
Abstract
Identifying genetic risk factors for parasitic infections such as the leishmaniases could provide important leads for improved therapies and vaccines. Until recently most genetic studies of human leishmaniasis were underpowered and/or not replicated. Here, we focus on recent genome-wide association studies of visceral leishmaniasis (VL) and cutaneous leishmaniasis (CL). For VL, analysis across 2287 cases and 2692 controls from three cohorts identified a single major peak of genome-wide significance (Pcombined = 2.76 × 10–17) at HLA-DRB1–HLA-DQA1. HLA-DRB1*1501 and DRB1*1404/DRB1*1301 were the most significant protective versus risk alleles, respectively, with specific residues at amino acid positions 11 and 13 unique to protective alleles. Epitope-binding studies showed higher frequency of basic AAs in DRB1*1404-/*1301-specific epitopes compared to hydrophobic and polar AAs in DRB1*1501-specific epitopes at anchor residues P4 and P6 which interact with residues at DRB1 positions 11 and 13. For CL, genome-wide significance was not achieved in combined analysis of 2066 cases and 2046 controls across 2 cohorts. Rather, multiple top hits at P < 5 × 10–5 were observed, amongst which IFNG-AS1 was of specific interest as a non-coding anti-sense RNA known to influence responses to pathogens by increasing IFN-γ secretion. Association at LAMP3 encoding dendritic cell lysosomal associated membrane protein 3 was also interesting. LAMP3 increases markedly upon activation of dendritic cells, localizing to the MHC Class II compartment immediately prior to translocation of Class II to the cell surface. Together these GWAS results provide firm confirmation for the importance of antigen presentation and the regulation of IFNγ in determining the outcome of Leishmania infections.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
The leishmaniases are a group of related diseases caused by parasites of the genus Leishmania (order Kinetoplastidae, family Trypansomatidae). The major clinical presentations of leishmaniasis are visceral leishmaniasis (VL) caused by members of the L. donovani species complex originating in the Old World, and various forms of cutaneous leishmaniasis (CL) caused by a wide variety of Old and New World species. All species of Leishmania are transmitted by sand flies, Phlebotomus spp. in the Old World and Lutzomyia longipalpis in the New World. Humans, wild animals and domestic animals are known to act as reservoir hosts. VL is characterized by fever, weight loss, epistaxis, pancytopenia, anorexia, abdominal pain, cough, diarrhoea, nausea and vomiting, weakness, fatigue, splenomegaly, hepatomegaly and lymphadenopathy. As well as being a severe systemic infection, a proportion of patients treated for VL go on to present with cutaneous disease termed post-Kala-Azar dermal leishmaniaia (PKDL) (Zijlstra and el-Hassan 2001a; Zijlstra et al. 1994). The World Health Organization estimates that 200,000–400,000 new cases occur annually, 90% of which occur in India, Bangladesh, Sudan, South Sudan, Ethiopia and Brazil (Alvar et al. 2012). The most common form of CL is characterized by localized skin lesions, mainly ulcers, on exposed parts of the body. While normally self-limiting, the degree of pathology and the rate of healing varies, and the lesions leave life-long scars. Mucosal leishmaniasis (ML) and disseminated leishmaniasis (DL) are generally preceded by localized CL, the same species of Leishmania causing both types of disease. ML leads to partial or total destruction of mucous membranes of the nose, mouth and throat. Over 90% of ML is associated with L. braziliensis infection in Bolivia, Brazil, Peru and Paraguay in the New World, and with L. aethiopica in Ethiopia in the Old World.
Only a small percentage of individuals infected with Leishmania parasites go on to develop clinical disease. This variability in rates of clinical disease is determined by a complex interplay between parasite, host and vector genetic factors, as well as environmental and socioeconomic risk factors (Badaro et al. 1986; Chakravarty et al. 2019; Hasker et al. 2018; Jeronimo et al. 1994; Peacock et al. 2001; Reithinger et al. 2007; Shaw et al. 1995; Zijlstra et al. 1994). Immunologically we know a Leishmania-specific cellular immune response is a correlate of infection control in resistant individuals, measured traditionally as a positive delayed type hypersensitivity skin test responses in vivo (Follador et al. 2002; Gomes-Silva et al. 2007; Jeronimo et al. 1994; Llanos Cuentas et al. 1984) or lymphocyte proliferation responses ex vivo (Castes et al. 1989; Ho et al. 1982). However, the different forms of CL are generally associated with exaggerated cellular immune responses as measured by these parameters, and for both CL and VL we know that a fine balance exists between the pro-inflammatory cytokines tumour necrosis factor (TNF) and interferon-γ (IFNγ) and anti-inflammatory interleukin-10 (IL-10) (Faria et al. 2005a; Gautam et al. 2011; Gomes-Silva et al. 2007; Kumar et al. 2014; Nylen et al. 2007; Nylen and Sacks 2007; Oliveira et al. 2014; Singh et al. 2012; Zijlstra and el-Hassan 2001b). Thus, it is the balance between antigen-specific pro-inflammatory and anti-inflammatory cytokine responses that are important in determining the outcome of infection with Leishmania parasites. A major interest has been in determining the extent to which host genetic factors determine these responses.
