
Abstract
We have investigated whether regions of the genome showing signs of positive selection in scans based on haplotype structure also show evidence of positive selection when sequence-based tests are applied, whether the target of selection can be localized more precisely, and whether such extra evidence can lead to increased biological insights. We used two tools: simulations under neutrality or selection, and experimental investigation of two regions identified by the HapMap2 project as putatively selected in human populations. Simulations suggested that neutral and selected regions should be readily distinguished and that it should be possible to localize the selected variant to within 40 kb at least half of the time. Re-sequencing of two ~300 kb regions (chr4:158Mb and chr10:22Mb) lacking known targets of selection in HapMap CHB individuals provided strong evidence for positive selection within each and suggested the micro-RNA gene hsa-miR-548c as the best candidate target in one region, and changes in regulation of the sperm protein gene SPAG6 in the other.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Positive or Darwinian selection is a process by which alleles that increase the fitness of the carrier increase in frequency in a population, and provides a basis for adaptive evolution. There has been considerable interest in documenting positive selection in the human lineage in order to extend our understanding of the emergence of modern humans as a species in Africa 50–200 thousand years ago (KYA) and their subsequent differentiation as they spread throughout the world and adapted to varied environments resulting from new geographical locations and climates or cultural innovations such as the development of pastoralism and farming (Jobling et al. 2004). Such investigations can be grouped into two types. In biological candidate studies, insights from phenotypic information have been used to investigate whether candidates of interest, usually single genes with a prior expectation of evolutionary significance, show genetic evidence of positive selection. These studies have successfully detected genetic signatures of positive selection at several candidates, such as high population differentiation at the Duffy blood group (DARC) O locus associated with resistance to vivax malaria in Africa but not elsewhere (Cavalli-Sforza et al. 1994), or unusual allele frequency spectra at the FOXP2 gene linked to speech/language development (Enard et al. 2002) and the inactive CASP12 gene associated with sepsis resistance (Xue et al. 2006), although other studies have also revealed the complexity of these selective events (Hamblin et al. 2002; Coop et al. 2008; Ptak et al. 2009). The alternative design has been a genome scan, in which the entire genome has been examined for evidence of positive selection. In some scans, such as when non-synonymous amino acid substitutions showing high levels of population differentiation were chosen (The International HapMap Consortium 2005), there has been a limited prior hypothesis about the target of selection. But genome scans can also be carried out in the absence of any such hypothesis. Such unbiased scans have the attractive feature that they can potentially lead to entirely unsuspected insights into evolutionary history, but in order to derive full benefit from them, the target of selection must be identified. In practice, most genome scans have been based on SNP genotyping, and methods for detecting potential selection have been primarily based on searching for unusual linkage disequilibrium (LD) or population differentiation patterns. Such scans have, in some senses, been highly successful. A review summarizing the combined results of nine such genome scans found that 5,110 distinct regions covering 14% of the genome and 4,243 (23%) RefSeq genes showed apparent evidence of positive selection (Akey 2009). However, although these findings are impressive for their yield of putatively selected regions, it was notable that there was limited overlap between the individual surveys and only 129 of the regions (2.5%) were identified in four or more studies. This poor concordance was described as “sobering” (Akey 2009) and pointed to the need for a better understanding of the false positive and false negative rates in such scans. Indeed, other analyses have suggested that the classic selective sweeps detected by these approaches are unlikely to have been frequent enough to dominate overall patterns of human genome diversity (Hernandez et al. 2011). A second feature of some of these scans, particularly those based on LD, is that the candidate regions identified could be very large. For example, the HapMap2 project listed 22 strong candidate regions with a combined length of ~16.7Mb and mean size of ~760 kb (Sabeti et al. 2007), making it difficult to identify the selected target and further investigate the biological implications of the selection.
We have set out to address three questions raised by genome scans that identify large candidate regions. First, do such candidates show evidence for selection if alternative criteria are used? Second, to what extent can the targets of selection be localized more precisely? And third, if more precise localization is possible, does this lead to increased insights into the possible biological basis of the selection? To achieve these aims, we reasoned that full re-sequence data would provide the most information. Indeed, only technical and cost limitations have previously hindered its use: re-sequencing complete genomes or even hundreds of kilobases (kb) to high accuracy in population samples has not been practical thus far (The 1000 Genomes Project Consortium 2010). We have thus explored experimentally the potential for enrichment of such regions followed by next-generation sequencing to generate suitable datasets. We chose for these trials two regions from the HapMap2 survey which were of intermediate size (~300 kb each) and where there was no obvious target for selection (Sabeti et al. 2007). We show using simulations that alternative tests for selection applied to sequence data from regions identified in such a way should readily distinguish between neutrality and likely selection, and will usually produce a more precise localization of the selected variant. We also show experimentally that suitable high-quality sequence data can be generated using next-gen technology, and finally that plausible biological candidates can then be proposed for these selective events.
