
Abstract
Activins and transforming growth factor (TGF)-βs, members of the TGF-β superfamily, affect numerous physiological processes, including apoptosis, in a variety of organs and tissues. Apoptotic functions of TGF-βs, in contrast to those of the activins, are well documented in the developing and adult nervous system. TGF-βs operate in a context-dependent manner and cooperate with other cytokines in the regulation of apoptosis. In this study, we show, for the first time, an apoptotic function of ActivinA in the nervous system, i.e. in oligodendroglial progenitor cells. Using the oligodendroglial cell line OLI-neu, we show that ActivinA acts autonomously, without cooperating with TGF-β. In contrast to the mechanism of TGF-β-mediated apoptosis involving Bcl-xl down-regulation, Bcl-xl in ActivinA-induced apoptosis is classically sequestered by the BH3-only protein Puma. Puma expression is controlled by the transcription factor p53 as demonstrated by experiments with the p53 inhibitor Pifithrin-α. Furthermore, in the apoptotic TGF-β pathway, caspase-3 is activated, whereas in the apoptotic ActivinA pathway, apoptosis-inducing factor is released to trigger DNA fragmentation. These data suggest that TGF-β and ActivinA induce apoptosis in oligodendrocytes by different apoptotic pathways.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Apoptosis plays a significant role in shaping tissues and organs during development by utilising many different intracellular signalling pathways leading to the ultimate demise of the cell. The mitochondrial apoptotic pathway is regulated by members of the Bcl-2 family comprising pro-apoptotic and anti-apoptotic proteins (Gross et al. 1999; Orrenius 2004). Classically, anti-apoptotic proteins such as Bcl-2, Bcl-xl and Mcl-1 protect mitochondrial integrity, whereas pro-apoptotic members promote the release of apoptogenic proteins such as cytochrome C and apoptosis-inducing factor (AIF) from the mitochondria (Krantic et al. 2007; Orrenius 2004). Pro-apoptotic proteins are further divided into the BH3-only proteins and the multidomain effectors Bax and Bak. BH3-only proteins are recognised as essential initiators of apoptosis because they act as death ligands to activate the pro-apoptotic effectors Bax and Bak or to inactivate anti-apoptotic members (Krantic et al. 2007; Orrenius 2004). Expression of some BH3-only proteins, including Puma, is promoted by the tumor suppressor p53 (Chipuk and Green 2006).
Caspases represent initiators and executioners of the classical apoptotic pathway. Initiator caspases are activated via extrinsic or intrinsic apoptotic signals to cleave other caspases and/or diverse effectors leading to apoptotic changes such as DNA fragmentation and cleavage of various intracellular substrates (Degterev et al. 2003; Krantic et al. 2007; Orrenius 2004; Wang 2000). AIF is a mediator of executioner caspase-independent apoptosis. Upon mitochondrial permeabilisation, AIF translocates from the mitochondria to the nucleus. In the nucleus, an AIF-containing complex triggers large-scale DNA fragmentation (Krantic et al. 2007; Modjtahedi et al. 2006).
The transforming growth factor (TGF)-β superfamily plays key roles in development and ontogenetic cell death. The family comprises, amongst other TGF-βs, bone morphogenetic proteins (BMPs), Activins and related proteins. TGF-βs act in context depending on the type of cell, its stage of differentiation and cooperating cytokines/molecules (Böttner et al. 2000; Sanchez-Capelo 2005; Schuster and Krieglstein 2002). Activins are important apoptotic players but have been described so far only in non-neural systems in adults, e.g. in the immune system, in the homeostasis of tissues and in tumorigenesis by way of various apoptotic pathways such as the Bcl-2 family and the caspase cascade. In neural systems, activins have been shown to promote survival rather than apoptosis (W. Chen et al. 2000; Y.G. Chen et al. 2002, 2006). TGF-βs are also involved in developmental and neuronal apoptosis and cooperate with other secreted cytokines such as BMPs or TNF-α (Böttner et al. 2000; Duenker and Krieglstein 2000, 2002, 2003; Duenker et al. 2002a, b; Franke et al. 2006; Sanchez-Capelo 2005; Schuster et al. 2003; Schuster and Krieglstein 2002; Unsicker and Strelau 2000). The importance of TGF-βs during neural ontogenetic cell death was first shown in chicken, where apoptosis is reduced in various neuron populations by immunoneutralisation of endogenous TGF-βs (Krieglstein et al. 2000). In retina cultures, TGF-βs mediate apoptosis by down-regulation of Bcl-xl protein (Schuster et al. 2002b). In the oligodendroglial cell line OLI-neu, TGF-β1-induced apoptosis is accompanied by down-regulation of caspases and Bcl-xl protein (Schuster et al. 2003).
In this study, we have analysed apoptotic TGF-β signalling with regard to possible cooperating factors. Analysis of oligodendroglial progenitor cells has shown, for the first time, that ActivinA alone is capable of mediating apoptosis in the nervous system. Therefore, ActivinA and TGF-β1 induce apoptosis without cooperation and by functioning in two different pathways. Previous studies of the TGF-β-responsive OLI-neu cells have demonstrated that TGF-β mediates DNA fragmentation through executioner caspases (Schuster et al. 2002a, 2003). ActivinA, as shown here, induces DNA fragmentation through AIF translocation from the mitochondria to the nucleus in an executioner-caspase-independent manner. The release of the various mediators of DNA fragmentation is the result of different regulation pathways on the mitochondria. In TGF-β-induced apoptosis, Bcl-xl protein is down-regulated (Schuster et al. 2002a), whereas in ActivinA-induced apoptosis, the amounts of Bcl-xl protein are unaffected, but Bcl-xl is sequestered through the BH3-only protein Puma.
