
Abstract
Completion of the sequencing of the Brassica rapa genome enabled us to undertake a genome-wide identification and functional study of the gene families related to the morphological diversity and agronomic traits of Brassica crops. In this study, we identified the auxin response factor (ARF) gene family, which is one of the key regulators of auxin-mediated plant growth and development in the B. rapa genome. A total of 31 ARF genes were identified in the genome. Phylogenetic and evolutionary analyses suggest that ARF genes fell into four major classes and were amplified in the B. rapa genome as a result of a recent whole genome triplication after speciation from Arabidopsis thaliana. Despite its recent hexaploid ancestry, B. rapa includes a relatively small number of ARF genes compared with the 23 members in A. thaliana, presumably due to a paralog reduction related to repetitive sequence insertion into promoter and non-coding transcribed region of the genes. Comparative genomic and mRNA sequencing analyses demonstrated that 27 of the 31 BrARF genes were transcriptionally active, and their expression was affected by either auxin treatment or floral development stage, although 4 genes were inactive, suggesting that the generation and pseudogenization of ARF members are likely to be an ongoing process. This study will provide a fundamental basis for the modification and evolution of the gene family after a polyploidy event, as well as a functional study of ARF genes in a polyploidy crop species.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Plant growth and development is regulated both by internal and external factors, including plant hormones and environmental stimuli. Auxin is one of the plant hormones that affect the genes involved in development processes at the cellular, tissue, and organ levels. Auxin treatment is known to increase the transcription of three early auxin response gene families, including SMALL AUXIN UP RNA (SAUR), Gretchen Hagen 3 (GH3), and indole-3-acetic acid-inducible gene (Aux/IAA), by modulating the interaction of transcription factors with auxin response elements (AuxREs) of the affected genes (reviewed in Hayashi 2012). A number of putative AuxREs have been defined within the upstream promoter regions of primary/early auxin responsive genes, such as one or more copies of a conserved motif, TGTCTC, or some variation of the motif (i.e., TGTCCC or TGTCAC), which confer auxin responsiveness (Ulmasov et al. 1997; Ulmasov et al. 1995). The transcription factors that bind specifically to these motifs are called auxin response factors (ARFs).
ARF is one of the plant protein families well known for their role in auxin-mediated responses. Most ARFs consist of three unique domains. The N-terminal region is a B3 DNA-binding domain and the C-terminal region has protein–protein interaction domains which are similar to those found in the C-terminal of Aux/IAAs and allow the dimerization of ARFs or ARF and Aux/IAA proteins. The variable middle region contains an activation or repression domain (reviewed in Guilfoyle and Hagen 2007). It has been reported that the ARF proteins are encoded by a larger gene family in Arabidopsis thaliana (Hagen and Guilfoyle 2002), maize (Xing et al. 2011), rice (Wang et al. 2007), poplar (Kalluri et al. 2007), and tomato (Kumar et al. 2011). A genome-wide comparison of the ARF gene family suggested that the whole genome and chromosomal segment duplication events are primarily responsible for the expansion of ARF genes in the plant genomes. In addition, the tandem duplication is an additional contributor that occurred in A. thaliana (Finet et al. 2010; Wang et al. 2007; Xing et al. 2011). Expression analysis demonstrated that these genes are, in general, transcribed in a wide variety of tissues and organs, with the exception of the ARF gene cluster on A. thaliana chromosome 1, which appears to be restricted to embryogenesis and seed development (Okushima et al. 2005).
Genetic studies have identified the well-established role of ARF genes in plant growth and development in A. thaliana. Mutations to ARF genes resulted in changes in the vascular strands and embryo axis formation (AtARF5) (Hardtke and Berleth 1998), suppression of hookless phenotype and hypocotyls bending (AtARF1 and AtARF2) (Ellis et al. 2005; Li et al. 2004), increased size and weight of seeds (AtARF2) (Schruff et al. 2005), abnormal gynoecium patterning (AtARF3) (Nishimura et al. 2005), abnormal development of floral organs and leaves (AtARF3 and AtARF4) (Finet et al. 2010), impaired hypocotyl response to blue light, growth, and auxin sensitivity (AtARF7) (Fukai et al. 2006; Sorin et al. 2005), and changed auxin homeostasis (AtARF8) (Goetz et al. 2006). Moreover, a double mutation showed a more severe phenotype than a single mutation, suggesting the unique and overlapping function of the ARF gene family members (AtARF7 and AtARF19) (Okushima et al. 2005). In addition, there is a growing body of information on the posttranscriptional regulation of ARF transcript abundance by micro-RNAs (miRNA) and trans-acting-small interfering RNAs (ta-siRNA). While AtARF6 and 8 are targets of miR167, and AtARF10, 16, and 17 are targeted by miR160, AtARF2, 3, and 4 are targets of TAS3 ta-siRNAs in A. thaliana (Guilfoyle and Hagen 2007). Mutation of the epigenetic regulators caused alterations in seed germination or leaf development, juvenile to adult phase changes, or heteroblasty (Fahlgren et al. 2006; Liu et al. 2007; Vaucheret et al. 2004).