Materials and methods
Simulations
Two-step simulations were performed to model both neutral and positively-selected scenarios, and are summarized in Fig. 1. In the first step, we carried out coalescent simulations using the cosi package to generate 1-Mb long haplotypes in a pair of ancestral populations 2,000 generations ago based on the best-fit demographic models for YRI and CHB populations (Schaffner et al. 2005). These haplotypes were then used as input for the second step, forward simulations using mpop (Pickrell et al. 2009). In some of these forward simulations in the CHB population, one allele with an initial frequency of 0.0006 (default initial frequency for new variants in the package), which will be under selection, was added in the middle of the simulated CHB haplotypes. Four different selection scenarios with selection coefficients of 0.001, 0.004, 0.007 and 0.01 were simulated; the selection start time was set at 2,000 generations ago. In total, 1,000 independent simulations were performed for each set of conditions. These used the genome-average recombination rate of 1 cM/Mb from the HapMap2, a mutation rate of 1.8 × 10−8/nucleotide/generation calculated from a comparison of human and chimpanzee sequences for the whole of chromosome 4, and a current effective population size of 100,000. The rest of the demographic parameters were as in Schaffner et al.’s best-fit demographic model (2005) from the package cosi. For computational efficiency, we re-scaled the parameters when performing the forward simulations: effective population size and time were reduced by a factor of 5, while mutation and recombination rates and selection coefficient were multiplied by 5 (Supplementary Materials). Fifty chromosomes were sampled from each simulation. We call this set of data the “simulated re-sequencing data”.

Simulation design. Dotted boxes represent simulated haplotype samples; the star indicates the presence of a positively selected SNP. Arrows show the performance of the analyses described in the oval boxes
The SNPs in the “simulated re-sequencing data” were subsampled to mimic the frequency spectrum of HapMap2 genotype data by matching the proportion of the SNPs of HapMap2 data in each frequency bin (bin size 0.1). We call this set of subsampled simulation data the “simulated genotype data”. XP-EHH scores (Sabeti et al. 2007) were calculated from the simulated genotype data and normalized using the mean and variance of the XP-EHH scores from the simulated genotype data in the neutral simulation in the CHB, using the YRI as the reference population. We only retained simulations when the XP-EHH score was above the 95th neutral percentile continuously for at least 100 kb surrounding the selected SNP, which mimics the experimentally investigated candidate regions from the survey based on the HapMap2 data.
We then returned to the corresponding simulated re-sequencing data for the retained simulations and calculated Tajima’s D (Tajima 1989), and Fay and Wu’s H (Fay and Wu 2000) as well as Nielsen et al.’s composite-likelihood ratio (CLR) (Nielsen et al. 2005). These were calculated in 10 kb non-overlapping windows, which provide a good balance between test power and genomic resolution, across the whole 300 kb region centered on the selected SNP (or equivalent location in neutral simulations) in each individual set. The significance levels for each of the neutrality tests were estimated based on the percentile of the test values in the null distribution from 1,000 neutral simulations with the same demographic model. The background frequency spectrum required by the CLR analysis was calculated in the 1,000 independent neutral simulations with the same recombination and mutation rate. Although Tajima’s D and Fay and Wu’s H are both frequency spectrum-based tests, and they were calculated using the same haplotype data, they test different aspect of the data. Tajima’s D measures the excess of rare variants while Fay and Wu’s H measures the excess of high-frequency derived alleles. So they can be regarded as independent tests, and indeed, the correlation coefficient between Tajima’s D and Fay and Wu’s H p values calculated from the neutral simulated data showed no correlation (r = 0.06). Therefore, a combined p value from Tajima’s D and Fay and Wu’s H for each 10 kb could be calculated using Fisher’s method (Fisher 1954), which is −2∑ln p (here: −2 × (ln p Tajiam’s D + ln p Fay and Wu’s H )); it has a χ2 distribution with degrees of freedom equal to two times the number of the tests (here, four), and we use this combined p value to present the results below.
Choice of regions for experimental investigation
Two regions were picked from the HapMap2 list of 22 regions showing strong evidence of selection (Sabeti et al. 2007) using the following criteria: no obvious candidate for the selected SNP or gene; selection at least in the CHB + JPT population; moderate size (0.2–1 Mb). The coordinates of the chosen regions were (March 2006, NCBI 36 assembly) chromosome 4: 158,702,285–159,016,211 (314 kb, called chr4:158Mb) and chromosome 10: 22,587,453–22,850,110 (263 kb, called chr10:22Mb). We also included a set of control regions, including CASP12 (13 kb) for which we had the Sanger capillary sequencing data from some of the same individuals (Xue et al. 2006) and 20 kb of unique sequence from the Y chromosome, where there should be no reads mapping in females and no heterozygote calls in males.