One problem in identifying genetic risk factors for complex parasitic infections [reviewed (Abel et al. 2014; Blackwell 2010; Blackwell et al. 2009; Burgner et al. 2006; Loeb 2013)] is that most studies undertaken to date have been underpowered. This makes it difficult to evaluate chance statistical events from real genetic associations. In previous reviews (Blackwell 2010; Burgner et al. 2006; Castellucci et al. 2014; Mohamed et al. 2019) we provided summary tables and/or comprehensive supplementary web tables that listed all the studies undertaken in humans up to that time looking for susceptibility genes for protozoan parasite infections, including leishmaniasis. It is not our intention to repeat that exercise here. Rather, we focus on how recent genome-wide approaches using well-powered sample sizes have uncovered a divergence in the way they highlight specific genes and pathways involved in susceptibility to the two major forms of visceral versus cutaneous disease.
Visceral leishmaniasis
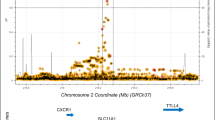
Complex segregation analysis of familial visceral leishmaniasis (VL) caused by L. chagasi in Brazil favored all genetic models over a sporadic model for disease susceptibility, with support for all single locus models over polygenic or multifactorial models of susceptibility (Peacock et al. 2001). While a single locus model might seem unlikely given the complex nature of a parasitic infectious disease, our recent GWAS of VL (Fakiola et al. 2013) (Fig. 1a), conducted as part of phase 2 of the Wellcome Trust Case–Control Consortium, highlighted the polymorphic HLA-DR-DQ region within the major histocompatibility complex of immune-related genes as the single major genetic determinant of VL (Pcombined = 2.76 × 1017; odds ratio = 1.41; 95% confidence interval = 1.30–1.52 over three cohorts). This replication across three independent cohorts crossed the geographical and epidemiological divides of continent and etiological parasite species, L. donovani in India and L. infantum/chagasi in Brazil. This robust demonstration that the most important genetic risk factor for VL lies at the heart of eliciting host CD4 + T-cell-mediated immunity against Leishmania parasites has major implications for vaccine design, allowing researchers to pursue single-minded post-GWAS functional analyses designed to understand the host–parasite interaction at a high resolution molecular level of antigen presenting cell function. Indeed, fine mapping (Fig. 1b) demonstrated that HLA-DRB1*1501 and DRB1*1404/DRB1*1301 were the most significant protective versus risk alleles, respectively, with specific residues at amino acid positions 11 and 13 unique to protective alleles (Singh et al. 2018). To determine the nature of epitopes binding to risk versus protective DRB1 alleles both in silico and in vitro experimental approaches were employed. NetMHCIIPan2.1 (Nielsen et al. 2010) was used to map epitopes and binding affinities across 49 Leishmania vaccine candidates, and across peptide epitopes captured from dendritic cells treated with crude Leishmania antigen and identified using mass spectrometry and alignment to a reference Leishmania genome (Singh et al. 2018). In these studies, greater peptide promiscuity was observed in sequence motifs for 9-mer core epitopes predicted to bind to risk (*1404/*1301) compared to protective (*1501) DRB1 alleles. In addition, there was a higher frequency of basic AAs in DRB1*1404-/*1301-specific epitopes compared to hydrophobic and polar AAs in DRB1*1501-specific epitopes (Fig. 1c) at anchor residues P4 and P6 which interact with residues at DRB1 position 11 and 13 (Fig. 1d). Peptides prepared based on sequences of epitopes captured from risk versus protective alleles were then used in whole blood cytokine assays to determine whether these differences in epitope-binding affected the important balance between pro- and anti-inflammatory cytokines. Cured VL patients made variable but robust IFN-γ, TNF and IL-10 responses to 20-mer peptides based on captured epitopes, with peptides based on DRB1*1501-captured epitopes resulting in a higher proportion (odds ratio 2.23; 95%CI 1.17–4.25; P = 0.017) of patients with IFN-γ:IL10 ratios > twofold compared to peptides based on DRB1*1301-captured epitopes (Fig. 1e). These genetic studies have, therefore, led to a greater understanding of the molecular interactions between host antigen presenting cell molecules and antigenic epitopes of the pathogen that drive the immune response towards either a protective or a disease-associated response.

