
Abstract.
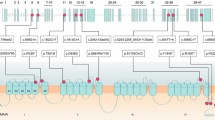
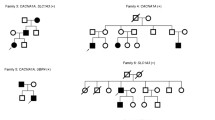
Mutations in the brain specific P/Q type Ca2+ channel alpha1 subunit gene, CACNA1A, have been identified in three clinically distinct disorders, viz. episodic ataxia type 2 (EA-2), familial hemiplegic migraine (FHM) and spinocerebellar ataxia 6 (SCA6). For individuals with EA-2, the mutations described thus far are presumed to result in a truncated protein product. Several different missense mutations have been identified in patients with FHM. At least two of these mutations have been identified on two different chromosome 19p13 haplotypes and thus represent recurrent mutations. In the present study, we have screened several individuals for mutations in all 47 exons in the CACNA1A gene by single-strand conformation analysis. We have characterised a novel missense mutation, G5260A, in exon 32 in a family segregating for EA-2. The consequence of this mutation is an amino acid substitution at a highly conserved position within the CACNA1A gene. This represents the first point mutation not resulting in a proposed truncated protein. Furthermore, this mutation has been detected in a family member with mild clinical signs including only migraine. Additionally, a second previously identified recurrent mutation, C2272T, in exon 16 has been discovered in a patient with FHM.
Similar content being viewed by others

Author information
Authors and Affiliations
Additional information
Electronic Publication
Rights and permissions
About this article
Cite this article
Friend, K., Crimmins, D., Phan, T. et al. Detection of a novel missense mutation and second recurrent mutation in the CACNA1A gene in individuals with EA-2 and FHM. Hum Genet 105, 261–265 (1999). https://doi.org/10.1007/s004399900101
Received:
Accepted:
Issue Date:
DOI: https://doi.org/10.1007/s004399900101