion (TPTX thyroparathyroidectomized)

Intestinal phosphatonins mediate changes in the renal fractional excretion (FE) of phosphate following the ingestion of meals containing increased amounts of phosphate (gray hatched areas). Long-term dietary ingestion of increased amounts of phosphate is associated with increased PTH secretion and reduced 1,25- dihydroxyvitamin D synthesis. The levels of phosphatonins (PTNs) may increase following chronic increases in dietary phosphate excretion in some experimental models. Excursions in the fractional excretion of phosphate mediated by the intestinal phosphatonins still occur in the presence of an elevated baseline fractional excretion of phosphate. When phosphorus intake is curtailed, the opposite series of events occurs
The phosphatonins and disorders of phosphate homeostasis in humans: The term “phosphatonin” was coined in 1994 to describe a circulating phosphaturic factor present in the serum of patients with oncogenic or tumor-induced osteomalacia (TIO) [10, 11]. Cai et al. described a patient with TIO in whom the biochemical phenotype of hypophosphatemia, renal phosphate wasting, reduced 1α,25-dihydroxyvitamin D (1α,25(OH)2D) concentrations and the osteomalacia resolved after removal of the tumor [10, 11]. X-linked hypophosphatemic rickets (XLH) [12], autosomal dominant hypophosphatemic rickets (ADHR) [13], and autosomal recessive hypophosphatemia (ARHP) [14] are disorders that are phenotypically similar to TIO and which demonstrate the presence of a circulating factor responsible for hypophosphatemia and renal phosphate wasting [14–17]. Conversely, concentrations of one of the phosphatonins, fibroblast growth factor 23, are reduced in patients with tumoral calcinosis (TC), a disorder characterized by hyperphosphatemia, reduced fractional excretion of phosphate and deposits of calcium phosphate in soft tissues [18–24]. Several proteins, such as fibroblast growth factor-23 (FGF-23), secreted frizzled-related protein (sFRP-4), matrix extracellular phosphoglycoprotein (MEPE) and fibroblast growth factor-7 (FGF-7) have been identified as potential phosphatonins and probably play a role in the pathogenesis of some of these disorders [3, 14, 16, 17, 25–28]. Table 1 summarizes the pathophysiology of some of these hypophosphatemic and hyperphosphatemic disorders. A more detailed discussion of each of these peptides and their physiology and pathophysiology follows.
The biology of phosphatonins
Fibroblast growth factor-23: FGF-23 is a secreted, circulating, 32-kDa protein that is predominantly expressed in osteocytes in the bone and in the endothelial cells that line the venous sinusoids of bone marrow and the thymus [29]. FGF-23 null mice have decreased bone mineral density, elevated plasma Pi and 1α,25(OH)2D3 concentrations and low PTH concentrations [30]. It is difficult to ascertain if the decreased bone mineralization is a direct effect of reduced FGF-23 or a consequence of elevated Pi and 1α,25(OH)2D3 concentrations. Ectopic calcification in FGF-23 null mice is greatly diminished by either ablation of the vitamin D receptor or by feeding the mice a low phosphate diet, suggesting that elevated calcium and phosphate levels are important in the formation of ectopic mineral deposits [31, 32]. Transgenic mice over-expressing FGF-23 have reduced plasma Pi concentration, phosphaturia and reduced renal sodium phosphate cotransporter [33].
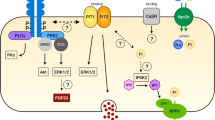
FGF-23 interacts with FGF receptors that belong to type 1 transmembrane phosphotyrosine kinase receptors to elicit a biological response in tissues [34]. Recent studies indicate that FGF-23 also requires Klotho, as a co-factor for receptor activation [34, 35]. In the mouse and human, the klotho/Klotho gene encodes a single-pass membrane protein which has homologies to β-glucosidases [36–39]. Two transcripts formed through alternative RNA splicing are transcribed from the gene and encode a membrane or secreted klotho protein [36]. A circulating and cerebrospinal fluid (CSF) form of klotho is also formed as a result of the cleavage of the membrane-bound form of the protein [36, 40]. Klotho is expressed in several tissues, including the kidney, reproductive tissues and brain [38]. The role of Klotho as a FGF-23 co-receptor is supported by the fact that Klotho-deficient mice have a phenotype similar to that of FGF-23 null mice [37].