Journey from GWAS to function in understanding the role of HLA-DRB1 as the single important genetic risk factor in VL. a Manhattan plot for meta-analysis across independent cohorts of VL caused by L. donovani in India and L. infantum/chagasi in Brazil (Fakiola et al. 2013). b Forest plot showing associations between VL and classical DRB1 alleles (Singh et al. 2018). c WebLogo plots for sequence motifs that characterize 9-mer cores for epitopes binding preferentially to the protective DRB1*1501 compared to the DRB1*1401 risk allele (Singh et al. 2018). Polar amino acids are green (G, S, T, Y, C) or purple (Q, N), basic amino acids are blue (K, R, H), acidic amino acids are red (D, E), and hydrophobic amino acids are black (A, V, L, I, P, W, F, M). The size of the letter indicates the frequency with which the amino acid is found in this position of core 9-mers, and the overall peak height on the y-axis indicates the degree of conservation for specific epitopes at this location. Greater promiscuity and a higher frequency of basic amino acids in DRB1*1401-specific epitopes, compared with hydrophobic and polar amino acids in DRB1*1501-specific epitopes, at anchor residues P4 and P6 which interact with residues at DRB1 positions 11 and 13. d Model of epitope binding to the DRA/DRB alpha/beta dimer demonstrating the specific interaction between amino acids at positions 4 and 6 in the 9-mer cores of Leishmania antigenic epitopes that bind to pockets 4 and 6 created by amino acids at positions 11 and 13 in the DRB1 molecule. e Shows the higher ratio of IFNG:IL10 in cytokine responses to peptides based on epitopes captured dendritic cells homozygous for the protective DRB1*1501 allele compared to epitopes captured from dendritic cells homozygous for the DRB1*1301 risk allele (Singh et al. 2018)
GWAS data for the Indian and Brazilian cohorts were also examined separately using a less stringent cutoff of P < 1 × 10–5 to look for potential population-specific associations. None of the regions associated at P < 1 × 10–5 in these separate analyses showed significant association in the combined analysis. Of the hits that were population-specific (one for India; 13 for Brazil), 8 were in intergenic regions and the remainder landed in genes with no obvious functional significance (Table S1).
Cutaneous leishmaniasis
Human family-based genetic epidemiology studies of CL disease caused by L. peruviana were consistent with a gene by environment multifactorial model, with a two-locus model of inheritance providing the best fit to the data (Shaw et al. 1995). This suggested that major genetic risk factors might also readily be found for CL disease. A GWAS was considered the best way to test this hypothesis. Genome-wide genetic analysis was undertaken (Castellucci et al. 2020) using DNAs from 2066 cases of CL caused by L. braziliensis and 2046 controls across two independent cohorts. The rarer forms of ML and DL disease caused by L. braziliensis were not used in this study due to lack of statistical power. Linear mixed models were employed in FastLMM (Lippert et al. 2011) to take account of relatedness, multiple testing and population heterogeneity. Combined analysis across these cohorts that were similarly powered to the VL study found no associations that achieved genome-wide significance, commonly accepted as P < 5 × 10–8 for studies employing variants with minor allele frequencies > 0.05 (Dudbridge and Gusnanto 2008; Fadista et al. 2016; Peer et al. 2008). Rather, multiple top hits at P < 5 × 10–5, consistent with complex disease inheritance, were observed that included some plausible novel candidate susceptibility genes. Amongst these were SERPINB10 (Pcombined = 2.67 × 10–6), CRLF3 (Pcombined = 5.12 × 10–6), STX7 (Pcombined = 6.06 × 10–6), KRT80 (Pcombined = 6.58 × 10–6), LAMP3 (Pcombined = 6.54 × 10–6) and IFNG-AS1 (Pcombined = 1.32 × 10–5). Effect sizes for these associations were small (odds ratios 1.07–1.21) (Castellucci et al. 2020) and lower than those observed for the top single nucleotides HLA variant associated with VL (odds ratio 1.49–1.50 Indian discovery; 1.38–1.67 Brazilian discovery) (Fakiola et al. 2013). To obtain functional support for these associations, published expression data (Novais et al. 2015) available on GEObase (GSE55664) was interrogated. LAMP3, STX7 and CRLF3 were all shown to be expressed more highly in CL biopsies compared to normal skin, whereas expression of KRT80 was strongly reduced (Castellucci et al. 2020). SERPINB10 was not expressed in skin, while probes were not available on the chip employed (Novais et al. 2015) for IFNG-AS1. Nevertheless, IFNG-AS1 was of specific interest as a non-coding anti-sense RNA known to influence responses to pathogens by increasing IFN-γ secretion in T cells and NK cells (Collier et al. 2012; Gomez et al. 2013; Petermann et al. 2019). Association at LAMP3 encoding dendritic cell lysosomal associated membrane protein 3 was also interesting. LAMP3 increases markedly upon activation of dendritic cells, localizing to the MHC Class II compartment immediately prior to translocation of Class II to the cell surface (de Saint-Vis et al. 1998). Together these GWAS results strengthen our knowledge of the importance of antigen presentation and the regulation of IFNγ in determining the outcome of Leishmania infections.
Do GWAS data provide support for previous candidate gene studies?