Materials and methods
Cell culture and culture conditions
Murine OLI-neu cells were cultured on poly-L-lysine (PLL)-coated dishes (Nunc) in Dulbecco’s modified Eagle’s medium with 1% horse serum, penicillin-streptomycin-neomycin (PSN), N2-supplement (GIBCO), 400 ng/ml L-thyroxine and 340 ng/ml tri-iodo-L-thyronine (Sigma Aldrich). Cultured cells were treated with either 5 ng/ml TGF-β1 or 50 ng/ml ActivinA (both: Peprotech) in serum-reduced media (0.2% horse serum) without N2-supplement and PSN. Inhibitors were added 2 h prior to the factors. The pan-caspase inhibitor Z-VAD-FMK (final concentration: 50 µM) and Pifithrin-α (PFT; final concentration: 20 nM) were purchased from Calbiochem/Merck Biosciences. The Alk-4/5/7 inhibitor SB 431542 was from Tocris, Germany, and was used at a concentration of 10 µM. Primary oligodendroglial cells were prepared as described by Schuster et al. (2002a).
RNA interference
The short interfering RNA (siRNA) constructs were designed with the RNAi pSuper.neo vector system (Oligoengine, USA) according to the manufacturer’s guidelines to interfere with the expression of AIF, the AIF siRNA sense sequence being 5′-ATGCAGAACTCCAAGCACG-3′. Cells were transfected with the siRNA vectors by using Fugene6 in accordance with the manufacturer’s guidelines at a ratio of 3:2 (Roche Diagnostics). After 3 days of incubation, cells were treated with cytokines as indicated.
Luciferase assays
Cells (3×104) were seeded in a PLL-coated 48-well plate. After 16 h of culture, the Smad-binding element (SBE)-luciferase reporter construct (Jonk et al. 1998) was transfected together with a β-GAL4 control construct by using Fugene6 (Roche Diagnostics) according to the manufacturer’s guidelines at a ratio of 3:2. After 16 h of incubation, samples were treated with the cytokines and then harvested in lysis buffer to measure the luciferase activity and the β-Gal activity with a luminometer (Lumat LB 9507, Berthold) by means of the Tropix Assay Kit of Nalgene (Bedford, Mass., USA) according to the manufacturer’s guidelines. Measurement of β-Gal activity served as a loading control to determine equal transfection rates. The luciferase activity of each sample was normalised to β-Gal activity.
The MLEC/PAI-luciferase assay was carried out according to Lacmann et al. (2007)
Immunofluorescence
Cells (2×105) were seeded onto PLL-coated 12-mm glass coverslips. Treated cells were fixed in 4% paraformaldehyde in phosphate-buffered saline (PFA/PBS) for 20 min and permeabilised with 0.1% Triton X-100 for 10 min at room temperature. Following several washing steps, blocking was performed with 5% bovine serum albumin in PBS for 1 h. Smad1/2/3 antibody (Santa Cruz) or AIF antibody (Chemicon) were detected by Cy3-conjugated secondary antibodies (Jackson ImmunoResearch Europe). Samples were mounted in Fluoromount-G mounting solution (Southern Biotechnology) containing 4,6-diamidino-2-phenylindole (DAPI). Whole cell analysis was performed by using a Nikon Eclipse E600 microscope with an AxioCamHR camera (Carl Zeiss). Images were examined with a Nikon PLAN APO 40x/0.95 lens at room temperature. Fluorochromes were visualised with filter sets for DAPI (364/454 nm) and Cy3 (552/570 nm).
Caspase activity assays
Samples were lysed with lysis buffer (10 mM TRIS-HCl, 10 mM NaH2PO4/NaHPO4, pH 7.5, 130 mM NaCl, 1% Triton X-100). Protein amounts were measured by using the Bradford assay. Caspase-2 (z-VDVAD-AFC) and caspase-9 (Ac-LEHD-AFC; Calbiochem), and caspase-8 (Ac-IETD-AFC) and caspase-3 (Ac-DEVD-AMC; BD Bioscience Europe) activity assays were employed according to the manufacturer’s guidelines and incubated together with 20 µg protein for 6 h at 37°C in an opaque 96-well plate (Greiner Labortechnik). Fluorometric probes were measured in the FLX800 Fluorescence Reader (BioTek Instruments) with suitable filters (AFC-fluorophores: excitation 400 nm, emission 505 nm; AMC-fluorophores: excitation 380 nm, emission 430–460 nm).
Western blotting
Total protein extracts were isolated with cell lysis buffer [10 mM TRIS-HCl, pH 7.4, 1.5 mM MgCl2, 0.5% Nonident P40, complete protease inhibitor mix (Roche Diagnostics)]. Following sonication (3×10 s), samples were centrifuged at 13,000 rpm for 10 min. Subcellular fractionation of proteins was performed according to the manufacturer’s guidelines by using the ProteoExtract® Subcellular Proteome Extraction Kit from Calbiochem. Protein samples were measured with the Bradford assay (Bio-Rad Laboratories). Equal amounts of protein were prepared with NuPage sample buffer (Invitrogen) and separated by SDS-gel-electrophoresis (BioRad Lab). Proteins were transferred onto nitrocellulose membrane (BioCat), blocked in 5% milk/TRIS-buffered saline with Triton X-100 for 1 h and probed with primary antibodies [Puma, Bcl-xl, CoxIV and Bax antibodies (Cell Signaling Tech), p53 (FL-393; Santa Cruz), AIF and Fractin antibodies (Chemicon Europe)]. As loading controls, D-glyceraldehyde-3-phosphate dehydrogenase (Gapdh) protein was detected by Gapdh antibody (Acris) and Histon1 by H1 (4112) antibody (kind gift of D. Doenecke, Department of Molecular Biology, University of Göttingen, Germany). Primary antibodies were detected by horseradish-peroxide-conjugated secondary antibodies (Jackson ImmunoResearch Europe) and immunoreactive signals were developed by chemiluminescence according to the manufacturer’s protocol (Millipore).