The presence of FGF-23 in the circulation of healthy human subjects suggests that it plays a role in the maintenance of Pi homeostasis. In humans, short-term alterations in dietary Pi intake do not alter FGF-23 concentrations [41], and long-term changes in Pi intake have modest or no effect on FGF-23 concentrations [42, 43]. In animals FGF-23 concentrations are suppressed by low Pi diets and are stimulated by high Pi diets [44, 45]. In the short term, however, in rats there are no changes in FGF-23 or sFRP-4 concentrations following increases in intestinal Pi [9]. Serum FGF-23 concentrations increase following the administration of exogenous 1α,25(OH)2D3 [46], and FGF-23 expression is increased in bone cells following 1α,25(OH)2D3 treatment [47]. It is possible that the resultant increase in serum Pi concentrations after the administration of 1α,25(OH)2D3 stimulates the release of FGF-23, which, in turn, reduces serum Pi by promoting phosphaturia (Fig. 6). In hyperphosphatemic states, elevated Pi and FGF-23 concentrations may jointly inhibit formation of 1α,25(OH)2D3.

Relationships between changes in Pi, 1α,25(OH)2D and FGF-23. Reprinted with permission [3]
Secreted frizzled-related protein, fibroblast growth factor-7 and matrix extracellular phosphoglycoprotein: Like FGF-23, sFRP-4 decreases renal Pi reabsorption by reducing sodium phosphate transporters in renal proximal tubules and inhibits formation of 1α,25(OH)2D3 [25]. FGF-7 inhibits sodium-dependent Pi transport in opossum kidney cells, and anti-FGF-7 antibodies attenuate the phosphate transport inhibition induced by FGF-7 [48]. We have recently shown that FGF-7 is phosphaturic in vivo [49]. MEPE has been shown to increase the fractional excretion of phosphate and to induce hypophosphatemia in vivo [50]. In addition, MEPE inhibits bone mineralization in vitro, and MEPE null mice have increased bone mineralization. Importantly, MEPE does not inhibit 1α,25(OH)2D3 formation.
We will briefly discuss some clinical disorders in which one or more of the phosphatonins play a key role in the pathogenesis of the disease.
Role of phosphatonins in clinical disorders
Tumor-induced osteomalacia
TIO is a syndrome due to the presence of mesenchymal tumors that is associated with hypophosphatemia, hyperphosphaturia, inappropriately low serum 1α,25(OH)2D3 concentrations and osteomalacia [10]. The resolution of these biochemical and bone abnormalities following tumor removal supports the notion of the presence of a circulating factor (phosphatonin) secreted by the tumor. Numerous reports show elevation of FGF-23 in some, but not all, patients with TIO [33, 51, 52]. Removal of the tumor is associated with reduction in serum FGF-23 concentrations, and there is a temporal association between reduction in FGF-23 concentration and elevation in serum Pi, decrease in renal Pi wasting and increase in 1α,25(OH)2D3 concentrations [33, 51, 52]. sFRP-4, MEPE and FGF-7 have also been shown to be expressed by tumors associated with TIO [25, 48, 53]. The presence of different phosphatonins in TIO points towards the complex pathogenesis of this clinical condition.
X-linked hypophosphatemic rickets
Patients with X-linked hypophosphatemic rickets (XLH) manifest phosphaturia, hypophosphatemia and rickets [12, 15, 54]. Parabiosis and kidney cross-transplantation experiments have shown that there is a circulating hypophosphatemic factor present in the serum of Hyp mice (the mouse homolog of human XLH) [55–57]. In XLH, there are mutations of the gene encoding the endopeptidase, PHEX [15]. Patients with XLH have elevated serum concentrations of FGF-23 [51, 58], thereby indicating that PHEX is involved in the processing of FGF-23. Some studies have demonstrated that PHEX is responsible for FGF-23 degradation in vitro [26], whereas others have failed to demonstrate such an effect [59–61].
Autosomal dominant hypophosphatemic rickets
Autosomal dominant hypophosphatemic rickets (ADHR) is an inherited disorder of Pi homeostasis characterized by phosphaturia, hypophosphatemia, osteomalacia and rickets [13]. The ADHR Consortium identified mutations in the FGF-23 gene that encodes a mutant FGF-23 protein that lacks a normal furin proconvertase site making it resistant to proteolysis [16]. A long-lived stable form of FGF-23 is responsible for the clinical manifestations of this disorder [62].