Target region enrichment
The target regions were amplified from 28 CHB and 2 YRI in a series of long-range PCRs of 5–11 kb, and data from >80% of the regions in more than 20 individuals were successfully generated (long-PCR details are described in the Supplementary Materials and the sequencing below). The PCR gaps were filled in using data from a hybridization enrichment experiment based on a Nimblegen custom array or solution pulldown (Supplementary Material), followed by sequencing in the same way. Data from the two YRI individuals were used for SNP data quality control, and not for population-genetic analyses.
Re-sequencing
Illumina paired-end libraries of ~200 bp fragments were constructed, and 37 bp from each end sequenced on an Illumina GAII (Quail et al. 2008), one sample per lane. After filtering out duplicate reads, the amount of mapped data ranged from 322 to 572 Mb, leading to a mean coverage per individual of ~500× to >1,000× for the parts which PCR amplified and ~35× to ~250× for pulldown regions.
The paired-end sequence reads were mapped back to the target reference sequences or the whole genome by SSAHA2 and candidate SNPs were called by SSAHASNP (Ning et al. 2001) for the PCR amplified regions, while MAQ (Li et al. 2008) and SAMtools (Li et al. 2009) were used for the data from the pulldown-enriched regions. By comparing the SNP calls based on Illumina data from CASP12 to the existing capillary sequence data and avoiding heterozygous Y chromosome SNP calls, we could set filtering criteria (Supplementary Material) and filter our complete set of genotype calls using these. The quality of the filtered SNP data was assessed by comparing the overlapping calls from our data with the genotypes from the same individuals in HapMap2. There were 43 discrepancies out of 2,981 comparisons for the chr4:158Mb and 5 of 857 for the chr10:22Mb, which suggested a low error rate for both regions (98.6 and 99.4% concordance, respectively) comparing with HapMap2 data. Simulations showed that such error rates would not affect the power of the sequence analyses (Supplementary Material).
We inferred the CHB individual’s haplotypes and occasional missing data using PHASE 2.1 on this single population (Stephens and Donnelly 2003). The ancestral allele of each site was assigned using chimpanzee–human alignment data from Ensembl. Then, the neutrality tests (Tajima’s D and Fay and Wu’s H) and Nielsen et al.’s CLR test were performed on non-overlapping 10–20 kb regions containing two or three PCR fragments chosen based on the size of each PCR fragment and the SNP densities.
Bioinformatic analysis
All miRBase (Release 13) mature miRNA sequences were scanned against the selected regions of the human genome using the MapMi algorithm (Guerra-Assunção and Enright 2010). This approach involves first scanning the regions for matches to mature miRNA sequences; regions with matches to known miRNAs (allowing one mismatch) were then excised and folded using RNAfold from the ViennaRNA package (Hofacker et al. 1994). These candidate regions were scored and filtered according to how well they fitted the stem–loop precursor structure common to miRNAs. We ran the pipeline in stand-alone mode, using non-repeat masked genomic sequence for increased sensitivity. The chr10:22Mb region had no significant hits for any known miRNA; however, the chr4:158Mb region had two hits to the miR-548 family of miRNAs, discussed below.
Results
Simulation of the power to detect and localize positive selection using genotype-based and sequence-based tests
We started by comparing the power of genotype-based and sequence-based analyses using simulations. The genotype-based tests modeled those in the HapMap2 study, and in particular, the selective events seen in the CHB + JPT by comparison with the YRI. To do this, we performed forward simulations under neutrality using the YRI and CHB demographic models, and with selection coefficients of 0.001, 0.004, 0.007 and 0.01 using the CHB demographic model, as described in “Materials and methods”. Of the 1,000 simulations in neutral and each selected CHB set, there were 16, 16, 233, 724 and 779, respectively, that met the XP-EHH filtering criteria. These were combined into two groups: 16 significant XP-EHH results under neutrality, and 1,752 under a range of selective conditions that would reflect the data that might be obtained from a population experiencing a variety of selective pressures.
We next applied the sequence-based tests to the 16 and 1,752 datasets. There were two simulations among the 16 retained neutral ones (12.5%) that showed at least one significant window for the combined p value (set at ≤0.01), and seven (44%) for Nielsen et al.’s CLR. These numbers represent the false positive rates for the two methods, and are significantly higher for the CLR (p = 0.048, Fisher exact test). In the retained selected simulations, 84% (1,469 out of 1,752) for combined p value and 85% (1,494 out of 1,752) for Nielsen et al.’s CLR showed at least one significant window. Thus there was good power to detect this form of selection using sequence-based tests.
To investigate the ability to localize the causal SNP using the sequence-based tests, we first examined the test statistics averaged over all retained simulations. The average values of both showed no pattern along the DNA in the neutral simulations, but a strong peak centered on the window containing the selected site in the selected set, with a gradual decrease on either side (Fig. 2a, b). This indicates that, on average, the neutrality or frequency spectrum-based tests can correctly identify the location of the causal SNP; in individual simulations, we observed sharp peaks (as in real data, see below), but with considerable variation in the location of the peak.