Data from the VL and CL GWAS provide the opportunity to evaluate earlier candidate gene studies which, as suggested above, were frequently compromised by lack of statistical power and/or were not replicated. Prior to the GWAS, family-based genome-wide linkage studies together with studies in mouse models had highlighted putative non-HLA candidate susceptibility genes for VL. These included CXCR2, CXCR1, SLC11A1, DLL1, CCL1, CCL16, and IL2RB (Bucheton et al. 2007; Fakiola et al. 2011; Jamieson et al. 2007; Mehrotra et al. 2011; Mohamed et al. 2004). None of these loci showed consistent association in the Indian or Brazilian cohorts used for the GWAS analysis, even when looking at precisely the same single nucleotide variant (Fakiola et al. 2013). Four of the original candidate gene studies were undertaken in The Sudan, which could indicate some geographic or ethnic heterogeneity in genetic risk factors for VL and/or differences in the parasite. On balance, however, it seems unlikely that variants in these genes remain strong candidates as genetic risk factors for VL.
For CL, candidate gene studies (Almeida et al. 2015; Cabrera et al. 1995; Castellucci et al. 2006, 2010, 2011, 2012, 2014; Ramasawmy et al. 2010; Salhi et al. 2008) undertaken in L. braziliensis endemic regions had demonstrated associations with polymorphisms at multiple genes associated with pro- and anti-inflammatory responses (TNFA, SLC11A1, CXCR1, IL6, IL10, CCL2/MCP1) and/or with wound healing (FLI1, CTGF, TGFBR2, SMAD2, SMAD3, SMAD7, COL1A1) in determining susceptibility to CL or ML disease. This included susceptibility genes identified from murine studies of leishmaniasis (SLC11A1, FLI1) (Castellucci et al. 2010, 2011). Although frequently underpinned by functional data (Castellucci et al. 2006; Ramasawmy et al. 2010; Salhi et al. 2008) and/or supported by prior immunological studies (Bacellar et al. 2002; Castes et al. 1993; D'Oliveira et al. 2002; Faria et al. 2005b; Lessa et al. 2001), these studies had also generally lacked statistical power. Given a priori evidence to look at these genes in the GWAS data, a value of Pcombined < 0.01 was used as a cutoff to identify possible associations. No variants were associated at Pcombined < 0.01 for TNFA, SLC11A1, CXCR1, IL6, IL10, CCL2/MCP1, FLI1, CTGF, COL1A1, or TGFBR2 in the CL GWAS data (Castellucci et al. 2020). This failure to replicate may, in some cases, be due to the fact that only the CL phenotype was examined in the GWAS, and not ML or DL disease. Associations were observed for variants at SMAD2 (P = 1.47 × 10–4), SMAD3 (P = 0.009) and SMAD7 (P = 0.005), wound healing genes that had previously been shown to be associated with CL (Castellucci et al. 2012). Some evidence for associations at related SMADs was also observed, specifically at SMAD1 (P = 7.49 × 10–4), SMAD4 (P = 1.90 × 10–4), SMAD6 (P = 0.001), and SMAD9 (P = 0.004). The major role for SMAD proteins is to transduce signals from receptors of the transforming growth factor beta superfamily. Although no association at Pcombined < 0.01 was observed at TGFBR2, association at the functionally related gene TGFBR3 (P = 3.98 × 10–4) was supported at multiple variants across the gene. Similarly, associations observed at collagen genes, COL24A1 (P = 2.06 × 10–4) and COL11A1 (P = 6.22 × 10–4), functionally related to wound healing gene COL1A1, were supported at multiple variants across each gene. Associations were also observed for variants at genes, IL6R (P = 7.14 × 10–4) and IL10R (P = 0.003), encoding receptors for cytokines IL-6 and IL-10 that have previously been shown to be genetically or functionally associated with CL. On balance, results of the CL GWAS provided more support for previously identified candidate genetic risk factors than had the VL GWAS data.
Conclusions
The results of these GWAS studies provide contrasting stories in relation to genetic risk factors for VL compared to CL disease. Whereas a single association at genome-wide significance was observed for VL (Fakiola et al. 2013), the results of the CL GWAS showed multiple potential genetic risk factors all with small effect sizes and none achieving genome-wide significance (Castellucci et al. 2020). The latter is consistent with polygenic inheritance for a multifactorial complex infectious disease. Much larger studies would be required to determine whether any of these putative susceptibility loci would ever achieve genome-wide significance. To better understand the pathogenesis of VL and CL disease, it is likely that a more cost-effective approach will be to use the current GWAS data in combination with functional expression studies that can be carried out on fewer individuals. This will certainly be true for the rarer forms of disease such as ML, DL or PKDL, where obtaining the sample size required to achieve genome-wide significance in genetic studies would be very difficult. This does not, however, preclude the possibility that future large-scale sequencing projects may become affordable and reveal yet uncovered genetic risk factors, especially in the context of these rarer forms of disease.