Fibrous dysplasia/McCune-Albright syndrome
Fibrous dysplasia is a genetic non-inherited disease caused by somatic activating missense mutations of GNAS 1 that lead to variable clinical features, including polyostotic fibrous dysplasia, with endocrine (precocious puberty, pituitary gigantism, Cushing’s syndrome, thyrotoxicosis) and cutaneous (pigment patches on the skin) abnormalities [63]. Pi wasting is seen in approximately 50% of these patients and is associated with defective bone mineralization. One study demonstrated that FGF-23 concentration was elevated in patients with hypophosphatemia but was not increased in patients with normal Pi concentrations [64]. It is possible that the fibrous dysplastic tissue secretes FGF-23, and that serum FGF-23 concentrations are reflective of the disease burden in these patients.
Tumor calcinosis
Patients with tumor calcinosis (TC) manifest hyperphosphatemia, reduced renal Pi excretion and elevated 1α,25(OH)2D3 concentrations [65]. Three different types of mutations account for this syndrome. The first type occurs in the gene Ga1NAc transferase 3 (GALNT3), which encodes a glycosyltransferase responsible for initiating mucin-type O-glycosylation [18]. Some patients with this syndrome have low concentrations of intact FGF-23 but high concentrations of FGF-23 fragments. It has been hypothesized that these FGF-23 fragments lack biological activity, and, therefore, the clinical picture is consistent with what would be seen with low intact FGF-23 concentrations. In vivo infusion studies with FGF-23 fragments, however, have shown that carboxyl-terminal fragments are biologically active [66]. At present, there is uncertainty as to the precise mechanism by which GALNT3 mutations cause the syndrome. The second class of mutations responsible for TC occur in the gene encoding FGF-23 [22, 24]. This mutation results in defective processing of FGF-23 and its retention in the Golgi apparatus. Failure to secrete FGF-23 results in low serum concentrations of FGF-23, which, in turn, results in hyperphosphatemia due reduced renal Pi excretion. A third class of mutations responsible for TC occurs in the gene for Klotho [67], which encodes the co-receptor for FGF-23.
Renal failure
Serum FGF-23 concentrations are elevated in patients with chronic renal failure (CRF), and the increase in FGF-23 correlates with the decline in glomerular filtration rate [68–70]. Elevated plasma Pi seen in renal failure could increase FGF-23 production, although it is possible that reduced clearance of the peptide might also be responsible. Whether or not the elevated serum FGF-23 concentrations found in chronic renal insufficiency are sufficient to correct the hyperphosphatemia of early and advanced CRF is not completely clear. Elevated FGF-23 could play a role in the suppression of 1,25(OH)2D production and the development of secondary hyperparathyroidism. The role of FGF-23 in renal osteodystrophy has not been established. Indeed, a recent study shows no effect of FGF-23 on bone histology in end-stage renal disease [71]. Recently, Fliser et al. [72] showed a correlation between increased FGF-23 concentrations and the progression of chronic renal failure in subjects with mild-to-moderate chronic renal disease, suggesting that FGF-23 may play a role in the progression of renal failure. It should be noted, however, that the number of other variables such as the calcium X phosphate product, parathyroid hormone, and vitamin D usage also correlated with progression in the subjects. Finally, there are no data available at present suggesting a direct role of FGF-23 on renal fibrogenesis.
Post-transplant hypophosphatemia
In some patients following transplantation, persistent hypophosphatemia is noted, despite relatively modest increases in concentrations of circulating parathyroid hormone [68, 70, 73, 74].
In such subjects, FGF-23 concentrations have been noted to be elevated, and it is possible that elevations in the concentrations of this growth factor are responsible for the hypophosphatemia seen in this situation.
Elevations in FGF-23 in patients with tumors
In patients with humoral hypercalcemia of malignancy, and with metastatic ovarian cancer, FGF-23 concentrations are elevated without significant hypophosphatemia [75–77]. This would suggest that tumors produce FGF-23, and that FGF-23 concentrations must reach a certain significant threshold in order to increase phosphate excretion in the kidney.
In conclusion, phosphatonins play a vital role in the pathogenesis of a wide array of disorders. The presence of several phosphatonins and their differential effects affirm the complexity of Pi regulation in both normal and disease states. Future studies are needed to better understand the role of these proteins.