Brassica rapa is of importance for agriculture and human nutrition. It is a member of the Brassicaceae family, like A. thaliana, and includes vegetables as well as oil crops. There are three well-defined groups of the cultivated species based on their morphological characteristics. The leafy type includes “Kimchi” cabbage (Korean heading form), Chinese cabbage, Pak choi, and celery mustard. The turnip type is comprised of turnip, rapini, and turnip broccoli. These two types are important as vegetable sources worldwide and are also useful as fodder. The oil type species are yellow sarson and toria, which are important oil crops in India and Canada. The diverse morphological variants of the species appear to be linked to the genomic changes associated with polyploidy (Lukens et al. 2004). Selection for different characteristics or hybridization between intraspecific types after domestication since 2,000 BC (Plant Biosafety Office 1999) might serve as an additional source of the morphological diversity of agronomically important cultivars. Morphological changes to the plant during genome evolution or domestication provides clues as to the evolutionary genetics of the genes related to structural diversification and the origin of crop species. One approach to understanding the fundamental basis of morphological diversity is the genomic evaluation of genes that are potentially involved in plant morphology, such as auxin response factors. Recent completion of sequencing of the euchromatic gene space of B. rapa allows for the genome-level identification of specific gene families in the genome.
A draft genome sequence of B. rapa, covering approximately 284 Mb of the genome, was assembled by Illumina GA short-read sequences and Sanger BAC-end sequences (The Brassica rapa Genome Sequencing Project Consortium 2011). This assembly was estimated to cover more than 98 % of the gene space. After the annotation and analysis, 41,174 protein-coding genes were identified in the genome, which are roughly 1.5 times as many genes as are found in A. thaliana (27,411, TAIR10), a difference that can be attributed to the whole-genome triplication (WGT, mesopolyploidy) thought to have occurred approximately 13 million years ago (Mya), followed by substantial gene loss (fractionation). This is consistent with our previous reports regarding BAC and A3 chromosome sequencing (Mun et al. 2009a, 2010). In particular, several hormone-related gene families, including auxin-related genes, were expanded or over-retained in the B. rapa genome compared with other gene families, which may contribute to the development of morphological variants of the species. The mesohexaploidy of B. rapa offers an opportunity to study the retention, subfunction, and neofunction of genes in the triplicated genome, as well as variation of the structure and function of the gene families. However, analyses of divergence as well as variation in the retained gene family have not yet been studied.
The main objective of this study is to identify ARF gene candidates of B. rapa (BrARFs) in the draft genome sequences. To the best of our knowledge, we determined the sequence characteristics of the ARF genes and examined their genomic distribution, along with phylogenetic and evolutionary analysis of the gene family to gain insight into the origin, divergence, and evolution of ARF genes in B. rapa. In addition, differentiation of expression between the paralogs in response to auxin treatment in seedlings, as well as during anther and pistil development, was investigated using next-generation mRNA sequencing technology (RNA-Seq). Understanding of the genomic organization of ARF genes in B. rapa will allow genome-level insights into the divergence, variation, and evolution of the auxin-related gene family, and will inform genetic and breeding strategies to engineer morphological plasticity in Brassica crops. In addition, this study will be beneficial to the understanding of the influence of polyploidy in the evolution of a gene family.