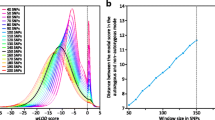
Simulation results. a Simulations were carried out under neutrality, and tests for selection [−ln combined p values for Tajima’s D and Fay and Wu’s H (top) or Nielsen et al.’s CLR (bottom)] were calculated in non-overlapping 10 kb windows across 300 kb. Values of the test were averaged over 16 independent neutral simulations that passed the XP-EHH filter. No departures from neutrality were seen. b 1,752 simulations with selection (selection coefficient 0.001, 0.004, 0.007, 0.01) that passed the XP-EHH filter and neutrality tests were averaged as in a. Departures from neutrality are seen most strongly in the window containing the selected SNP. c. The distribution of the top signal (lowest combined p value) or highest CLR in each simulation is shown across the 300-kb region. d. Probability that the known selected variant is found at each distance from the peak test value
We investigated this variation in location further by counting the occurrence of the most significant signals in each window in the different simulations. For the combined p value, the most significant window lay within the 40 kb region (i.e. ±20 kb) surrounding the selected allele in 46% of the simulations, compared with 68% for the CLR (Fig. 2c). These results show that Nielsen et al.’s CLR performs better for localizing the selection signal, as previously reported (Nielsen et al. 2005). Since the combined p value and Nielsen et al.’s CLR have similar power for detecting selection (84 and 85%), the combined p value has a lower false positive rate, but Nielsen et al.’s CLR test provides better localization, we investigated the benefits of further combining these signals. We tried using the combined p value to detect selection and then the CLR to localize the signal. This approach did systematically increase the accuracy of localization, although only by a small amount (Fig. 2d). We also considered the subset of simulations where the combined p value and Nielsen et al.’s CLR signals lie within the same 10 kb window. Although the proportion is low (11.3%, or 198 out of 1,752 simulations), these might represent a favorable situation with the best chance to localize the selection signal. Indeed, this subset of simulations has about 90% chance to localize the selection to a 40-kb region and 80% to 20 kb. These results provide an overall view of the power for localizing the signals in different scenarios and can guide the search for the biological basis of the selection.
Detection and localization of positive selection signals in experimental data
We re-sequenced two ~300 kb regions that had shown strong signals of positive selection in the HapMap2 study in 25 (chr4:158Mb) or 24 (chr10:22Mb) CHB individuals. The combined p value and Nielsen et al.’s CLR were calculated in chunks spanning either two or three PCR fragments, and are plotted in Fig. 3a and b. In both cases, a single window carries the most significant signal from each test: a combined p value of 0.00036 for chr4:158Mb, and 0.000015 for chr10:22Mb, and corresponding CLR values of 47 and 62. The two windows are located at 158,971,591–158,985,262 of chr4, and 22,755,918–22,776,116 of chr10, with sizes of ~13 and ~20 kb, respectively. Based on the simulations, this is a particularly favorable situation for localizing the selected variant, and we can have 80% confidence that the target of selection should lie in a 20-kb region centered on these windows.

Experimental results: localization of likely selection targets in the chr4 and chr10 regions. a. -log e of combined p values from Tajima’s D and Fay and Wu’s H (top) and Nielsen et al.’s CLR (bottom) calculated from re-sequencing data in windows corresponding to two or three PCR fragments (10–20 kb). The most significant statistics are shown in red, and fall into the same window at ~158.98 Mb (blue highlight). b Corresponding analysis of the chr10:22Mb region, where the most significant signals again fall into the same window, this time at ~22.78 Mb. c, d. Protein-coding genes from the Vega annotation, non-coding RNA and miRNA genes, and relevant ENCODE chromatin modifications in the two regions. e. Predicted miRNA in the chr4:158Mb target region. Two SNPs are present, including a G > A at the end of the miRNA carried on the major haplotype (49/50 chromosomes, selected in CHB) that may influence the strand forming the mature miRNA. f. H3K4me1 chromatin modifications indicating enhancer regions in GM12878 (second) and K562 (third) cells, SNPs with high derived allele frequencies (fourth), predicted regulatory potential (fifth) and 28 species conservation (bottom). Three high-frequency derived SNPs lie within candidate enhancers in one or other of the cell lines, but high-frequency derived SNPs do not lie within regions with high predicted regulatory potential or conservation
Biological targets of selection
The final stage of our analysis was to search for possible targets of selection. Such targets should most likely lie within the narrowed interval, and carry a biologically relevant difference between the selected and non-selected haplotypes. The 314-kb region on chromosome 4 consists entirely of intergenic sequence, and the nearest annotated protein-coding gene is located more than 50 kb outside this region. No histone modifications indicative of promoters, insulators or enhancers were apparent in publically available data (Fig. 3c). However, using the MapMi approach (Guerra-Assunção and Enright 2010), we found two predicted micro-RNAs (miRNAs) belonging to the mir-548 family (Fig. 3c). One of these lay far from the selection signal but the other, hsa-miR-548c, lay at 158.982 Mb, within the narrowed region (Fig. 3c). Strikingly, two SNPs are present within this predicted miRNA and both show high derived allele frequencies in the CHB. One of these SNPs lies within a loop in the predicted RNA and is not predicted to have functional consequences. However, the other is the first nucleotide of the miRNA precursor and could, therefore, determine which strand is processed to form the mature miRNA and consequently change the set of target genes (Fig. 3e).