References
Abel L, Alcais A, Schurr E (2014) The dissection of complex susceptibility to infectious disease: bacterial, viral and parasitic infections. Curr Opin Immunol 30C:72–78. https://doi.org/10.1016/j.coi.2014.07.002
Almeida L, Oliveira J, Guimaraes LH, Carvalho EM, Blackwell JM, Castellucci L (2015) Wound healing genes and susceptibility to cutaneous leishmaniasis in Brazil: role of COL1A1. Infect Genet Evol 30:225–229. https://doi.org/10.1016/j.meegid.2014.12.034
Alvar J, Velez ID, Bern C, Herrero M, Desjeux P, Cano J, Jannin J, den Boer M (2012) Leishmaniasis worldwide and global estimates of its incidence. PLoS ONE 7:e35671. https://doi.org/10.1371/journal.pone.0035671
Bacellar O, Lessa H, Schriefer A, Machado P, Ribeiro de Jesus A, Dutra WO, Gollob KJ, Carvalho EM (2002) Up-regulation of Th1-type responses in mucosal leishmaniasis patients. Infect Immun 70:6734–6740
Badaro R, Jones TC, Lorenco R, Cerf BJ, Sampaio D, Carvalho EM, Rocha H, Teixeira R, Johnson WD Jr (1986) A prospective study of visceral leishmaniasis in an endemic area of Brazil. J Infect Dis 154:639–649
Blackwell JM (2010) Chapter 35: Immunogenetics of host response to parasites in humans. In: Kaufmann SHE, Rouse B, Sacks D (eds) Immunology of infectious diseases. ASM Publications, Washington, pp 483–490
Blackwell JM, Jamieson SE, Burgner D (2009) HLA and infectious diseases. Clin Microbiol Rev 22:370–385
Bucheton B, Argiro L, Chevillard C, Marquet S, Kheir MM, Mergani A, El-Safi SH, Dessein AJ (2007) Identification of a novel G245R polymorphism in the IL-2 receptor beta membrane proximal domain associated with human visceral leishmaniasis. Genes Immun 8:79–83
Burgner D, Jamieson SE, Blackwell JM (2006) Genetic susceptibility to infectious diseases: big is beautiful, but will bigger be even better? Lancet Infect Dis 6:653–663
Cabrera M, Shaw M-A, Sharples C, Williams H, Castes M, Convit J, Blackwell JM (1995) Polymorphism in TNF genes associated with mucocutaneous leishmaniasis. J Exp Med 182:1259–1264
Castellucci L, Menezes E, Oliveira J, Magalhaes A, Guimaraes LH, Lessa M, Ribeiro S, Reale J, Noronha EF, Wilson ME, Duggal P, Beaty TH, Jeronimo S, Jamieson SE, Bales A, Blackwell JM, de Jesus AR, Carvalho EM (2006) IL6 -174 G/C promoter polymorphism influences susceptibility to mucosal but not localized cutaneous leishmaniasis in Brazil. J Infect Dis 194:519–527
Castellucci L, Jamieson SE, Miller EN, Menezes E, Oliveira J, Magalhaes A, Guimaraes LH, Lessa M, de Jesus AR, Carvalho EM, Blackwell JM (2010) CXCR1 and SLC11A1 polymorphisms affect susceptibility to cutaneous leishmaniasis in Brazil: a case-control and family-based study. BMC Med Genet 11:10
Castellucci L, Jamieson SE, Miller EN, de Almeida LF, Oliveira J, Magalhaes A, Guimaraes LH, Lessa M, Lago E, de Jesus AR, Carvalho EM, Blackwell JM (2011) FLI1 polymorphism affects susceptibility to cutaneous leishmaniasis in Brazil. Genes Immun 12:589–594. https://doi.org/10.1038/gene.2011.37
Castellucci L, Jamieson SE, Almeida L, Oliveira J, Guimaraes LH, Lessa M, Fakiola M, Jesus AR, Nancy Miller E, Carvalho EM, Blackwell JM (2012) Wound healing genes and susceptibility to cutaneous leishmaniasis in Brazil. Infect Genet Evol 12:1102–1110. https://doi.org/10.1016/j.meegid.2012.03.017
Castellucci LC, Almeida LF, Jamieson SE, Fakiola M, Carvalho EM, Blackwell JM (2014) Host genetic factors in American cutaneous leishmaniasis: a critical appraisal of studies conducted in an endemic area of Brazil. Mem Inst Oswaldo Cruz 109:279–288
Castellucci LC, Almeida L, Cherllin S, Fakiola M, Carvalho E, Figueriedo AB, Cavalcanti CM, Alves NS, Gollob KJ, Dutra WO, Cordell HJ, Blackwell JM (2020) A genome-wide association study highlights a regulatory role for IFNG-AS1 contributing to cutaneous leishmaniasis in Brazil. J Infect Dis 10.1101/2020.01.13.903989
Castes M, Moros Z, Martinez A, Trujillo D, Castellanos PL, Rondon AJ, Convit J (1989) Cell-mediated immunity in localized cutaneous leishmaniasis patients before and after treatment with immunotherapy or chemotherapy. Parasite Immunol 11:211–222