Materials and methods
Identification of ARF gene family in the B. rapa genome
We used 41,174 gene models of B. rapa ssp. pekinensis cv. Chiifu to search for ARF gene candidates in the genome. All publicly known A. thaliana ARF genes (AtARF1 to AtARF23) were used in the initial protein queries and candidate genes were identified based on a BLASTP search at a cut-off value of <E −20 (Altschul et al. 1997). To ensure that there were no additional related genes missing from the gene models, a TBLASTN search was also used against the 284 Mb assembled genome sequences at a cut-off value of <E −10. A conserved domain and motif search was performed using the Conserved Domain Database (CDD) of the National Center for Biotechnology Information (NCBI) to confirm each candidate protein sequence as an auxin response factor protein (Finn et al. 2008). To identify expressed sequence tag (EST) matches of the candidate ARF genes of B. rapa, 147,217 EST sequences of B. rapa ssp. pekinensis cv. Chiifu that we generated were used (Yu et al. 2011). All ESTs were compared with the coding sequences of candidate ARF genes. We considered ESTs to have a genome match if more than 90 % of the sequences matched with at least 95 % identity in a BLASTN search. If one EST matched with multiple candidate genes, only the gene with the best alignment score was considered.
A promoter search was performed to identify auxin response elements within the approximately 1 kb region upstream from the 5′-untranslated region (5′-UTR) of the BrARF genes, based on search of the PLACE database (www.dna.affrc.go.jp/PLACE). To determine whether specific repetitive elements drive the sequence divergence of BrARF, the tandem repeat (TR), inverted repeat (IR), transposable element (TE), low complexity repeat (LCR), and simple sequence repeats (SSRs) were also investigated in the region 1 kb upstream of the 5′-UTR to the 3′-UTR of the genes. Since the draft B. rapa genome did not provide information on the 5′- and 3′-UTR regions of the gene models, we used in-house trained FGENESH (www.softberry.com) to predict the 5′- and 3′-UTR regions of the BrARF genes. For those genes that have an intergenic sequence shorter than 1 kb from the end of the former gene, only the region between the 3′-UTR of the former gene to the 5′-UTR of the BrARF genes was considered as a putative promoter region. TRs and IRs were searched using Tandem Repeat Finder version 4.04 (Benson 1999) and Inverted Repeats Finder version 3.05 (Warburton et al. 2004), using default parameter values, respectively. TEs and LCRs were detected by RepeatMasker (http://www.repeatmasker.org) using B. rapa repetitive sequences. For identification of SSRs longer than 20 bp with mono- to deca-nucleotide motifs, the SSRIT (Temnykh et al. 2001) was used.
Phylogenetic analysis of the sequences
For phylogenetic reconstruction of the ARF gene family, we downloaded the sequenced plant genomes, as of November 2011, from the NCBI Entrez Genome database (http://www.ncbi.nlm.nih.gov/genome/PLANTS/PlantList.html). We identified putative ARF genes from the sequenced genomes, which included A. thaliana (23), papaya (13), cucumber (16), strawberry (17), soybean (54), poplar (36), castor bean (18), cacao (19), grape (19), Brachypodium distachyon (24), rice (28), sorghum (25), and maize (37), based on a BLASTP search as described earlier. Amino acid sequences of the ARF genes were aligned using ClustalW (Thompson et al. 1994), using the default options, and the alignment was manually corrected using the alignment editor Jalview (Waterhouse et al. 2009). Aligned sequences were trimmed at both ends to eliminate regions of poor alignment. Phylogenetic trees were constructed using the neighbor-joining (NJ) and the maximum likelihood (ML) methods in MEGA5 (Tamura et al. 2011). The stability of tree nodes was tested by bootstrap analysis with 1,000 replicates.
Genome block comparison and synonymous substitution rate analysis of homologous sequences between B. rapa and A. thaliana
Syntenic regions between the B. rapa and A. thaliana genomes were identified by a proteome comparison based on BLASTP analysis (Altschul et al. 1997). The entire proteomes of the two genomes were compared, and only the top reciprocal BLASTP matches per chromosome pair were selected (minimum of 50 % alignment coverage at a cutoff of <E −20). Chromosome scale synteny blocks were inferred by visual inspection of dot-plots using DiagHunter with parameters as described in previous reports (Cannon et al. 2003; Mun et al. 2009a). Assignment of 24 building blocks of ancestral karyotype (AK) to both genomes was carried out according to previous comparative genome mapping studies (Lysak et al. 2006; Schranz et al. 2006), and homologous gene pairs between the genomes were illustrated by Circos (Krzywinski et al. 2009).
The timings of a duplication event or divergence of homologous genes and selective pressure on duplicated genes were estimated by calculating the synonymous (K s) and non-synonymous substitution (K a) per site between homologous genes. To calculate the K s and K a values, the protein sequences of genes were aligned and the resulting alignment was used as a reference to align the nucleotide sequences. After the removal of gaps, the K s and K a values were determined using the maximum likelihood method implemented in the CODEML (Goldman and Yang 1994) module of the PAML (Yang 2007) package, as previously described (Mun et al. 2009b). The K s age distribution was represented at an interval of 0.1 and the calculation of divergence time was based on the neutral substitution rate of 1.5 × 10−8 substitutions per site per year for chalcone synthase (Chs) (Koch et al. 2000).