References
Diem K, Lentner C (1970) Scientific tables, Documenta Geigy. Ciba-Geigy Pharmaceuticals, New York
Fleisch H (1980) Homeostasis of inorganic phosphate. In: Urist MR (ed) Fundamental and clinical bone physiology. Lippincott, Philadelphia
Berndt T, Kumar R (2007) Phosphatonins and the regulation of phosphate homeostasis. Annu Rev Physiol 69:341–359
Forster IC, Virkki L, Bossi E, Murer H, Biber J (2006) Electrogenic kinetics of a mammalian intestinal type IIb Na(+)/P(I) cotransporter. J Membr Biol 212:177–190
Werner A, Kinne RK (2001) Evolution of the Na-P(I) cotransport systems. Am J Physiol Regul Integr Comp Physiol 280:R301–R312
Forster IC, Hernando N, Biber J, Murer H (2006) Proximal tubular handling of phosphate: a molecular perspective. Kidney Int 70:1548–1559
Berndt TJ, Schiavi S, Kumar R (2005) “Phosphatonins” and the regulation of phosphorus homeostasis. Am J Physiol Renal Physiol 289:F1170–F1182
Berndt T, Knox F (1992) Renal regulation of phosphate excretion. In: Giebisch G (ed) The kidney: physiology and pathophysiology. Raven Press, New York, pp 2511–2532
Berndt T, Thomas LF, Craig TA, Sommer S, Li X, Bergstralh EJ, Kumar R (2007) Evidence for a signaling axis by which intestinal phosphate rapidly modulates renal phosphate reabsorption. Proc Natl Acad Sci U S A 104:11085–11090
Cai Q, Hodgson SF, Kao PC, Lennon VA, Klee GG, Zinsmiester AR, Kumar R (1994) Brief report: inhibition of renal phosphate transport by a tumor product in a patient with oncogenic osteomalacia. N Engl J Med 330:1645–1649
Econs MJ, Drezner MK (1994) Tumor-induced osteomalacia—unveiling a new hormone. N Engl J Med 330:1679–1681
Drezner MK (2000) PHEX gene and hypophosphatemia. Kidney Int 57:9–18
Econs MJ, McEnery PT (1997) Autosomal dominant hypophosphatemic rickets/osteomalacia: clinical characterization of a novel renal phosphate-wasting disorder. J Clin Endocrinol Metab 82:674–681
Lorenz-Depiereux B, Bastepe M, Benet-Pages A, Amyere M, Wagenstaller J, Muller-Barth U, Badenhoop K, Kaiser SM, Rittmaster RS, Shlossberg AH, Olivares JL, Loris C, Ramos FJ, Glorieux F, Vikkula M, Juppner H, Strom TM (2006) DMP1 mutations in autosomal recessive hypophosphatemia implicate a bone matrix protein in the regulation of phosphate homeostasis. Nat Genet 38:1248–1250
The HYP Consortium (1995) A gene (PEX) with homologies to endopeptidases is mutated in patients with X-linked hypophosphatemic rickets. The HYP Consortium. Nat Genet 11:130–136
ADHR Consortium (2000) Autosomal dominant hypophosphataemic rickets is associated with mutations in FGF23. Nat Genet 26:345–348
Feng JQ, Ward LM, Liu S, Lu Y, Xie Y, Yuan B, Yu X, Rauch F, Davis SI, Zhang S, Rios H, Drezner MK, Quarles LD, Bonewald LF, White KE (2006) Loss of DMP1 causes rickets and osteomalacia and identifies a role for osteocytes in mineral metabolism. Nat Genet 38:1310–1315
Topaz O, Shurman DL, Bergman R, Indelman M, Ratajczak P, Mizrachi M, Khamaysi Z, Behar D, Petronius D, Friedman V, Zelikovic I, Raimer S, Metzker A, Richard G, Sprecher E (2004) Mutations in GALNT3, encoding a protein involved in O-linked glycosylation, cause familial tumoral calcinosis. Nat Genet 36:579–581
Larsson T, Yu X, Davis SI, Draman MS, Mooney SD, Cullen MJ, White KE (2005) A novel recessive mutation in fibroblast growth factor-23 causes familial tumoral calcinosis. J Clin Endocrinol Metab 90:2424–2427