The chromosome 10 region contains three annotated protein-coding genes, COMMD3, BMI1 and SPAG6, and no miRNA genes (Fig. 3d). SPAG6 transcripts (e.g. SPAG6-002, OTTHUMT00000047185: http://vega.sanger.ac.uk/Homo_sapiens/index.html) extend into the narrowed region (Fig. 3d). ChIP-seq experiments reveal extensive chromatin modification within the 263-kb region, including H3K4me1, H3K4me2, H3K4me3, H3K9ac, H3K27me3 and H3K27ac (http://genome.ucsc.edu/ ENCODE Histone Mods, Broad ChIP-seq; Fig. 3d), as would be expected for a region containing several protein-coding genes. The narrowed region contains two peaks of H3K4me1 which could indicate an enhancer (Heintzman et al. 2009). Thus, SPAG6 provides a good candidate on the basis of its location relative to the signal of selection. Although SPAG6 contains a relatively high-frequency derived non-synonymous SNP (rs7074847) in the YRI (Sabeti et al. 2007), there are no non-synonymous differences between the selected and non-selected CHB haplotypes, and no support for positive selection on SPAG6 from analysis of the dN/dS ratio within primates (results not shown), suggesting that selection is more likely to be acting on an aspect of transcription than on a change in the protein sequence.
Discussion
Although a recent study has suggested that classic sweeps acting on non-synonymous amino acid variants have been rare (but still present) in human history (Hernandez et al. 2011), these unusual events are of exceptional interest, and thus merit further investigation. True positive and false discovery rates of genome-wide scans for positive selection can be difficult to estimate (Teshima et al. 2006; Thornton and Jensen 2007) and so further studies, such as the current one, are needed.
The first question we addressed was whether or not candidate regions identified in genome scans for positive selection using LD-based tests on genotype data, such as that performed by the HapMap2 project, would show supporting evidence for selection when frequency spectrum-based neutrality tests were applied to re-sequencing data. Such tests are sometimes considered most suitable for detecting complete sweeps, in contrast to the partial sweeps detected by LD-based methods, but are also highly effective in detecting partial sweeps (Xue et al. 2009). However, the timescales of selective events most effectively detected by the two kinds of tests differ, with the LD-based tests more sensitive to recent events and the frequency spectrum-based tests more sensitive to older events (Sabeti et al. 2006). Despite this reservation, the answer to this question, from both our simulations and the two experimental examples investigated, was a clear “yes”. Significant departures from neutrality (combined p value from Tajima’s D and Fay and Wu’s H) were seen in 84% of the 1,752 simulations that passed the XP-EHH threshold, contrasted with just two of the 16 neutral simulations that by chance passed (not significantly different from 0 out of 16, Fisher exact test). A similar result was seen with Nielsen et al.’s CLR, although the false positive rate was higher. This correspondence is unsurprising, given the similar underlying basis for the two tests, but there was value in combining the two (see “Results”). In the two regions investigated experimentally, significant values were seen in both with all the tests applied.
The second question was the extent to which targets of selection could be localized more precisely when using re-sequencing data. From the simulations, we found that re-sequencing data do provide valuable additional information about the localization of selection targets. Higher SNP density and the presence of more rare variants make a higher resolution of signals possible. One of the disadvantages of LD-based test is that they detect large LD blocks, which are often several hundred kb in length. Although some frequency spectrum-based tests can also be used on genotype data, for example Nielsen et al.’s CLR, the window size often has to be relatively large because information from many SNPs needs to be combined to get enough power. We applied Nielsen et al.’s CLR using HapMap2 genotype data, and for chr4:158Mb, it detected a signal of selection localized to ~40 kb, while for the chr10:22Mb region, where the HapMap2 SNP density was high in the critical interval, the selected region was narrowed down to a similar length to the sequencing data (Fig. 4). A method for combining multiple signals derived from genotype data has been described (Grossman et al. 2010), which provides a median localization to a 55-kb interval. That method identified a chr4:158Mb interval spanning ~60 kb (158,862,019–158,921,890, with top SNP at 158,904,521), but failed to find any significant signal at chr10:22Mb (Grossman et al. 2010) (Fig. 4). We repeated the CMS analysis using the HapMap2 genotype data and similarly localized the chr4:158Mb signal to a similar ~58-kb interval, although with a different peak SNP (158,862,019–158,920,326, with top SNP at 158,920,326), and also found no signal in the chr10:22Mb interval (Fig. 4; supplementary figure). In contrast, re-sequencing followed by the application of the tests used here provided localization to a <20-kb interval in both cases.