Plant material and library construction for mRNA sequencing
Seeds of B. rapa ssp. pekinensis cv. Chiifu were surface-sterilized in 12 % sodium hypochlorite and were germinated on 0.5× Murashige and Skoog (MS) agar plates (0.7 %) in a growth chamber at 22 °C with a 16 h light/8 h dark cycle and 60 % humidity. Seedlings, 5 days after germination, were placed on 0.7 % agar plates containing 1 μM 1-naphthalene acetic acid (NAA) in 0.5× MS agar media for 0, 0.5, 1, and 2 h under the same growth conditions as described earlier and then were harvested. To collect floral tissues, 1-month-old plants were vernalized for 4 weeks at 4 °C and then were grown in a growth chamber. After inflorescences induction, the anthers and pistils at floral development stages 9–13 were collected. Floral development stage was determined according to that of A. thaliana (Smyth et al. 1990; Yu et al. 2005).
Total RNA was isolated from the plant tissues using the GeneAll HybridR+ kit (GeneAll, Seoul, Korea), and messenger RNA was purified using the TruSeqTM RNA sample preparation kit (Illumina, San Diego, CA) following the manufacturer’s instruction. Purified mRNA was chemically fragmented to 200 bp fragments and the first- and second-strand cDNAs were synthesized, followed by end repair and index adapter ligation. Next, the resulting libraries were pooled with up to 12 samples per flow cell lane for cluster generation on the Illumina flow cell using the cBot.
mRNA sequencing and data analysis
Gene expression analysis (three samples per treatment or tissue as biological replicates) was conducted by Illumina RNA-Seq technology. The library clusters were sequenced on the Illumina HiSeq1000 sequencer using the TruSeq™ SBS kit v3-HS to generate paired-end sequences (2×100 bp). Real-time analysis and base calling were performed using the CASAVA 1.8.2 software package (Illumina). The approximate number of reads collected from seedling, anther, and pistil libraries were 38, 25, and 24 million reads, respectively, with a minimum Illumina quality score of 31.
The paired-end sequence reads were aligned to the B. rapa ssp. pekinensis cv. Chiifu reference gene models using CLC Assembly Cell 3.2 (CLC Bio, Aarhus, Denmark), with a minimum similarity fraction of 0.9, a minimum length fraction of 0.9, a maximum of two mismatches, and paired-read mode options. The reads that mapped to multiple locations were excluded. The resulting mapped reads were normalized using the TMM (Trimmed Mean of M component) method for each gene to report the expression of genes. The TMM process estimates scale factors from the raw data that can be used in downstream statistical analysis procedures such as the detection of differential expression (Robinson and Oshlack 2010). The data of three biological replicates were pooled and the average expression levels of BrARF genes were extracted and analyzed.
Results
Identification of BrARF genes in the B. rapa genome
The BrARF genes were identified based on a BLAST search of all reference gene models against the A. thaliana ARF gene family, followed by an hmmpfam search through the CDD database. A total of 31 BrARF genes were identified from the genome (Table 1). They were distributed on all 10 chromosomes and no tandem clusters of the genes in 1 Mb intervals were identified, unlike the cluster of AtARF genes on chromosome 1 of A. thaliana (AtARF 12–15 and 20–23). The gene structure of BrARFs is similar to that of AtARFs with an average of 11 exons in the BrARF genes. However, BrARF10, 16-1, 16-2, 17-1, and 17-2 have only 2–5 exons, like their A. thaliana homologs (Fig. 1). The genes encode weak acidic proteins ranging in size from approximately 60 to 121 kDa. Like other transcription factors, BrARFs contain conserved domains that can function in DNA binding. All the BrARFs consist of an N-terminal plant-specific B3 DNA-binding domain and an auxin response super family domain at the middle region. However, a C-terminal AUX/IAA super family domain was missing in several proteins, including BrARF3-1, 3-2, 17-1, and 17-2. In A. thaliana, AtARF3, 13, and 17 also lack the C-terminal AUX/IAA domain, which functions as a dimerization domain with AUX/IAA proteins. In addition, AtARF23 is a partial-length gene with a stop codon in its B3 DNA-binding domain (Guilfoyle and Hagen 2007), but no such truncated genes were identified in the B. rapa genome. In silico analysis of gene expression indicated that 18 genes (58 %) have EST support, with an average of 3.1 ESTs per expressed BrARF gene. These genes were expressed in a narrow range of cDNA libraries, including those constructed from root, floral tissues, or stress-induced condition, suggesting the specialized expression of BrARF gene members in B. rapa plants. The most abundantly expressed gene was BrARF2-2, which displayed 9 ESTs, both in vegetative and floral tissues. Alternative transcription was observed in BrARF6, of which two ESTs had lengths of an additional 36 bp due to differential splicing of the 11th exon–intron junction compared with the coding sequence. The remaining 13 genes had no ESTs collected. They might play a unique role in specific tissue or developmental conditions, or be silenced due to redundancy.