Ichikawa S, Lyles KW, Econs MJ (2005) A novel GALNT3 mutation in a pseudoautosomal dominant form of tumoral calcinosis: evidence that the disorder is autosomal recessive. J Clin Endocrinol Metab 90:2420–2423
Frishberg Y, Topaz O, Bergman R, Behar D, Fisher D, Gordon D, Richard G, Sprecher E (2005) Identification of a recurrent mutation in GALNT3 demonstrates that hyperostosis-hyperphosphatemia syndrome and familial tumoral calcinosis are allelic disorders. J Mol Med 83:33–38
Araya K, Fukumoto S, Backenroth R, Takeuchi Y, Nakayama K, Ito N, Yoshii N, Yamazaki Y, Yamashita T, Silver J, Igarashi T, Fujita T (2005) A novel mutation in fibroblast growth factor (FGF)23 gene as a cause of tumoral calcinosis. J Clin Endocrinol Metab 90:5523–5527
Benet-Pages A, Orlik P, Strom TM, Lorenz-Depiereux B (2005) An FGF23 missense mutation causes familial tumoral calcinosis with hyperphosphatemia. Hum Mol Genet 14:385–390
Larsson T, Davis SI, Garringer HJ, Mooney SD, Draman MS, Cullen MJ, White KE (2005) Fibroblast growth factor-23 mutants causing familial tumoral calcinosis are differentially processed. Endocrinology 146:3883–3891
Berndt T, Craig TA, Bowe AE, Vassiliadis J, Reczek D, Finnegan R, Jan De Beur SM, Schiavi SC, Kumar R (2003) Secreted frizzled-related protein 4 is a potent tumor-derived phosphaturic agent. J Clin Invest 112:785–794
Bowe AE, Finnegan R, Jan de Beur SM, Cho J, Levine MA, Kumar R, Schiavi SC (2001) FGF-23 inhibits renal tubular phosphate transport and is a PHEX substrate. Biochem Biophys Res Commun 284:977–981
Shimada T, Mizutani S, Muto T, Yoneya T, Hino R, Takeda S, Takeuchi Y, Fujita T, Fukumoto S, Yamashita T (2001) Cloning and characterization of FGF23 as a causative factor of tumor-induced osteomalacia. Proc Natl Acad Sci U S A 98:6500–6505
De Beur SM, Finnegan RB, Vassiliadis J, Cook B, Barberio D, Estes S, Manavalan P, Petroziello J, Madden SL, Cho JY, Kumar R, Levine MA, Schiavi SC (2002) Tumors associated with oncogenic osteomalacia express genes important in bone and mineral metabolism. J Bone Miner Res 17:1102–1110
Liu S, Zhou J, Tang W, Jiang X, Rowe DW, Quarles LD (2006) Pathogenic role of FGF23 in Hyp mice. Am J Physiol Endocrinol Metab 291:E38–E49
Shimada T, Kakitani M, Yamazaki Y, Hasegawa H, Takeuchi Y, Fujita T, Fukumoto S, Tomizuka K, Yamashita T (2004) Targeted ablation of Fgf23 demonstrates an essential physiological role of FGF23 in phosphate and vitamin D metabolism. J Clin Invest 113:561–568
Razzaque MS, Sitara D, Taguchi T, St-Arnaud R, Lanske B (2006) Premature aging-like phenotype in fibroblast growth factor 23 null mice is a vitamin D-mediated process. FASEB J 20:720–722
Stubbs JR, Liu S, Tang W, Zhou J, Wang Y, Yao X, Quarles LD (2007) Role of hyperphosphatemia and 1,25-dihydroxyvitamin D in vascular calcification and mortality in fibroblastic growth factor 23 null mice. J Am Soc Nephrol 18:2116–2124
Larsson T, Marsell R, Schipani E, Ohlsson C, Ljunggren O, Tenenhouse HS, Juppner H, Jonsson KB (2004) Transgenic mice expressing fibroblast growth factor 23 under the control of the alpha1(I) collagen promoter exhibit growth retardation, osteomalacia, and disturbed phosphate homeostasis. Endocrinology 145:3087–3094
Kurosu H, Ogawa Y, Miyoshi M, Yamamoto M, Nandi A, Rosenblatt KP, Baum MG, Schiavi S, Hu MC, Moe OW, Kuro-o M (2006) Regulation of fibroblast growth factor-23 signaling by klotho. J Biol Chem 281:6120–6123