Localization of the signal of selection within the chr4 and chr10 regions using different approaches. The two starting regions are shown at the top (Sabeti et al. 2007), localizations using sequence data (gray bars) or HapMap2 genotype data (white bars) by this study in the middle, and the localization by the CMS statistic (Grossman et al. 2010 or this work) at the bottom
Although in simulations we know the true target of selection, therefore, have a firm basis for evaluating any test, simulations never capture the complexities of real data. It is thus important also to evaluate and compare tests using real data. This requires a set of targets where we know that selection has acted, independently of the signal from the test. The timescale should also be appropriate. There are few such loci, but EDAR, TRPV6, MATP (SLC45A2) and CASP12 are strong candidates because they carry both a functional variant, and the alleles of this variant are associated with an experimentally measured phenotypic difference (Saleh et al. 2004; Graf et al. 2005; Fujimoto et al. 2008; Suzuki et al. 2008; Kimura et al. 2009). We, therefore, applied our approach to these examples using 1000 Genomes Pilot data (The 1000 Genomes Project Consortium 2010) and found that the combined tests could localize the signal of selection to the 10-kb containing the functional variant, or the adjacent window, in three cases (Supplementary Figure 3). The exception was EDAR, where the strongest signal lay ~50 kb upstream of the Val370Ala amino acid variant, within an intron; a pattern noted before (The 1000 Genomes Project Consortium 2010) and awaiting full explanation. It was particularly striking that a signal was identified at CASP12, where selection has previously been detected by targeted investigation using sequence-based tests, but not by genotype-based tests (Xue et al. 2006). These findings thus validate, albeit on a small scale, the ability of our approach to detect and localize targets of selection.
The final question was whether increased insights into the possible biological basis for the selection could be obtained. Due to our inability to predict the phenotypic consequences of most DNA variants, particularly when these lie outside protein-coding regions, it is often still difficult to identify the causal variant. Nevertheless, the narrowed region provides the best starting point for investigation. It is, in principle, possible that variants in a region could be acting on distant genes, but this in practice seems rare: a study of human eQTLs, for example, found that most lie either within or close to the genes they affect, with only 5% lying >20 kb away (Veyrieras et al. 2008). On this basis, we therefore focus on targets close to the narrowed regions in the following discussion.
For chr4:158Mb, the above considerations and the lack of any annotated protein-coding genes in the vicinity make a direct effect on a protein-coding gene unlikely. Predicted miRNA hsa-miR-548c, however, provides an intriguing candidate. Members of the hsa-miR-548 family are derived from the transposable element Made1, present in multiple [~30] copies in the human genome (Piriyapongsa and Jordan 2007). Made1 elements are found only in primates, and hsa-miR-548 sequences have been documented only in the human, chimpanzee and macaque genomes, where they appear to be evolving rapidly. Since miRNAs function as post-transcriptional regulators, a change in the sequence of a mature miRNA could influence the expression of a large number of genes, and a change in the strand present in the miRNA could have even greater regulatory effects. More than 3,500 genes have been listed as predicted hsa-miR-548 targets (Piriyapongsa and Jordan 2007). We can thus speculate that a variant hsa-miR-548c might have been selected because of altered target gene regulation, but the large number of hsa-miR-548 family members and potential targets makes it difficult to formulate or test more precise predictions. Nevertheless, a link to changes in gene regulation fits well with general thinking about the importance of regulatory mutations in human evolution (King and Wilson 1975) and the inference of recent positive selection acting on a miRNA-rich region on chromosome 14 devoid of annotated protein-coding genes (Quach et al. 2009).
For chr10:22Mb, similar considerations lead to the suggestion that SPAG6 is the most likely target of selection, and a change in the level, timing or location of its expression as the most likely mechanism. In particular, two H3K4me1 signals indicative of enhancers are located within the narrowed region, and three high-frequency derived SNPs (rs16922285 at 22,773,002, rs11012996 at 22,773,902 and rs11012997 at 22,774,094) specific to the selected haplotype overlapped with them (Fig. 3f). An altered enhancer activity thus provides the most plausible biological mechanism. SPAG6 is a component of sperm (Neilson et al. 1999), and mouse knockout models have been investigated: homozygous Spag6 −/− mice showed abnormalities of sperm structure and mobility, and were infertile (Sapiro et al. 2002). Heterozygous Spag6 +/− animals were fertile, but their sperm swam more slowly, suggesting that a reduced level of SPAG6 protein can have a detectable effect on the sperm phenotype. An effect on reproduction in humans would be consistent with both the inference of recent positive selection on another sperm protein gene, SPAG4, in the CHB among other populations (Voight et al. 2006), and the high frequency with which genes linked to reproduction are found more generally in surveys of positive selection (e.g. Bustamante et al. 2005; Voight et al. 2006).