Gene structure and conserved protein domain of BrARF genes. Exon–intron organization of the BrARF genes and conserved domains identified in the encoded protein were displayed together with those of their AtARF homologs. a Exon–intron structure organization of BrARF and AtARF genes. The exons and introns are represented by black boxes and gray lines, respectively. b Conserved domain structure of BrARF and AtARF proteins. The conserved domains are denoted by colored boxes
Phylogenetic relationship of the BrARF gene family
A phylogenetic analysis of 31 BrARF sequences was performed to classify subgroups and to identify relationships between genes. Both neighbor-joining and maximum likelihood algorithms produced similar trees (Supplementary Figs. 1, 2), and the phylogenetic patterns of the subgroups in both trees were consistent with the previously reported trees using A. thaliana and rice ARF proteins (Okushima et al. 2005; Wang et al. 2007). Figure 2a shows a phylogeny of BrARF genes based on the neighbor-joining method, indicating that the genes fall into four subgroups: Group I (BrARF1, 2-1, 2-2, 2-3, 9-1, 9-2, 11, 18-1, 18-2, 24, 25, 26, 27), II (BrARF 3-1, 3-2, 4), III (BrARF 5-1, 5-2, 5-3, 6, 7-1, 7-2, 8-1, 8-2, 19-1, 19-2), and IV (BrARF10, 16-1, 16-2, 17-1, 17-2). A composite tree for the 23 AtARF (Okushima et al. 2005) and 31 BrARF genes was consistent with that of the BrARF gene tree (Fig. 2b). All BrARF subgroups present in the BrARF gene tree were present in the composite phylogenetic tree for both genes, which support the categorization of BrARF genes into four subgroups. In addition, the tree indicates homologous relationships between the genes of B. rapa and A. thaliana. Most of the AtARF genes have one to three homologous counterparts of the BrARFs, and the exon–intron structures of the genes in the same subgroup were similar (Fig. 1). The phylogenetic distribution of A. thaliana and B. rapa ARF genes detected a local clustering of particular types. Four genes of Group I (BrARF24–27) formed a local cluster separated from the AtARF12–15 and 20–23 subclade. Unlike the Arabidopsis genes that are tandem clustered in the upper arm of A. thaliana chromosome 1 and appear to be specifically expressed in the seed and embryo (Okushima et al. 2005), these genes might have been generated by dispersed duplication of BrARF27 recently after the divergence of A. thaliana and B. rapa, followed by negative selection based on the location of genes on the B. rapa chromosomes and K s–K a comparison between the paralogous gene pairs (Table 1, Supplementary Tables 1 and 2). In addition, they are not expressed in any of the tissues investigated.

Phylogenetic tree of B. rapa and A. thaliana ARF genes. Phylogenetic trees of 31 BrARF genes alone (a) and together with 23 AtARF genes (b) were generated using MEGA5 program by neighbor-joining analysis. Bootstrap values (above 50 %) are indicated on the branches and the branch length reflects the estimated number of substitutions per 100 sites. Genes in the same group are represented by color bars. A lineage-specific local cluster of the Group I genes is indicated with an asterisk
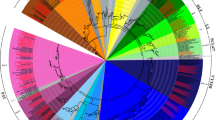
Further phylogenetic reconstruction using ARF genes from the sequenced plant genomes was performed to assess the support for the ARF gene subgroups and to investigate the evolution of BrARF genes (Fig. 3). A phylogenetic tree constructed using the protein sequences of the ARF genes from the sequenced plant genomes shows that plant ARF genes are divided into five major clades. All the dicot genes, including BrARFs and AtARFs, were clustered into four subgroups (Group I–IV) that show a similar tree topology to the composite tree for AtARF and BrARF genes in Fig. 2b. Interestingly, Group V contains only monocot genes, suggesting that this group arose in monocots or was lost from dicots after the divergence of monocots and dicots. Taken together, these results suggest that BrARF genes shared the same origin with the AtARF genes, and they were more amplified after speciation from A. thaliana, presumably by WGT.