Urakawa I, Yamazaki Y, Shimada T, Iijima K, Hasegawa H, Okawa K, Fujita T, Fukumoto S, Yamashita T (2006) Klotho converts canonical FGF receptor into a specific receptor for FGF23. Nature 444:770–774
Shiraki-Iida T, Aizawa H, Matsumura Y, Sekine S, Iida A, Anazawa H, Nagai R, Kuro-o M, Nabeshima Y (1998) Structure of the mouse klotho gene and its two transcripts encoding membrane and secreted protein. FEBS Lett 424:6–10
Kuro-o M, Matsumura Y, Aizawa H, Kawaguchi H, Suga T, Utsugi T, Ohyama Y, Kurabayashi M, Kaname T, Kume E, Iwasaki H, Iida A, Shiraki-Iida T, Nishikawa S, Nagai R, Nabeshima YI (1997) Mutation of the mouse klotho gene leads to a syndrome resembling ageing. Nature 390:45–51
Kato Y, Arakawa E, Kinoshita S, Shirai A, Furuya A, Yamano K, Nakamura K, Iida A, Anazawa H, Koh N, Iwano A, Imura A, Fujimori T, Kuro-o M, Hanai N, Takeshige K, Nabeshima Y (2000) Establishment of the anti-Klotho monoclonal antibodies and detection of Klotho protein in kidneys. Biochem Biophys Res Commun 267:597–602
Torres PU, Prie D, Molina-Bletry V, Beck L, Silve C, Friedlander G (2007) Klotho: an antiaging protein involved in mineral and vitamin D metabolism. Kidney Int 71:730–737
Imura A, Iwano A, Tohyama O, Tsuji Y, Nozaki K, Hashimoto N, Fujimori T, Nabeshima Y (2004) Secreted Klotho protein in sera and CSF: implication for post-translational cleavage in release of Klotho protein from cell membrane. FEBS Lett 565:143–147
Nishida Y, Taketani Y, Yamanaka-Okumura H, Imamura F, Taniguchi A, Sato T, Shuto E, Nashiki K, Arai H, Yamamoto H, Takeda E (2006) Acute effect of oral phosphate loading on serum fibroblast growth factor 23 levels in healthy men. Kidney Int 70:2141–2147
Ferrari SL, Bonjour JP, Rizzoli R (2005) Fibroblast growth factor-23 relationship to dietary phosphate and renal phosphate handling in healthy young men. J Clin Endocrinol Metab 90:1519–1524
Burnett SM, Gunawardene SC, Bringhurst FR, Juppner H, Lee H, Finkelstein JS (2006) Regulation of C-terminal and intact FGF-23 by dietary phosphate in men and women. J Bone Miner Res 21:1187–1196
Sommer S, Berndt T, Craig T, Kumar R (2007) The phosphatonins and the regulation of phosphate transport and vitamin D metabolism. J Steroid Biochem Mol Biol 103:497–503
Perwad F, Azam N, Zhang MY, Yamashita T, Tenenhouse HS, Portale AA (2005) Dietary and serum phosphorus regulate fibroblast growth factor 23 expression and 1,25-dihydroxyvitamin D metabolism in mice. Endocrinology 146:5358–5364
Saito H, Maeda A, Ohtomo S, Hirata M, Kusano K, Kato S, Ogata E, Segawa H, Miyamoto K, Fukushima N (2005) Circulating FGF-23 is regulated by 1α,25-dihydroxyvitamin D3 and phosphorus in vivo. J Biol Chem 280:2543–2549
Kolek OI, Hines ER, Jones MD, LeSueur LK, Lipko MA, Kiela PR, Collins JF, Haussler MR, Ghishan FK (2005) 1α,25-Dihydroxyvitamin D3 upregulates FGF23 gene expression in bone: the final link in a renal-gastrointestinal-skeletal axis that controls phosphate transport. Am J Physiol Gastrointest Liver Physiol 289:G1036–G1042
Carpenter TO, Ellis BK, Insogna KL, Philbrick WM, Sterpka J, Shimkets R (2005) Fibroblast growth factor 7: an inhibitor of phosphate transport derived from oncogenic osteomalacia-causing tumors. J Clin Endocrinol Metab 90:1012–1020
Shaikh A, Berndt T, Kumar R (2007) FGF-7 is a potent in vivo phosphaturic agent in rats. J Bone Miner Res 22:S106
Rowe PS, Kumagai Y, Gutierrez G, Garrett IR, Blacher R, Rosen D, Cundy J, Navvab S, Chen D, Drezner MK, Quarles LD, Mundy GR (2004) MEPE has the properties of an osteoblastic phosphatonin and minhibin. Bone 34:303–319