Castes M, Trujillo D, Rojas ME, Fernandez CT, Araya L, Cabrera M, Blackwell J, Convit J (1993) Serum levels of tumor necrosis factor in patients with American cutaneous leishmaniasis. Biol Res 26:233–238
Chakravarty J, Hasker E, Kansal S, Singh OP, Malaviya P, Singh AK, Chourasia A, Singh T, Sudarshan M, Singh AP, Singh B, Singh RP, Ostyn B, Fakiola M, Picado A, Menten J, Blackwell JM, Wilson ME, Sacks D, Boelaert M, Sundar S (2019) Determinants for progression from asymptomatic infection to symptomatic visceral leishmaniasis: a cohort study. PLoS Negl Trop Dis 13:e0007216. https://doi.org/10.1371/journal.pntd.0007216
Collier SP, Collins PL, Williams CL, Boothby MR, Aune TM (2012) Cutting edge: influence of Tmevpg1, a long intergenic noncoding RNA, on the expression of Ifng by Th1 cells. J Immunol 189:2084–2088. https://doi.org/10.4049/jimmunol.1200774
de Saint-Vis B, Vincent J, Vandenabeele S, Vanbervliet B, Pin JJ, Ait-Yahia S, Patel S, Mattei MG, Banchereau J, Zurawski S, Davoust J, Caux C, Lebecque S (1998) A novel lysosome-associated membrane glycoprotein, DC-LAMP, induced upon DC maturation, is transiently expressed in MHC class II compartment. Immunity 9:325–336. https://doi.org/10.1016/s1074-7613(00)80615-9
D'Oliveira A Jr, Machado P, Bacellar O, Cheng LH, Almeida RP, Carvalho EM (2002) Evaluation of IFN-gamma and TNF-alpha as immunological markers of clinical outcome in cutaneous leishmaniasis. Rev Soc Bras Med Trop 35:7–10
Dudbridge F, Gusnanto A (2008) Estimation of significance thresholds for genomewide association scans. Genet Epidemiol 32:227–234. https://doi.org/10.1002/gepi.20297
Fadista J, Manning AK, Florez JC, Groop L (2016) The (in)famous GWAS P-value threshold revisited and updated for low-frequency variants. Eur J Hum Genet 24:1202–1205. https://doi.org/10.1038/ejhg.2015.269
Fakiola M, Miller EN, Fadl M, Mohamed HS, Jamieson SE, Francis RW, Cordell HJ, Peacock CS, Raju M, Khalil EA, Elhassan A, Musa AM, Silveira F, Shaw JJ, Sundar S, Jeronimo SM, Ibrahim ME, Blackwell JM (2011) Genetic and functional evidence implicating DLL1 as the gene that influences susceptibility to visceral leishmaniasis at chromosome 6q27. J Infect Dis 204:467–477. https://doi.org/10.1093/infdis/jir284
Fakiola M, Strange A, Cordell HJ, Miller EN, Pirinen M, Su Z, Mishra A, Mehrotra S, Monteiro GR, Band G, Bellenguez C, Dronov S, Edkins S, Freeman C, Giannoulatou E, Gray E, Hunt SE, Lacerda HG, Langford C, Pearson R, Pontes NN, Rai M, Singh SP, Smith L, Sousa O, Vukcevic D, Bramon E, Brown MA, Casas JP, Corvin A, Duncanson A, Jankowski J, Markus HS, Mathew CG, Palmer CN, Plomin R, Rautanen A, Sawcer SJ, Trembath RC, Viswanathan AC, Wood NW, Wilson ME, Deloukas P, Peltonen L, Christiansen F, Witt C, Jeronimo SM, Sundar S, Spencer CC, Blackwell JM, Donnelly P (2013) Common variants in the HLA-DRB1-HLA-DQA1 HLA class II region are associated with susceptibility to visceral leishmaniasis. Nat Genet 45:208–213. https://doi.org/10.1038/ng.2518
Faria DR, Gollob KJ, Barbosa J Jr, Schriefer A, Machado PR, Lessa H, Carvalho LP, Romano-Silva MA, de Jesus AR, Carvalho EM, Dutra WO (2005a) Decreased in situ expression of interleukin-10 receptor is correlated with the exacerbated inflammatory and cytotoxic responses observed in mucosal leishmaniasis. Infect Immun 73:7853–7859. https://doi.org/10.1128/IAI.73.12.7853-7859.2005
Faria DR, Gollob KJ, Barbosa JJ, Schriefer A, Machado PR, Lessa H, Carvalho LP, Romano-Silva MA, De Jesus AR, Carvalho EM, Dutra WO (2005b) Decreased in situ expression of interleukin-10 receptor is correlated with the exacerbated inflammatory and cytotoxic responses observed in mucosal leishmaniasis. Infect Immun 73:7853–7859
Follador I, Araujo C, Bacellar O, Araujo CB, Carvalho LP, Almeida RP, Carvalho EM (2002) Epidemiologic and immunologic findings for the subclinical form of Leishmania braziliensis infection. Clin Infect Dis 34:E54–E58