Immunoprecipitation
Treated cells (2×106) were harvested and resuspended in cell lysis buffer [50 mM TRIS pH 7.5, 500 mM NaCl, 1% Nonident P40, complete protease inhibitor mix (Roche Diagnostics)]. After sonication (3×10 s), samples were centrifuged at 13,000 rpm for 10 min and 50 µl ProteinA sepharose 4 Fast Flow beads (50% slurry; Amersham Bioscience Europe) pre-cleared the cell lysates. Equal protein amounts were determined by the Bradford assay. IP antibodies (1–2 µg) were used for antigen coupling. Bcl-xl was purchased from Cell Signaling Tech. Precipitation of immune complexes was carried out by adding 20 µl ProteinA sepharose. The immune complexes were washed and mixed with 2xNuPage sample buffer (Invitrogen) to analyse the supernatants by SDS-gel-electrophoresis as described (above).
TGF-β release assay
Specific quantification of released TGF-β was as described elsewhere (Krieglstein et al. 2000).
Flow cytometry
Cells (2×105) were trypsinised, washed in PBS, and fixed in 70% ethanol at -20°C overnight. Samples were washed in PBS before being staining with propidium iodide solution (100 µg/ml propidium iodide, 100 µg/ml RNase in PBS) for 30 min at 37°C. Fluorescence-activated cell sorting (FACS) samples were counted with a FACScan flow cytometer and analysed with CellQuest software (Becton Dickinson). The percentage of cells present in the sub-G1 peak, representing apoptotic cells, was calculated after exclusion of cell doublets.
Detection of cell death by TUNEL technology
Cells (2×105) were cultured on PLL-coated 12-mm glass slides, treated and fixed in 4% PFA/PBS for 60 min. Fragmented nuclei were detected with the In Situ Cell Death Detection Kit, TMR red according to the manufacturer’s guidelines (Roche Diagnostics). Samples were mounted in DAPI/Fluoromount-G (Southern Biotech) staining solution. Whole cell analysis was performed by using a Nikon Eclipse E600 microscope with an AxioCamHR camera (Carl Zeiss). To quantify the number of dead cells, red fluorescence (filter: 552/570 nm) was determined in relation to DAPI-stained nuclei.
Densitometric measurement and statistical analysis
Densitometric quantification of signals was carried out with the AlphaEaseFC program. Measurements of experimental bands were compared with loading control, by calculating the relative values (±SEM). Statistical analysis of the data was performed with Prism4 GraphPad Software using Student’s t-test.
Results
ActivinA induces apoptosis in oligodendroglial progenitor cells
TGF-β is known as a contextually acting molecule that cooperates with other cytokines, including TNF-α and BMPs, to induce apoptosis (Franke et al. 2006; Schuster et al. 2003). In the present study, we show that another cytokine of the TGF-β superfamily, ActivinA, also induces apoptosis in oligodendroglial progenitor cells (Fig. 1a,b).
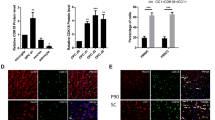
ActivinA mediated apoptosis as analysed by flow cytometry and the counting of apoptotic nuclei (sub-G1 peak). A dose of 50 ng/ml ActivinA proved to be an optimal apoptotic condition (Fig. 1a). TGF-β1 (5 ng/ml) served as positive apoptotic control. To test whether ActivinA and TGF-β1 cooperated in their capacity to induce apoptosis, we co-treated OLI-neu cells with 5 ng/ml TGF-β1 and 50 ng/ml ActivinA. This combination resulted in the same apoptosis rate as treatment with TGF-β1 alone, excluding a possible cooperation between TGF-β1 and ActivinA (Fig. 1a). Further, TUNEL staining showed a significant increase of fragmented DNA following ActivinA treatment (Fig. 1b). TGF-β-mediated apoptosis seemed to be more potent than ActivinA-mediated apoptosis, as seen in flow cytometry and in TUNEL staining (Fig. 1a,b). To confirm that ActivinA induced apoptosis in oligodendrocytes, we performed TUNEL staining in primary oligodendroglial cultures (Fig. 1e). By counting TUNEL-positive nuclei in relation to DAPI-positive nuclei, we showed that ActivinA also significantly induced fragmented DNA in primary oligodendroglial cultures.