There are two other protein-coding genes in the interval, both >100 kb from the strongest selection signal. Little is known about COMMD3 itself, but diverse functions have been ascribed to other COMMD family members, including copper metabolism and regulation of the activity of the transcription factor NF-κB and cell proliferation, perhaps through the ubiquitin pathway (Maine and Burstein 2007). BMI1, in contrast, has been studied extensively. It is a polycomb protein, involved in DNA repair, chromatin remodeling and stem cell renewal, and its inappropriate over-expression can lead to tumor formation (Shakhova et al. 2005; Schuringa and Vellenga 2010; Ginjala et al. 2011). Knockout mice are viable and homozygotes show hematopoietic, skeletal and neurological abnormalities, but phenotypic effects in the heterozygotes were not noted (van der Lugt et al. 1994). In humans, a cysteine to tyrosine substitution at position 18 leads to substantially lower levels of BMI1 protein, and is present in the general population, including in the YRI and CEU (but not CHB) HapMap samples (Zhang and Sarge 2009). Since increased expression of BMI1 leads to cancer, and a decreased expression phenotype is present in HapMap populations but has not been positively selected, both COMMD3 and BMI1 seem less strong candidates than SPAG6 for the target of chr10:22Mb selection.
From these examples, we can conclude that the approach used here, of re-sequencing large target regions, refining the target location and making inferences about the biology of the selection events, is fruitful. However, it could be improved in several ways. Re-sequencing technology is still imperfect and data quality needs to be improved. This study required a combination of two enrichment strategies, PCR and pulldown, to generate adequate coverage, and such intensive effort is impractical for large-scale studies. Most urgently, however, better statistics for localizing the target of selection using re-sequencing are needed, and improved methods for interpreting the biological consequences of DNA variants discovered are especially needed. But even with the present tools, specific topics to follow-up experimentally can be suggested, e.g., comparison of sperm mobility and other sperm characteristics between carriers of selected and non-selected haplotypes in the chr10:22Mb region. More generally, the availability of population-scale re-sequence data from both the increasing number of personal genome projects (Yngvadottir et al. 2009) and projects such as the 1000 Genomes Project (2010) will make the approach used here applicable across the genome.
References
Akey JM (2009) Constructing genomic maps of positive selection in humans: where do we go from here? Genome Res 19:711–722
Bustamante CD et al (2005) Natural selection on protein-coding genes in the human genome. Nature 437:1153–1157
Cavalli-Sforza LL, Menozzi P, Piazza A (1994) The history and geography of human genes. Princeton University Press, Princeton
Coop G, Bullaughey K, Luca F, Przeworski M (2008) The timing of selection at the human FOXP2 gene. Mol Biol Evol 25:1257–1259
Enard W et al (2002) Molecular evolution of FOXP2, a gene involved in speech and language. Nature 418:869–872
Fay JC, Wu CI (2000) Hitchhiking under positive Darwinian selection. Genetics 155:1405–1413
Fisher RA (1954) Statistical methods for research workers. Oliver and Boyd, Edinburgh
Fujimoto A et al (2008) A scan for genetic determinants of human hair morphology: EDAR is associated with Asian hair thickness. Hum Mol Genet 17:835–843
Ginjala V et al (2011) BMI1 is recruited to DNA breaks and contributes to DNA damage induced H2A ubiquitination and repair. Mol Cell Biol 31:1972–1982
Graf J, Hodgson R, van Daal A (2005) Single nucleotide polymorphisms in the MATP gene are associated with normal human pigmentation variation. Hum Mutat 25:278–284
Grossman SR et al (2010) A composite of multiple signals distinguishes causal variants in regions of positive selection. Science 327:883–886
Guerra-Assunção JA, Enright AJ (2010) MapMi: automated mapping of microRNA loci. BMC Bioinformatics 11:133
Hamblin MT, Thompson EE, Di Rienzo A (2002) Complex signatures of natural selection at the Duffy blood group locus. Am J Hum Genet 70:369–383
Heintzman ND et al (2009) Histone modifications at human enhancers reflect global cell-type-specific gene expression. Nature 459:108–112
Hernandez RD et al (2011) Classic selective sweeps were rare in recent human evolution. Science 331:920–924
Hofacker IL et al (1994) Fast folding and comparison of RNA secondary structures. Monatshefte für Chemie 125:167–188
Jobling MA, Hurles ME, Tyler-Smith C (2004) Human evolutionary genetics. Garland Science, New York and Abingdon
Kimura R et al (2009) A common variation in EDAR is a genetic determinant of shovel-shaped incisors. Am J Hum Genet 85:528–535
King MC, Wilson AC (1975) Evolution at two levels in humans and chimpanzees. Science 188:107–116
