
Abstract
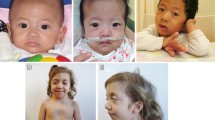
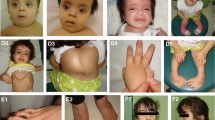
Defects leading to impaired intracellular trafficking have recently been shown to play an important role in the pathogenesis of genodermatoses, such as the Ehlers–Danlos and the cutis laxa syndromes. A new genodermatosis, termed macrocephaly, alopecia, cutis laxa and scoliosis (MACS) syndrome has been described, resulting from a homozygous 1-bp deletion in RIN2. RIN2 encodes the Ras and Rab interactor 2, involved in the regulation of Rab5-mediated early endocytosis. We performed a clinical, ultrastructural and molecular study in a consanguineous Algerian family with three siblings affected by a distinctive autosomal recessive genodermatosis, reported in 2005 by Verloes et al. The most striking clinical features include progressive facial coarsening, gingival hypertrophy, severe scoliosis, sparse hair and skin and joint hyperlaxity. Ultrastructural studies of the skin revealed important abnormalities in the collagen fibril morphology, and fibroblasts exhibited a dilated endoplasmic reticulum and an abnormal Golgi apparatus with rarefied and dilated cisternae. Molecular analysis of RIN2 revealed a novel homozygous 2-bp deletion in all affected individuals. The c.1914_1915delGC mutation introduces a frameshift and creates a premature termination codon, leading to nonsense-mediated mRNA decay. These findings confirm that RIN2 defects are associated with a distinct genodermatosis and underscore the involvement of RIN2 and its associated pathways in the pathogenesis of connective tissue disorders. The current family displays considerable phenotypic overlap with MACS syndrome. However, our family shows a dermatological and ultrastructural phenotype belonging to the Ehlers–Danlos rather than the cutis laxa spectrum. Therefore, the MACS acronym is not entirely appropriate for the current family.





Similar content being viewed by others

Abbreviations
- EDS:
-
Ehlers–Danlos syndrome
- GO:
-
Gerodermia osteodysplastica
- MACS:
-
Macrocephaly, alopecia, cutis laxa and scoliosis
- RIN2:
-
Ras and Rab interactor 2
- GEF:
-
Guanine nucleotide exchange factor
- Array-CGH:
-
Array comparative genome hybridization
- BW:
-
Birth weight
- BL:
-
Birth length
- OFC:
-
Occipitofrontal circumferences
- ER:
-
Endoplasmic reticulum
- NMD:
-
Nonsense-mediated RNA decay
- CHX:
-
Cycloheximide
- HCTD:
-
Heritable connective tissue disorders
- GAPO:
-
Growth retardation, alopecia, pseudo-anodontia and optic atrophy
- GTP:
-
Guanosine triphosphate
- GDP:
-
Guanosine diphosphate
- HGF:
-
Hepatocyte growth factor
References
Aoki Y, Niihori T, Narumi Y, Kure S, Matsubara Y (2008) The RAS/MAPK syndromes: novel roles of the RAS pathway in human genetic disorders. Hum Mutat 29:992–1006
Basel-Vanagaite L, Sarig O, Hershkovitz D, Fuchs-Telem D, Rapaport D, Gat A, Isman G, Shirazi I, Shohat M, Enk CD, Birk E, Kohlhase J, Matysiak-Scholze U, Maya I, Knopf C, Peffekoven A, Hennies HC, Bergman R, Horowitz M, Ishida-Yamamoto A, Sprecher E (2009) RIN2 deficiency results in macrocephaly, alopecia, cutis laxa, and scoliosis: MACS syndrome. Am J Hum Genet 85:254–263
Carter MS, Doskow J, Morris P, Li S, Nhim RP, Sandstedt S, Wilkinson MF (1995) A regulatory mechanism that detects premature nonsense codons in T-cell receptor transcripts in vivo is reversed by protein synthesis inhibitors in vitro. J Biol Chem 270:28995–29003
Fukada T, Civic N, Furuichi T, Shimoda S, Mishima K, Higashiyama H, Idaira Y, Asada Y, Kitamura H, Yamasaki S, Hojyo S, Nakayama M, Ohara O, Koseki H, Dos Santos HG, Bonafe L, Ha-Vinh R, Zankl A, Unger S, Kraenzlin ME, Beckmann JS, Saito I, Rivolta C, Ikegawa S, Superti-Furga A, Hirano T (2008) The zinc transporter SLC39A13/ZIP13 is required for connective tissue development; its involvement in BMP/TGF-beta signaling pathways. PLoS One 3:e3642
Giunta C, Elcioglu NH, Albrecht B, Eich G, Chambaz C, Janecke AR, Yeowell H, Weis M, Eyre DR, Kraenzlin M, Steinmann B (2008) Spondylocheiro dysplastic form of the Ehlers–Danlos syndrome—an autosomal-recessive entity caused by mutations in the zinc transporter gene SLC39A13. Am J Hum Genet 82:1290–1305
Goloni-Bertollo EM, Ruiz MT, Goloni CB, Muniz MP, Valerio NI, Pavarino-Bertelli EC (2008) GAPO syndrome: three new Brazilian cases, additional osseous manifestations, and review of the literature. Am J Med Genet A 146A:1523–1529