ActivinA induces apoptosis in oligodendrocytes, autonomously of TGF-β. a ActivinA (ActA)-mediated DNA fragmentation. OLI-neu cells were treated with ActivinA at various concentrations for 20 h. TGF-β1-treated cells served as an apoptotic control. Cells were harvested and fixed in 70% ethanol and nuclei were stained with propidium iodide for flow cytometry. The intensity of the stained nuclei showed the stage of the cell. The sub-G1 peak (DNA<n2) represented apoptotic nuclei. Values are means ±SEM of three separate experiments. ***Statistically significant difference at P<0.001, calculated by Student’s t-test. b OLI-neu cells were seeded on glass slides and treated with ActivinA (50 ng/ml) for 20 h. Fixed cells were prepared for staining of fragmented DNA by enzymatic fluorescence labelling. Counts of fragmented DNA were compared with phase-contrast images. c ActivinA (ActA) acts autonomously of TGF-β. OLI-neu cells were pre-treated with neutralising TGF-β1/2/3 antibody and ActivinA (50 ng/ml) was added for 20 h. Apoptotic cells were analysed by flow cytometry as in a. d ActivinA does not induce TGF-β release. OLI-neu cells were cultured and treated with 50 ng/ml ActivinA for 12 h. Supernatants were quantified with the MLEC/PAI-luciferase assay. Data are given as amounts of TGF-β release in the supernatant, as means±SEM of two separate experiments, each with two replicates (RLU relative light units). Statistical significance was tested with Student’s t-test. e DNA fragmentation in primary cultures. Primary oligodendroglial cells were seeded on glass slides and treated with ActivinA (50 ng/ml) for 20 h. Fixed cells were prepared for staining of fragmented DNA by enzymatic fluorescence labelling with subsequent DAPI staining. Positive-fragmented cells were counted in relation to DAPI staining and the percentage of apoptotic cells was calculated. Values are given as means ±SEM of three separate experiments, counting 3000 cells. ***Statistcially significant at P<0.001, calculated by Student’s t-test
To exclude the possibility that apoptosis is an effect of ActivinA-induced TGF-β release, we performed TGF-β release assays. OLI-neu cells were pre-treated with the neutralising antibody against TGF-β1/2/3 to absorb all released TGF-β isoforms. After additional ActivinA treatment (20 h), cells were analysed by flow cytometry, apoptotic nuclei being counted. As shown in Fig. 1c, in spite of co-treatment with neutralising antibodies, ActivinA induced DNA fragmentation. We additionally determined possible TGF-β release by using a highly specific and sensitive TGF-β bioassay, the MLEC/PAI-luciferase assay (Lacmann et al. 2007). As seen in Fig. 1d, after ActivinA treatment (12 h), no increased amounts of TGF-β were detectable in the cell culture supernatants. Together, these results indicate that two independent cytokine-induced apoptotic pathways exist in the same cell type.
The signal transduction of the TGF-β superfamily is mediated through Smad proteins, which translocate into the nucleus (Derynck and Zhang 2003). Using a Smad-binding element (SBE)-luciferase reporter assay, we demonstrated that the Smad cascade was significantly activated by ActivinA and was blocked following co-treatment with the Alk4/5/7 inhibitor SB421542 (Fig. 2a). Immunocytochemistry experiments with a Smad1/2/3 antibody confirmed Smad activation by showing Smad translocation into the nucleus after ActivinA treatment (2 h; Fig. 2b).

a Analysis of Smad activation in ActivinA-mediated apoptosis. OLI-neu cells were cultured and transfected with an SBE-luciferase construct for 16 h. SB431542 was pre-incubated for 2 h, and 50 ng/ml ActivinA was added for 12 h. After cell lysis, Smad activation was monitored by measurement of luciferase expression. Luciferase activity was compared with β-Gal activity to calculate relative values. Values are given as means ±SEM of two separate experiments, each with two replicates; **P<0.01 (Student’s t-test). b Immunofluorescence staining of Smad-1/2/3 together with DAPI staining. OLI-neu cells were treated with ActivinA for 2 h. TGF-β1 served as a positive control. Nuclear Smad staining occurred in ActivinA-treated and in TGF-β-treated samples. Bar 8 µm
ActivinA leads to an AIF-mediated DNA fragmentation, whereas TGF-β-mediated apoptosis occurs via executioner caspases
To characterise the apoptotic pathway in ActivinA-induced cell death, we attempted to identify the caspase cascade that might be involved in the ActivinA network. In TGF-β-induced apoptosis, initiator and executioner caspases are activated (Schuster et al. 2002a, 2003). To test whether caspases per se were involved in apoptotic processes after ActivinA treatment, we inhibited all caspases by an irreversible broad-spectrum caspase inhibitor (Z-VAD-FMK). After pre-treatment with the pan-caspase inhibitor, we added ActivinA and analysed apoptotic nuclei by flow cytometry. TGF-β1-treated samples served as a positive control. As shown in Fig. 3a, the apoptotic cell population was reduced after co-treatment with ActivinA and pan-caspase inhibitor revealing that ActivinA-induced apoptosis was caspase-dependent. To identify which specific caspases were involved in the ActivinA-induced pathway, we performed caspase activity assays for initiator caspase-2 and caspase-8, for mitochondrial caspase-9 and for executioner caspase-3 (Fig. 3b-e). After treatment with ActivinA or TGF-β1 for various time periods (8–20 h), cells were harvested and proteins were used for fluorometric activity assays. Initiator caspase-2 and caspase-8 were significantly activated following ActivinA or TGF-β1 treatment (Fig. 3b,c). Mitochondrial caspase-9 and executioner caspase-3 were only significantly involved in TGF-β1-induced samples (Fig. 3d,e). From these experiments, we conclude that the classical caspase cascade involving caspase-3 to induce DNA fragmentation is only activated in TGF-β-mediated apoptosis. In the ActivinA pathway, another DNA-fragmentation-inducing molecule must be involved.

Analysis of the caspase cascade in ActivinA (ActA)-induced apoptosis. a OLI-neu cells were pre-treated with 50 µM of the pan-caspase inhibitor Z-VAD-ZMK, and ActivinA (50 ng/ml) or TGF-β1 (5 ng/ml) was added for 20 h. Percentage of apoptotic cells (sub-G1 peak, DNA<n2) was determined by flow cytometry. Values represent means±SEM from three separate experiments; ***P<0.001 (Student’s t-test). b–e OLI-neu cells were treated with ActivinA (50 ng/ml) or TGF-β1 (5 ng/ml) at various time points and lysed; equal protein amounts were assayed for the activity of caspase-2 (b), caspase-8 (c), caspase-3 (d) and caspase-9 (e). Treated samples were normalised to control cells. Values are given as means±SEM from three separate experiments, each with two replicates; *P<0.05, **P<0.01, ***P<0.001
Several studies have shown that AIF is a mediator of caspase-independent cell death or, more exactly, of executioner-caspase-independent cell death (Krantic et al. 2007). To analyse whether AIF is involved in ActivinA-induced DNA fragmentation, we performed AIF release assays. Isolated mitochondrial and nuclear protein fractions were analysed by Western blot and showed a 3.6-fold AIF translocation into the nucleus only after ActivinA treatment (Fig. 4a). In TGF-β-treated samples, no AIF translocation into the nucleus could be observed. Immunocytochemical experiments validated the AIF translocation into the nucleus (Fig. 4b). In ActivinA-induced OLI-neu cells (16 h), we detected strong nuclear AIF staining if the nuclei were condensed or fragmented, as shown by DAPI staining. In control cells, the AIF staining appeared granular in the cytosol. In primary oligodendroglial cultures, nuclear AIF staining was also observed after ActivinA treatment (Fig. 4d).