Gautam S, Kumar R, Maurya R, Nylen S, Ansari N, Rai M, Sundar S, Sacks D (2011) IL-10 neutralization promotes parasite clearance in splenic aspirate cells from patients with visceral leishmaniasis. J Infect Dis 204:1134–1137. https://doi.org/10.1093/infdis/jir461
Gomes-Silva A, de Cassia BR, Dos Santos NR, Amato VS, da Silva MM, Oliveira-Neto MP, Coutinho SG, Da-Cruz AM (2007) Can interferon-gamma and interleukin-10 balance be associated with severity of human Leishmania (Viannia) braziliensis infection? Clin Exp Immunol 149:440–444. https://doi.org/10.1111/j.1365-2249.2007.03436.x
Gomez JA, Wapinski OL, Yang YW, Bureau JF, Gopinath S, Monack DM, Chang HY, Brahic M, Kirkegaard K (2013) The NeST long ncRNA controls microbial susceptibility and epigenetic activation of the interferon-gamma locus. Cell 152:743–754. https://doi.org/10.1016/j.cell.2013.01.015
Hasker E, Malaviya P, Cloots K, Picado A, Singh OP, Kansal S, Boelaert M, Sundar S (2018) Visceral Leishmaniasis in the Muzaffapur Demographic Surveillance Site: A Spatiotemporal Analysis. Am J Trop Med Hyg 99:1555–1561. https://doi.org/10.4269/ajtmh.18-0448
Ho M, Siongok TK, Lyerly WH, Smith DH (1982) Prevalence and disease spectrum in a new focus of visceral leishmaniasis in Kenya. Trans R Soc Trop Med Hyg 76:741–746
Jamieson SE, Miller EN, Peacock CS, Fakiola M, Wilson ME, Bales-Holst A, Shaw MA, Silveira F, Shaw JJ, Jeronimo SM, Blackwell JM (2007) Genome-wide scan for visceral leishmaniasis susceptibility genes in Brazil. Genes Immun 8:84–90
Jeronimo SM, Oliveira RM, Mackay S, Costa RM, Sweet J, Nascimento ET, Luz KG, Fernandes MZ, Jernigan J, Pearson RD (1994) An urban outbreak of visceral leishmaniasis in Natal. Brazil Trans R Soc Trop Med Hyg 88:386–388
Kumar R, Singh N, Gautam S, Singh OP, Gidwani K, Rai M, Sacks D, Sundar S, Nylen S (2014) Leishmania specific CD4 T cells release IFNgamma that limits parasite replication in patients with visceral leishmaniasis. PLoS Negl Trop Dis 8:e3198. https://doi.org/10.1371/journal.pntd.0003198
Lessa HA, Machado P, Lima F, Cruz AA, Bacellar O, Guerreiro J, Carvalho EM (2001) Successful treatment of refractory mucosal leishmaniasis with pentoxifylline plus antimony. Am J Trop Med Hyg 65:87–89
Lippert C, Listgarten J, Liu Y, Kadie CM, Davidson RI, Heckerman D (2011) FaST linear mixed models for genome-wide association studies. Nat Methods 8:833–835. https://doi.org/10.1038/nmeth.1681
Llanos Cuentas EA, Cuba CC, Barreto AC, Marsden PD (1984) Clinical characteristics of human Leishmania braziliensis braziliensis infections. Trans R Soc Trop Med Hyg 78:845–846. https://doi.org/10.1016/0035-9203(84)90043-9
Loeb M (2013) Host genomics in infectious diseases. Infect Chemother 45:253–259. https://doi.org/10.3947/ic.2013.45.3.253
Mehrotra S, Fakiola M, Oommen J, Jamieson SE, Mishra A, Sudarshan M, Tiwary P, Rani DS, Thangaraj K, Rai M, Sundar S, Blackwell JM (2011) Genetic and functional evaluation of the role of CXCR1 and CXCR2 in susceptibility to visceral leishmaniasis in north-east India. BMC Med Genet 12:162. https://doi.org/10.1186/1471-2350-12-162
Mohamed HS, Ibrahim ME, Miller EN, White JK, Cordell HJ, Howson JMM, Peacock CS, Khalil EAG, Elhassan AM, Blackwell JM (2004) SLC11A1 (formerly NRAMP1) and susceptibility to visceral leishmaniasis in the Sudan. Eur J Hum Genet 12:66–74
Mohamed HS, Ibrahim ME, Blackwell JM (2019) 4. Genetic susceptibility to visceral leishmaniasis. In: Ibrahim ME, Rotimi CN (eds) The genetics of african populations in health and disease. Cambridge University Press, Cambridge, pp 71–85
Nielsen M, Justesen S, Lund O, Lundegaard C, Buus S (2010) NetMHCIIpan-2.0 improved pan-specific HLA-DR predictions using a novel concurrent alignment and weight optimization training procedure. Immunome Res 6:9. https://doi.org/10.1186/1745-7580-6-9
Novais FO, Carvalho LP, Passos S, Roos DS, Carvalho EM, Scott P, Beiting DP (2015) Genomic profiling of human Leishmania braziliensis lesions identifies transcriptional modules associated with cutaneous immunopathology. J Invest Dermatol 135:94–101. https://doi.org/10.1038/jid.2014.305