Grosshans BL, Ortiz D, Novick P (2006) Rabs and their effectors: achieving specificity in membrane traffic. Proc Natl Acad Sci USA 103:11821–11827
Hennies HC, Kornak U, Zhang H, Egerer J, Zhang X, Seifert W, Kuhnisch J, Budde B, Natebus M, Brancati F, Wilcox WR, Muller D, Kaplan PB, Rajab A, Zampino G, Fodale V, Dallapiccola B, Newman W, Metcalfe K, Clayton-Smith J, Tassabehji M, Steinmann B, Barr FA, Nurnberg P, Wieacker P, Mundlos S (2008) Gerodermia osteodysplastica is caused by mutations in SCYL1BP1, a Rab-6 interacting golgin. Nat Genet 40:1410–1412
Hucthagowder V, Morava E, Kornak U, Lefeber DJ, Fischer B, Dimopoulou A, Aldinger A, Choi J, Davis EC, Abuelo DN, Adamowicz M, Al-Aama J, Basel-Vanagaite L, Fernandez B, Greally MT, Gillessen-Kaesbach G, Kayserili H, Lemyre E, Tekin M, Turkmen S, Tuysuz B, Yuksel-Konuk B, Mundlos S, Van Maldergem L, Wevers RA, Urban Z (2009) Loss-of-function mutations in ATP6V0A2 impair vesicular trafficking, tropoelastin secretion and cell survival. Hum Mol Genet 18:2149–2165
Kimura T, Sakisaka T, Baba T, Yamada T, Takai Y (2006) Involvement of the Ras–Ras-activated Rab5 guanine nucleotide exchange factor RIN2–Rab5 pathway in the hepatocyte growth factor-induced endocytosis of E-cadherin. J Biol Chem 281:10598–10609
Kornak U, Reynders E, Dimopoulou A, van Reeuwijk J, Fischer B, Rajab A, Budde B, Nurnberg P, Foulquier F, Lefeber D, Urban Z, Gruenewald S, Annaert W, Brunner HG, van Bokhoven H, Wevers R, Morava E, Matthijs G, Van Maldergem L, Mundlos S (2008) Impaired glycosylation and cutis laxa caused by mutations in the vesicular H+-ATPase subunit ATP6V0A2. Nat Genet 40:32–34
Malfait F, De Coster P, Hausser I, van Essen AJ, Franck P, Colige A, Nusgens B, Martens L, De Paepe A (2004) The natural history, including orofacial features of three patients with Ehlers–Danlos syndrome, dermatosparaxis type (EDS type VIIC). Am J Med Genet A 131:18–28
Nielsen E, Severin F, Backer JM, Hyman AA, Zerial M (1999) Rab5 regulates motility of early endosomes on microtubules. Nat Cell Biol 1:376–382
Nuytinck L, Dalgleish R, Spotila L, Renard JP, Van Regemorter N, De Paepe A (1996) Substitution of glycine-661 by serine in the alpha1(I) and alpha2(I) chains of type I collagen results in different clinical and biochemical phenotypes. Hum Genet 97:324–329
Quaglino D Jr, Bergamini G, Boraldi F, Pasquali Ronchetti I (1996) Ultrastructural and morphometrical evaluations on normal human dermal connective tissue—the influence of age, sex and body region. Br J Dermatol 134:1013–1022
Saito K, Murai J, Kajiho H, Kontani K, Kurosu H, Katada T (2002) A novel binding protein composed of homophilic tetramer exhibits unique properties for the small GTPase Rab5. J Biol Chem 277:3412–3418
Tidyman WE, Rauen KA (2009) The RASopathies: developmental syndromes of Ras/MAPK pathway dysregulation. Curr Opin Genet Dev 19:230–236
Verloes A, Benmansour A, Mortier G, Pierard GE, Le Merrer M (2005) A new recessive connective tissue disorder with fleshy swelling of lips, lid and cheeks, macrocephaly, hyperextensible skin and severe scoliosis. In: 55th annual meeting of the American Society of Human Genetics, vol 674, Salt Lake City
Yeowell HN, Walker LC (2000) Mutations in the lysyl hydroxylase 1 gene that result in enzyme deficiency and the clinical phenotype of Ehlers–Danlos syndrome type VI. Mol Genet Metab 71:212–224
Acknowledgments
We would like to thank Wim Annaert for helpful comments. DS, FM and JH are fellows of the Fund for Scientific Research (FWO), Flanders (Belgium). This work was supported by Methusalem grant 08/01M01108 from the Ghent University to A. De Paepe and grant G.0171.05 from the Fund for Scientific Research, Flanders to A. De Paepe. The experiments comply with the current Belgian laws. This study has been approved by the Ethics Committee of the Ghent University Hospital, Ghent, Belgium.
Conflicts of interest statement
The authors declare that they have no conflict of interest.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article
Cite this article
Syx, D., Malfait, F., Van Laer, L. et al. The RIN2 syndrome: a new autosomal recessive connective tissue disorder caused by deficiency of Ras and Rab interactor 2 (RIN2). Hum Genet 128, 79–88 (2010). https://doi.org/10.1007/s00439-010-0829-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00439-010-0829-0