AIF translocates into the nucleus only after ActivinA treatment. a Analysis of subcellular localisation of AIF in fractionated samples after treatment with ActivinA (50 ng/ml) and TGF-β1 (5 ng/ml; 16 or 24 h). Subcellular fractioning was verified by characteristic markers [CoxIV mitochondrial (mtc) fraction, H1 nuclear (nc) fraction] as loading control. Measurements of experimental bands were normalised to loading control. b, d ActivinA (ActA)-induced nuclear AIF staining. OLI-neu cells (b) or primary oligodendroglial cultures (d) were treated with 50 ng/ml ActivinA for 16 h. After fixation, immunofluorescence staining of AIF together with DAPI staining was performed. Bars 8 µm. c Analysis of AIF silencing. OLI-neu cells were transfected with green fluorescent protein (GFP)/short interfering RNA (siRNA) expression vectors for AIF (siAF) or scramble (Scramble) for 3 days. AIF expression was tested by Western blotting. D-glyceraldehyde-3-phosphate dehydrogenase (Gapdh) served as loading control. AIF expression was normalised to Gapdh expression and compared with the control. e ActivinA (ActA)-induced apoptosis is dependent on AIF. OLI-neu cells were transfected with GFP/siRNA expression vectors for AIF or scramble for 3 days. After down-regulation of AIF expression, ActivinA (50 ng/ml) or TGF-β1 (5 ng/ml) was added for 20 h. Apoptotic cells were identified by TUNEL staining. Positive-fragmented cells were counted in relation to GFP-stained cells and the percentage of apoptotic cells was calculated. Values are given as means±SEM (three separate experiments counting 3000 cells). **Statistically significant difference at P<0.01, calculated by Student’s t-test
To delimit ActivinA-induced apoptosis from TGF-β-induced apoptosis, we silenced AIF expression with a green fluorescent protein (GFP)-tagged siAIF expression vector system. After transfection of OLI-neu cells with the GFP/siAIF vector for 3 days, we analysed AIF silencing by Western blot (Fig. 4c). AIF expression was reduced to approximately one-third, in agreement with the AIF knock-down Harlequin mouse strain (Modjtahedi et al. 2006). To verify the AIF dependence in apoptosis, GFP/siAIF-transfected OLI-neu cells (3 days incubation) were treated either with ActivinA or with TGF-β1 for 20 h and DNA fragmentation was analysed by TUNEL staining (Fig. 4e). The AIF knock-down could only block ActivinA-induced DNA fragmentation, whereas TGF-β1-mediated DNA fragmentation also occurred, despite AIF knock-down. These results show that ActivinA induces apoptosis via mitochondrial AIF release, whereas TGF-β activates executioner caspases to induce DNA fragmentation.
ActivinA induces Bcl-xl sequestering by Puma to release AIF, whereas TGF-β reduces Bcl-xl protein amounts
We assumed that different regulatory mechanisms were acting on mitochondria to trigger DNA fragmentation by AIF or caspase-3. Therefore, we investigated the regulation of Bcl-xl. In TGF-β1-induced apoptosis, Bcl-xl protein amounts are decreased (Schuster et al. 2002a). In contrast, Bcl-xl protein amounts in ActivinA-treated cells were unaltered (Fig. 5a). Because of this important difference in Bcl-2 family regulation, we investigated other Bcl-2 family members known to be involved in mitochondrial events. Thus, we analysed BH3-only proteins and discovered an increase in Puma expression after ActivinA treatment (Fig. 5b). In primary oligodendroglial cultures, we also detected Puma activation after ActivinA treatment (Fig. 5c). Compared with ActivinA, TGF-β1 was not able to mediate an increase in Puma expression (Fig. 5b). The tumor suppressor p53 is the most important transcription factor of Puma (Nakano and Vousden 2001). We used the p53 inhibitor PFT to explore p53 function in ActivinA-induced apoptosis. OLI-neu cells were pre-treated with PFT and either ActivinA or TGF-β1 was added for 20 h. Cells were harvested and prepared for flow cytometry, apoptotic nuclei being counted (Fig. 5d). PFT significantly prevented ActivinA-induced, but not TGF-β-induced apoptosis. To analyse the effect of PFT on ActivinA-mediated AIF translocation into the nucleus, we isolated mitochondrial and nuclear protein phases from treated cells and performed Western blots with an AIF antibody. As shown in Fig. 5e, PFT inhibited the ActivinA-induced AIF translocation. These results clearly indicate that ActivinA-mediated AIF translocation depends on p53.