Nylen S, Sacks D (2007) Interleukin-10 and the pathogenesis of human visceral leishmaniasis. Trends Immunol 28:378–384. https://doi.org/10.1016/j.it.2007.07.004
Nylen S, Maurya R, Eidsmo L, Manandhar KD, Sundar S, Sacks D (2007) Splenic accumulation of IL-10 mRNA in T cells distinct from CD4+CD25+ (Foxp3) regulatory T cells in human visceral leishmaniasis. J Exp Med 204:805–817. https://doi.org/10.1084/jem.20061141
Oliveira WN, Ribeiro LE, Schrieffer A, Machado P, Carvalho EM, Bacellar O (2014) The role of inflammatory and anti-inflammatory cytokines in the pathogenesis of human tegumentary leishmaniasis. Cytokine 66:127–132. https://doi.org/10.1016/j.cyto.2013.12.016
Peacock CS, Collins A, Shaw MA, Silveira F, Costa J, Coste CH, Nascimento MD, Siddiqui R, Shaw JJ, Blackwell JM (2001) Genetic epidemiology of visceral leishmaniasis in northeastern Brazil. Genet Epidemiol 20:383–396
Pe'er I, Yelensky R, Altshuler D, Daly MJ (2008) Estimation of the multiple testing burden for genomewide association studies of nearly all common variants. Genet Epidemiol 32:381–385. https://doi.org/10.1002/gepi.20303
Petermann F, Pekowska A, Johnson CA, Jankovic D, Shih HY, Jiang K, Hudson WH, Brooks SR, Sun HW, Villarino AV, Yao C, Singleton K, Akondy RS, Kanno Y, Sher A, Casellas R, Ahmed R, O'Shea JJ (2019) The magnitude of IFN-gamma responses is fine-tuned by DNA architecture and the non-coding transcript of Ifng-as1. Mol Cell 75(1229–1242):e5. https://doi.org/10.1016/j.molcel.2019.06.025
Ramasawmy R, Menezes E, Magalhaes A, Oliveira J, Castellucci L, Almeida R, Rosa ME, Guimaraes LH, Lessa M, Noronha E, Wilson ME, Jamieson SE, Kalil J, Blackwell JM, Carvalho EM, de Jesus AR (2010) The -2518bp promoter polymorphism at CCL2/MCP1 influences susceptibility to mucosal but not localized cutaneous leishmaniasis in Brazil. Infect Genet Evol 10:607–613
Reithinger R, Dujardin JC, Louzir H, Pirmez C, Alexander B, Brooker S (2007) Cutaneous leishmaniasis. Lancet Infect Dis 7:581–596. https://doi.org/10.1016/S1473-3099(07)70209-8
Salhi A, Rodrigues V Jr, Santoro F, Dessein H, Romano A, Castellano LR, Sertorio M, Rafati S, Chevillard C, Prata A, Alcais A, Argiro L, Dessein A (2008) Immunological and genetic evidence for a crucial role of IL-10 in cutaneous lesions in humans infected with Leishmania braziliensis. J Immunol 180:6139–6148
Shaw MA, Davies CR, Llanos-Cuentas EA, Collins A (1995) Human genetic susceptibility and infection with Leishmania peruviana. Am J Hum Genet 57:1159–1168
Singh OP, Stober CB, Singh AK, Blackwell JM, Sundar S (2012) Cytokine responses to novel antigens in an Indian population living in an area endemic for visceral leishmaniasis. PLoS Negl Trop Dis 6:e1874. https://doi.org/10.1371/journal.pntd.0001874
Singh T, Fakiola M, Oommen J, Singh AP, Singh AK, Smith N, Chakravarty J, Sundar S, Blackwell JM (2018) Epitope-binding characteristics for risk versus protective DRB1 alleles for visceral leishmaniasis. J Immunol 200:2727–2737. https://doi.org/10.4049/jimmunol.1701764
Zijlstra EE, El-Hassan AM (2001a) Leishmaniasis in Sudan. Post kala-azar dermal leishmaniasis. Trans R Soc Trop Med Hyg 95:S59–76
Zijlstra EE, El-Hassan AM (2001b) Leishmaniasis in Sudan. Visceral leishmaniasis. Trans R Soc Trop Med Hyg 95:S27–S58
Zijlstra EE, El Hassan AM, Ismael A, Ghalib HW (1994) Endemic kala-azar in Eastern Sudan, a longitudinal study on the incidence of clinical and subclinical infection and post-kala-azar dermal leishmaniasis. Am J Trop Med Hyg 51:826–836
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflicts of Interest
The authors declare that they have no conflicts of interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Blackwell, J.M., Fakiola, M. & Castellucci, L.C. Human genetics of leishmania infections. Hum Genet 139, 813–819 (2020). https://doi.org/10.1007/s00439-020-02130-w
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00439-020-02130-w