Li H, Ruan J, Durbin R (2008) Mapping short DNA sequencing reads and calling variants using mapping quality scores. Genome Res 18:1851–1858
Li H et al (2009) The sequence alignment/map format and SAMtools. Bioinformatics 25:2078–2079
Maine GN, Burstein E (2007) COMMD proteins: COMMing to the scene. Cell Mol Life Sci 64:1997–2005
Neilson LI et al (1999) cDNA cloning and characterization of a human sperm antigen (SPAG6) with homology to the product of the Chlamydomonas PF16 locus. Genomics 60:272–280
Nielsen R et al (2005) Genomic scans for selective sweeps using SNP data. Genome Res 15:1566–1575
Ning Z, Cox AJ, Mullikin JC (2001) SSAHA: a fast search method for large DNA databases. Genome Res 11:1725–1729
Pickrell JK et al (2009) Signals of recent positive selection in a worldwide sample of human populations. Genome Res 19:826–837
Piriyapongsa J, Jordan IK (2007) A family of human microRNA genes from miniature inverted-repeat transposable elements. PLoS One 2:e203
Ptak S et al (2009) Linkage disequilibrium extends across putative selected sites in FOXP2. Mol Biol Evol 26:2181–2184
Quach H et al (2009) Signatures of purifying and local positive selection in human miRNAs. Am J Hum Genet 84:316–327
Quail MA et al (2008) A large genome center’s improvements to the Illumina sequencing system. Nat Methods 5:1005–1010
Sabeti PC et al (2006) Positive natural selection in the human lineage. Science 312:1614–1620
Sabeti PC et al (2007) Genome-wide detection and characterization of positive selection in human populations. Nature 449:913–918
Saleh M et al (2004) Differential modulation of endotoxin responsiveness by human caspase-12 polymorphisms. Nature 429:75–79
Sapiro R et al (2002) Male infertility, impaired sperm motility, and hydrocephalus in mice deficient in sperm-associated antigen 6. Mol Cell Biol 22:6298–6305
Schaffner SF et al (2005) Calibrating a coalescent simulation of human genome sequence variation. Genome Res 15:1576–1583
Schuringa JJ, Vellenga E (2010) Role of the polycomb group gene BMI1 in normal and leukemic hematopoietic stem and progenitor cells. Curr Opin Hematol 17:294–299
Shakhova O, Leung C, Marino S (2005) Bmi1 in development and tumorigenesis of the central nervous system. J Mol Med 83:596–600
Stephens M, Donnelly P (2003) A comparison of bayesian methods for haplotype reconstruction from population genotype data. Am J Hum Genet 73:1162–1169
Suzuki Y et al (2008) Gain-of-function haplotype in the epithelial calcium channel TRPV6 is a risk factor for renal calcium stone formation. Hum Mol Genet 17:1613–1618
Tajima F (1989) Statistical method for testing the neutral mutation hypothesis by DNA polymorphism. Genetics 123:585–595
Teshima KM, Coop G, Przeworski M (2006) How reliable are empirical genomic scans for selective sweeps? Genome Res 16:702–712
The 1000 Genomes Project Consortium (2010) A map of human genome variation from population-scale sequencing. Nature 467:1061–1073
The International HapMap Consortium (2005) A haplotype map of the human genome. Nature 437:1299–1320
Thornton KR, Jensen JD (2007) Controlling the false-positive rate in multilocus genome scans for selection. Genetics 175:737–750
van der Lugt NM et al (1994) Posterior transformation, neurological abnormalities, and severe hematopoietic defects in mice with a targeted deletion of the bmi-1 proto-oncogene. Genes Dev 8:757–769
Veyrieras JB et al (2008) High-resolution mapping of expression-QTLs yields insight into human gene regulation. PLoS Genet 4:e1000214
Voight BF, Kudaravalli S, Wen X, Pritchard JK (2006) A map of recent positive selection in the human genome. PLoS Biol 4:e72
Xue Y et al (2006) Spread of an inactive form of caspase-12 in humans is due to recent positive selection. Am J Hum Genet 78:659–670
Xue Y et al (2009) Population differentiation as an indicator of recent positive selection in humans: an empirical evaluation. Genetics 183:1065–1077
Yngvadottir B, MacArthur DG, Jin H, Tyler-Smith C (2009) The promise and reality of personal genomics. Genome Biol 10:237
Zhang J, Sarge KD (2009) Identification of a polymorphism in the RING finger of human Bmi-1 that causes its degradation by the ubiquitin-proteasome system. FEBS Lett 583:960–964
Acknowledgments
We thank Nancy Hamlin for managing the Illumina sequencing project, all members of the Sanger Illumina sequencing and library construction teams, and Daniel G. MacArthur for helpful suggestions on XP-EHH calculation. This work was supported by The Wellcome Trust.
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License (https://creativecommons.org/licenses/by-nc/2.0), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
Hu, M., Ayub, Q., Guerra-Assunção, J.A. et al. Exploration of signals of positive selection derived from genotype-based human genome scans using re-sequencing data. Hum Genet 131, 665–674 (2012). https://doi.org/10.1007/s00439-011-1111-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00439-011-1111-9