a Analysis of Bcl-xl expression. ActivinA (ActA)-treated (50 ng/ml) or TGF-β1-treated (5 ng/ml) cells were lysed and prepared for Western blotting with a Bcl-xl antibody. Gapdh served as loading control. b, c Puma activation in ActivinA (ActA)-induced cell death in oligodendrocytes. OLI-neu cells (b) or primary oligodendroglial cells (c) were treated with ActivinA (50 ng/ml) or TGF-β1 (5 ng/ml) for various time spans, lysed and prepared for Western blotting with an antibody against Puma. Gapdh served as loading control. d ActivinA-induced apoptosis is dependent on p53. OLI-neu cells were pre-treated with 20 µM p53 inhibitor Pifithrin-α (PFT) and ActivinA (50 ng/ml) or TGF-β1 (5 ng/ml) was added for 20 h. Percentage of apoptotic cells (sub-G1 peak) was determined by flow cytometry. Values represent means±SEM (three separate experiments); **P<0.01 (Student’s t-test). e AIF translocation is dependent on p53. After a pre-incubation in 20 µM PFT, ActivinA-treated cells (16 h) and non-treated cells were fractionated into their subcellular components to determine the subcellular fraction of AIF. AIF expression was normalised to loading controls of the subcellular fractions [CoxIV mitochondrial (mtc) fraction, H1 nuclear (nc) fraction]
The regulatory context, including Puma, nuclear and cytosolic p53 and other Bcl-2 family members, can vary and is dependent on cell type (Chipuk and Green 2006; Mihara et al. 2003). To ascertain the various possibilities, we decided to analyse whether Puma expression was dependent on nuclear p53 in our OLI-neu model. For this purpose, OLI-neu cells were pre-treated with PFT and ActivinA was added for 8 h. TGF-β1-treated samples served as a negative control. As shown in Fig. 6a, PFT reduced Puma activation in ActivinA-treated cells revealing a role for p53 as a nuclear factor to activate Puma expression. Puma is known to interact only with pro-survival proteins such as Bcl-xl and Bcl-2 (L. Chen et al. 2005) and, therefore, we investigated these two possible binding partners for Puma. Immunoprecipitations showed that Bcl-xl served as interaction partner for Puma following ActivinA treatment in our cell model (Fig. 6b). In contrast, Puma failed to interact with Bcl-xl in TGF-β-induced apoptosis (Fig. 6b). Further, we could not detect any binding of Bcl-2 to Puma, neither in TGF-β- nor in ActivinA-mediated apoptosis (data not shown). Because of the PFT-mediated reduction of ActivinA-induced DNA fragmentation, we analysed whether cytoplasmic p53 could also interact with Bcl-xl or Bax (Chipuk and Green 2006; Kuwana et al. 2005; Mihara et al. 2003). Immunoprecipitation experiments excluded Bcl-xl and Bax as p53 interaction partners in OLI-neu cells (data not shown).

Activated Puma sequesters Bcl-xl resulting in Bax translocation to mitochondria in ActivinA-induced apoptosis. a Nuclear p53 induces Puma expression. OLI-neu cells were pretreated with 20 µM PFT, and ActivinA (50 ng/ml) or TGF-β1 (5 ng/ml) was added for 8 h. By Western blotting, Puma expression was detected in lysed samples with a Puma antibody. Gapdh served as loading control. b Puma sequesters Bcl-xl following ActivinA treatment. For immunoprecipitation, equal amounts of ActivinA-treated and TGF-β1-treated proteins (8 h) were tested with Bcl-xl antibody. A rabbit IgG antibody served as a control for unspecific binding. As shown in an immunoblot (IB) for Puma protein, the Bcl-xl protein is sequestered by Puma represented by a 22-kDa band. c Subcellular fractionation experiments of ActivinA-induced OLI-neu cells for Bax translocation. Cells were pretreated with PFT and then treated with 50 ng/ml ActivinA for 8 h. Samples were fractionated into a cytosolic (cyto) phase and a mitochondrial (mtc) phase and analysed by Western blotting. Bax translocation to mitochondria was detected by a Bax antibody. Gapdh served as loading control for the cytosolic fraction and CoxIV as loading control for the mitochondrial fraction
The consequence of Bcl-xl sequestering by a BH3-only protein is the translocation of pro-apoptotic effectors, such as Bax, to the mitochondria (Gross et al. 1999; Orrenius 2004). As shown in subcellular fragmentations, Bax translocation to mitochondria was increased after ActivinA treatment (Fig. 6c). PFT inhibited Bax translocation showing that Bax activation lay downstream of p53 in the ActivinA signalling pathway (Fig. 6c).
These data show that apoptosis in oligodendrocytes occurs through TGF-β and ActivinA, independently of each other and via different mechanisms. TGF-β mediates apoptosis involving Bcl-xl protein down-regulation to imbalance anti-apoptotic and pro-apoptotic factors on the mitochondria; this leads to the activation of executioner caspases to trigger DNA fragmentation. ActivinA induces p53-dependent Puma expression to sequester Bcl-xl and to activate Bax. The Bax-mediated pore formation on the mitochondria leads to the release of AIF and subsequent DNA fragmentation.
Discussion
In the present study, we show that TGF-β1 and ActivinA induce apoptosis in oligodendrocytes. Whereas apoptotic-acting TGF-βs are well described in ontogenetic cell death and in the nervous system (Böttner et al. 2000; Duenker and Krieglstein 2000, 2002, 2003; Duenker et al. 2002a, b; Krieglstein et al. 2000; Sanchez-Capelo 2005; Schuster and Krieglstein 2002; Unsicker and Strelau 2000), apoptotic-acting Activins have so far only been described in non-neural and adult systems (W. Chen et al. 2000; Y.G. Chen et al. 2002, 2006). Thus, we present, for the first time, evidence that ActivinA also induces apoptosis in the nervous system, especially in oligodendroglial progenitors. Further analysis with the established oligodendroglial precursor cell line OLI-neu has excluded cooperative effects between ActivinA and TGF-β, which is known to induce apoptosis in this model system (Schuster et al. 2002a). The autonomy of these two cytokine-mediated pathways is reflected by their different effects on mitochondria resulting in separate DNA fragmentation mechanisms. Whereas TGF-β down-regulates Bcl-xl protein (Schuster et al. 2002a), Bcl-xl is sequestered, in ActivinA-induced apoptosis, by the pro-apoptotic BH3-only protein Puma that regulates apoptotic pathways by interacting with anti-apoptotic proteins to release Bax (L. Chen et al. 2005; Kuwana et al. 2005). Thereby, Puma expression is dependent on its transcription factor p53 (Nakano and Vousden 2001). In contrast to TGF-β, ActivinA-induced apoptosis and Bax translocation are prevented by the p53 inhibitor PFT.