Yamazaki Y, Okazaki R, Shibata M, Hasegawa Y, Satoh K, Tajima T, Takeuchi Y, Fujita T, Nakahara K, Yamashita T, Fukumoto S (2002) Increased circulatory level of biologically active full-length FGF-23 in patients with hypophosphatemic rickets/osteomalacia. J Clin Endocrinol Metab 87:4957–4960
Takeuchi Y, Suzuki H, Ogura S, Imai R, Yamazaki Y, Yamashita T, Miyamoto Y, Okazaki H, Nakamura K, Nakahara K, Fukumoto S, Fujita T (2004) Venous sampling for fibroblast growth factor-23 confirms preoperative diagnosis of tumor-induced osteomalacia. J Clin Endocrinol Metab 89:3979–3982
Rowe PS, de Zoysa PA, Dong R, Wang HR, White KE, Econs MJ, Oudet CL (2000) MEPE, a new gene expressed in bone marrow and tumors causing osteomalacia. Genomics 67:54–68
Drezner MK (2003) Hypophosphatemic rickets. Endocr Dev 6:126–155
Meyer RA Jr, Meyer MH, Gray RW (1989) Parabiosis suggests a humoral factor is involved in X-linked hypophosphatemia in mice. J Bone Miner Res 4:493–500
Meyer RA Jr, Tenenhouse HS, Meyer MH, Klugerman AH (1989) The renal phosphate transport defect in normal mice parabiosed to X-linked hypophosphatemic mice persists after parathyroidectomy. J Bone Miner Res 4:523–532
Nesbitt T, Coffman TM, Griffiths R, Drezner MK (1992) Crosstransplantation of kidneys in normal and Hyp mice. Evidence that the Hyp mouse phenotype is unrelated to an intrinsic renal defect. J Clin Invest 89:1453–1459
Jonsson KB, Zahradnik R, Larsson T, White KE, Sugimoto T, Imanishi Y, Yamamoto T, Hampson G, Koshiyama H, Ljunggren O, Oba K, Yang IM, Miyauchi A, Econs MJ, Lavigne J, Juppner H (2003) Fibroblast growth factor 23 in oncogenic osteomalacia and X-linked hypophosphatemia. N Engl J Med 348:1656–1663
Guo R, Liu S, Spurney RF, Quarles LD (2001) Analysis of recombinant Phex: an endopeptidase in search of a substrate. Am J Physiol Endocrinol Metab 281:E837–E847
Liu S, Guo R, Tu Q, Quarles LD (2002) Overexpression of Phex in osteoblasts fails to rescue the Hyp mouse phenotype. J Biol Chem 277:3686–3697
Benet-Pages A, Lorenz-Depiereux B, Zischka H, White KE, Econs MJ, Strom TM (2004) FGF23 is processed by proprotein convertases but not by PHEX. Bone 35:455–462
Shimada T, Muto T, Urakawa I, Yoneya T, Yamazaki Y, Okawa K, Takeuchi Y, Fujita T, Fukumoto S, Yamashita T (2002) Mutant FGF-23 responsible for autosomal dominant hypophosphatemic rickets is resistant to proteolytic cleavage and causes hypophosphatemia in vivo. Endocrinology 143:3179–3182
Levine MA (1991) The McCune-Albright syndrome. The whys and wherefores of abnormal signal transduction. N Engl J Med 325:1738–1740
Riminucci M, Collins MT, Fedarko NS, Cherman N, Corsi A, White KE, Waguespack S, Gupta A, Hannon T, Econs MJ, Bianco P, Gehron Robey P (2003) FGF-23 in fibrous dysplasia of bone and its relationship to renal phosphate wasting. J Clin Invest 112:683–692
Lufkin EG, Kumar R, Heath H 3rd (1983) Hyperphosphatemic tumoral calcinosis: effects of phosphate depletion on vitamin D metabolism, and of acute hypocalcemia on parathyroid hormone secretion and action. J Clin Endocrinol Metab 56:1319–1322
Berndt TJ, Craig TA, McCormick DJ, Lanske B, Sitara D, Razzaque MS, Pragnell M, Bowe AE, O’Brien SP, Schiavi SC, Kumar R (2007) Biological activity of FGF-23 fragments. Pflugers Arch 454:615–623
Ichikawa S, Imel EA, Kreiter ML, Yu X, Mackenzie DS, Sorenson AH, Goetz R, Mohammadi M, White KE, Econs MJ (2007) A homozygous missense mutation in human KLOTHO causes severe tumoral calcinosis. J Clin Invest 117:2684–2691