The different DNA fragmentation processes in the ActivinA and TGF-β pathways confirm two existing apoptotic pathways in oligodendrocytes. Whereas caspase-3 is activated in TGF-β-induced apoptosis, AIF triggers DNA fragmentation in ActivinA-induced apoptosis. AIF was firstly described as a caspase-independent mediator of DNA fragmentation, but further studies showed an involvement of caspases such as caspase-2 (Arnould et al. 2003; Seth et al. 2005). We have demonstrated that ActivinA-induced apoptosis and AIF release are dependent on caspases based on results with the pan-caspase inhibitor Z-VAD-FMK. Caspase-dependent AIF release has also been shown to be the possible consequence of calpain inhibition by Z-VAD-FMK, because calpains are important proteases of AIF cleavage (Modjtahedi et al. 2006). We have excluded this possibility with calpain activity assays and specific calpain inhibitors, which have failed to detect calpain activation in ActivinA-induced apoptosis. Indeed, caspases play a role in ActivinA-induced DNA fragmentation and AIF release, but their involvement in the ActivinA network is not yet fully understood. Caspase-2 and caspase-8 are activated in both cytokine-mediated pathways, in contrast to caspase-3. How these same caspases lead to differences in signalling pathways has, until now, been unclear. Possibly, p53 involvement, which has been demonstrated only in the ActivinA pathway, distinguishes between the two signal branches. Hence, it is shown that caspase-2 activation mediates a functional connection to p53 to release AIF, as described in other cell models (Seth et al. 2005; Vakifahmetoglu et al. 2006).
Further, the way that TGF-β superfamily signalling is discriminated intracellularly, leading to the different events of p53-dependent Puma expression (ActivinA) or Bcl-xl protein down-regulation (TGF-β) is of interest. In OLI-neu cells, general Smad activation is stimulated by ActivinA and by TGF-β, but without further clarification of the diverse TGF-β superfamily regulation regarding combinatorial interactions, heterodimeric receptors, Smad complexes, receptor-interacting proteins and Smad-interacting proteins (Derynck and Zhang 2003). Different regulatory Smads or diverse phosphorylation sites in Smads might mediate various types of transcriptional regulation. However, further studies are required to complete our understanding of the role of Smads in these apoptotic pathways.
An important question is why the cell possesses two different apoptotic pathways that are inducible by different cytokines. In the present study, we have excluded the possibility of cooperating effects between TGF-β and ActivinA and have demonstrated that the two pathways are indeed different with respect to the involved signalling. Another explanation could be that different cellular stages occur in primary oligodendroglial cultures and in our cellular model OLI-neu. In thymocytes, Activin and TGF-β have been shown to act individually at different maturation stages (Rosendahl et al. 2003). However, we clearly demonstrate that OLI-neu cells are responsive to either TGF-β or ActivinA, at the same time showing double-responsive cells that might be in a specific maturation stage. Double-responsive cells suggest that a developmental switch occurs depending on the available cytokines during development. However, further studies are required to answer this question.
Thus, our results have revealed the presence of two different apoptotic pathways in the same cellular context induced by either ActivinA or TGF-β, both members of the TGF-β superfamily. Thereby, we have shown, for the first time, that ActivinA induces apoptosis in neural cells. In our glial model, ActivinA promotes apoptosis through the neutralisation of the Bcl-xl function, whereas TGF-β triggers apoptosis by the down-regulation of the Bcl-xl protein. These diverging regulation patterns of the same Bcl-2 family member lead to different DNA fragmentation mechanisms.
References
Arnould D, Gaume B, Karbowski M, Sharpe JC, Cecconi F, Youle RJ (2003) Mitochondrial release of AIF and EndoG requires caspase activation downstream of Bax/Bak-mediated permeabilization. EMBO J 22:4385–4399
Böttner M, Krieglstein K, Unsicker K (2000) The transforming growth factor-betas: structure, signaling, and roles in nervous system development and functions. J Neurochem 75:2227–2240
Chen L, Willis SN, Wei A, Smith BJ, Fletcher JI, Hinds MG, Colman PM, Day CL, Adams JM, Huang DCS (2005) Differential targeting of prosurvival Bcl-2 proteins by their BH3-only ligands allow complementary apoptotic function. Mol Cell 17:393–403
Chen W, Woodruff TK, Mayo KE (2000) Activin A-induced HepG2 liver cell apoptosis: involvement of activin receptors and smad proteins. Endocrinology 141:1263–1272
Chen YG, Lui HM, Lin SL, Lee JM, Ying SY (2002) Regulation of cell proliferation, apoptosis, and carcinogenesis by activin. Exp Biol Med (Maywood) 227:75–87
Chen YG, Wang Q, Lin SL, Chang CD, Chuang J, Ying SY (2006) Activin signaling and its role in regulation of cell proliferation, apoptosis, and carcinogenesis. Exp Biol Med (Maywood) 231:534–544
Chipuk JE, Green DR (2006) Dissecting p53-dependent apoptosis. Cell Death Differ 13:994–1002
Degterev A, Boyce M, Yuan J (2003) A decade of caspases. Oncogene 22:8543–8567
Derynck R, Zhang YE (2003) Smad-dependent and Smad-independent pathways in TGF-beta family signalling. Nature 425:577–584