Pande S, Ritter CS, Rothstein M, Wiesen K, Vassiliadis J, Kumar R, Schiavi SC, Slatapolsky E, Brown AJ (2006) FGF-23 and sFRP-4 in chronic kidney disease and post-renal transplantation. Nephron Physiol 104:p23–p32
Imanishi Y, Inaba M, Nakatsuka K, Nagasue K, Okuno S, Yoshihara A, Miura M, Miyauchi A, Kobayashi K, Miki T, Shoji T, Ishimura E, Nishizawa Y (2004) FGF-23 in patients with end-stage renal disease on hemodialysis. Kidney Int 65:1943–1946
Larsson T, Nisbeth U, Ljunggren O, Juppner H, Jonsson KB (2003) Circulating concentration of FGF-23 increases as renal function declines in patients with chronic kidney disease, but does not change in response to variation in phosphate intake in healthy volunteers. Kidney Int 64:2272–2279
Urena Torres P, Friedlander G, de Vernejoul MC, Silve C, Prie D (2008) Bone mass does not correlate with the serum fibroblast growth factor 23 in hemodialysis patients. Kidney Int 73:102–107
Fliser D, Kollerits B, Neyer U, Ankerst DP, Lhotta K, Lingenhel A, Ritz E, Kronenberg F, Kuen E, Konig P, Kraatz G, Mann JF, Muller GA, Kohler H, Riegler P (2007) Fibroblast growth factor 23 (FGF23) predicts progression of chronic kidney disease: the Mild to Moderate Kidney Disease (MMKD) study. J Am Soc Nephrol 18:2600–2608
Evenepoel P, Naesens M, Claes K, Kuypers D, Vanrenterghem Y (2007) Tertiary ‘hyperphosphatoninism’ accentuates hypophosphatemia and suppresses calcitriol levels in renal transplant recipients. Am J Transplant 7:1193–1200
Bhan I, Shah A, Holmes J, Isakova T, Gutierrez O, Burnett SA, Juppner H, Wolf M (2006) Post-transplant hypophosphatemia: tertiary ‘hyper-phosphatoninism’? Kidney Int 70:1486–1494
Singh RJ, Kumar R (2003) Fibroblast growth factor 23 concentrations in humoral hypercalcemia of malignancy and hyperparathyroidism. Mayo Clin Proc 78:826–829
Tebben PJ, Kalli KR, Cliby WA, Hartmann LC, Grande JP, Singh RJ, Kumar R (2005) Elevated fibroblast growth factor 23 in women with malignant ovarian tumors. Mayo Clin Proc 80:745–751
Tebben PJ, Singh RJ, Clarke BL, Kumar R (2004) Fibroblast growth factor 23, parathyroid hormone, and 1α,25-dihydroxyvitamin D in surgically treated primary hyperparathyroidism. Mayo Clin Proc 79:1508–1513
Carpenter TO, Ellis BK, Insogna KL, Philbrick WM, Sterpka J, Shimkets R (2005) FGF7—an inhibitor of phosphate transport derived from oncogenic osteomalacia-causing tumors. J Clin Endocrinol Metab 90:1012–1020
Yuan B, Takaiwa M, Clemens T, Feng J, Kumar R, Rowe P, Xie Y, Drezner M (2008) Aberrant Phex function in osteoblasts and osteocytes alone underlines murine X-linked hypophosphatemia. J Clin Invest 118:722–734
Araya K, Fukumoto S, Backenroth R, Takeuchi Y, Nakayama K, Ito N, Yoshii N, Yamazaki Y, Yamashita T, Silver J, Igarashi T, Fujita T (2005) A novel mutation in fibroblast growth factor 23 gene as a cause of tumoral calcinosis. J Clin Endocrinol Metab 90:5523–5527
Acknowledgments
Supported by National Institutes of Health (NIH) grants DK 65830, DK 76829, DK 77669 and DK 79858.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article
Cite this article
Shaikh, A., Berndt, T. & Kumar, R. Regulation of phosphate homeostasis by the phosphatonins and other novel mediators. Pediatr Nephrol 23, 1203–1210 (2008). https://doi.org/10.1007/s00467-008-0751-z
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00467-008-0751-z