Duenker N, Krieglstein K (2000) Targeted mutations of transforming growth factor-beta genes reveal important roles in mouse development and adult homeostasis. Eur J Biochem 267:6982–6988
Duenker N, Krieglstein K (2002) Tgfbeta2-/- Tgfbeta3-/- double knockout mice display severe midline fusion defects and early embryonic lethality. Anat Embryol (Berl) 206:73–83
Duenker N, Krieglstein K (2003) Reduced programmed cell death in the retina and defects in lens and cornea of Tgfbeta2(-/-) Tgfbeta3(-/-) double-deficient mice. Cell Tissue Res 313:1–10
Duenker N, Schmitt K, Krieglstein K (2002a) TGF-beta is required for programmed cell death in interdigital webs of the developing mouse limb. Mech Dev 113:111–120
Duenker N, Schmitt K, Schuster N, Krieglstein K (2002b) The role of transforming growth factor beta-2, beta-3 in mediating apoptosis in the murine intestinal mucosa. Gastroenterology 122:1364–1375
Franke AG, Gubbe C, Beier M, Duenker N (2006) Transforming growth factor-beta and bone morphogenetic proteins: cooperative players in chick and murine programmed retinal cell death. J Comp Neurol 495:263–278
Gross A, McDonnell JM, Korsmeyer SJ (1999) BCL-2 family members and the mitochondria in apoptosis. Genes Dev 13:1899–1911
Jonk LJ, Itoh S, Heldin CH, Dijke P ten, Kruijer W (1998) Identification and functional characterization of a Smad binding element (SBE) in the JunB promotor that acts as a TGF-beta, activin, and BMP-inducible enhancer. J Biol Chem 273:21145–21152
Krantic S, Mechawar N, Reix S, Quirion R (2007) Apoptosis-inducing factor: a matter of neuron life and death. Neurobiology 81:179–199
Krieglstein K, Richter S, Farkas L, Schuster N, Dunker N, Oppenheim RW, Unsicker K (2000) Reduction of endogenous transforming growth factors beta prevents ontogenetic neuron death. Nat Neurosci 3:1085–1090
Kuwana T, Bouchier-Hayes L, Chipuk JE, Bonzon C, Sullivan BA, Green DR, Newmeyer DD (2005) BH3 domains of BH3-only proteins differentially regulate Bax-mediated mitochondrial membrane permeabilization both directly and indirectly. Mol Cell 17:525–535
Lacmann A, Hess D, Gohla G, Roussa E, Krieglstein K (2007) Activity-dependent release of transforming growth factor-beta in a neural network in vitro. Neuroscience 150:647–657
Mihara M, Erster S, Zaika A, Petrenko O, Chittenden T, Pancoska P, Moll UM (2003) p53 has a direct apoptogenic role at the mitochondria. Mol Cell 11:577–590
Modjtahedi N, Giordanetto F, Madeo F, Kroemer G (2006) Apoptosis-inducing factor: vital and lethal. Trends Cell Biol 16:264–272
Nakano K, Vousden KH (2001) Puma, a novel proapoptotic gene, is induced by p53. Mol Cell 7:683–694
Orrenius S (2004) Mitochondrial regulation of apoptotic cell death. Toxicol Lett 149:19–23
Rosendahl A, Speletas M, Leandersson K, Ivars F, Sideras P (2003) Transforming growth factor-beta and Activin-Smad signaling pathways are activated at distinct maturation stages of the thymopoeisis. Internat Immunol 15:1401–1414
Sanchez-Capelo A (2005) Dual role for TGF-beta1 in apoptosis. Cytokine Growth Factor Rev 16:15–34
Schuster N, Krieglstein K (2002) Mechanisms of TGF-beta-mediated apoptosis. Cell Tissue Res 307:1–14
Schuster N, Bender H, Philippi A, Subramaniam S, Strelau J, Wang Z, Krieglstein K (2002a) TGF-beta induces cell death in the oligodendroglial cell line OLI-neu. Glia 40:95–108
Schuster N, Dunker N, Krieglstein K (2002b) Transforming growth factor-beta induced cell death in the developing chick retina is mediated via activation of c-jun N-terminal kinase and downregulation of the anti-apoptotic protein Bcl-X(L). Neurosci Lett 330:239–242
Schuster N, Bender H, Rossler OG, Philippi A, Dunker N, Thiel G, Krieglstein K (2003) Transforming growth factor-beta and tumor necrosis factor-alpha cooperate to induce apoptosis in the oligodendroglial cell line OLI-neu. J Neurosci Res 73:324–333
Seth R, Yang C, Kaushal V, Shah SV, Kaushal GP (2005) p53-dependent caspase-2 activation in mitochondrial release of apoptosis-inducing factor and its role in renal tubular epithelial cell injury. J Biol Chem 280:31230–31239
Unsicker K, Strelau J (2000) Functions of transforming growth factor-beta isoforms in the nervous system. Cues based on localization and experimental in vitro and in vivo evidence. Eur J Biochem 267:6972–6975
Vakifahmetoglu H, Olsson M, Orrenius S, Zhivotovsky B (2006) Functional connection between p53 and caspase-2 is essential for apoptosis induced by DNA damage. Oncogene 25:5683–5692
Wang KK (2000) Calpain and caspases: can you tell the difference? Trends Neurosci 23:20–26
Acknowledgements
We thank Dr. D. Doenecke, Department of Molecular Biology, University of Göttingen, Germany for providing the H1 antibody.
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Author information
Authors and Affiliations
Corresponding author
Additional information
This work was funded by the Deutsche Forschungsgemeinschaft (Kr1477/11–1).
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License (https://creativecommons.org/licenses/by-nc/2.0), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
Schulz, R., Vogel, T., Dressel, R. et al. TGF-β superfamily members, ActivinA and TGF-β1, induce apoptosis in oligodendrocytes by different pathways. Cell Tissue Res 334, 327–338 (2008). https://doi.org/10.1007/s00441-008-0714-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00441-008-0714-5