Phylogenetic relationships of plant ARF gene family. The neighbor-joining tree was constructed using 31 B. rapa, 23 A. thaliana, 13 papaya, 16 cucumber, 17 strawberry, 54 soybean, 36 poplar, 18 castor bean, 19 cacao, 19 grape, 24 B. distachyon, 28 rice, 25 sorghum, and 37 maize putative ARF genes. Groups of genes are represented by color arcs. Bootstrap values (above 50 %) are indicated on the branches and the branch length reflects the estimated number of substitution per 100 sites. BrARFs and AtARFs were color coded according to their subgroup in Fig. 2. At Arabidopsis thaliana, Br Brassica rapa, Cp Carica papaya, Cs Cucumis sativus, Fv Fragaria vesca, Gm Glycine max, Pt Populus trichocarpa, Rc Ricinus communis, Tc Theobroma cacao, Vv Vitis vinifera, Bd Brachypodium distachyon, Os Oryza sativa, Sb Sorghum bicolor, Zm Zea mays
Evolutionary origin and divergence of BrARF genes
A genome-level comparison between the B. rapa and A. thaliana chromosomes presented an insight into the origin and evolution of the BrARF gene family. Considering the recent WGT event, the B. rapa genome is expected to have approximately 60 ARF genes. However, we could identify only 31 BrARF genes in the genome, suggesting the loss of redundant genes through the diploidization process after WGT. To determine the evolutionary origin of BrARF genes, we compared their genomic positions with those of their A. thaliana counterparts with the view of 24 AK genome building blocks. A synteny block comparison between homologous gene pairs revealed that all AtARF genes, with the exception of the tandem duplicated genes on chromosome 1 (AtARF12–15, 20–23), have their BrARF homologs on the same AK genome building blocks (Table 2; Fig. 4). For example, AtARF2 in Group I has their three homologs on the triplicated X blocks of the B. rapa chromosome. Only AtARF5 and AtARF8 have additional homologs on the distinct AK blocks of B. rapa genome suggesting genomic rearrangement. Furthermore, most of the BrARF genes, except the BrARF2 and 5 genes, which have triplicated paralogs, were duplicates or single members, suggesting a loss of paralogs throughout the genome.

Circos diagrams of ARF gene pairs. BrARF genes in each group are plotted against their homologous AtARF counterparts. Individual chromosomes of B. rapa (Br) and A. thaliana (At) are shown as ancestral karyotype genome building blocks to represent the shared ancestral origin of their genomes
The age distributions of the orthologous pairs between the AtARF and BrARF genes displayed a single major peak at K s = 0.35–0.40, indicating that the ARF genes of the two genomes were separated approximately 13 Mya (Fig. 5a). This is in good agreement with our previous study of speciation between the Brassica and Arabidopsis genomes (Mun et al. 2009a). A comparison of the K s mode for the paralogs in both genomes identified overall similar K s distribution patterns above K s = 1.3 in both genomes, representing the origin of the genes from ancient polyploidy events. However, distinct peaks for each genome were found at K s = 0.05–0.10 for AtARFs and K s = 0.25–0.35 for BrARFs, suggesting the occurrence of additional members for each genome after speciation (Fig. 5b, c). In particular, note that a peak at K s = 0.30–0.40 for B. rapa is responsible for the K s between the paralog pairs generated by WGT, whereas a peak for A. thaliana paralogs at K s = 0.05–0.10 is responsible for K s between the tandem clusters of AtARF12–15 and 20–23 (Supplementary Tables 2 and 3). Interestingly, duplicated or triplicated BrARFs had higher K s values for their AtARF orthologs than those of single-member BrARFs in a two-tailed t test (P < 0.05), indicating a higher sequence divergence of multiplicated members of the BrARF genes (Table 2). This finding proposes a hypothesis that BrARF genes, which shared the same origin from the ancestral genome blocks with AtARF genes, were amplified by WGT, but the redundant genes might have been lost during genome diploidization due to events such as the accumulation of mutations (sequence divergence), loss of redundant genes, recombination, or epigenetic marking.

Age distribution of ARF genes in the genome of A. thaliana (At) and B. rapa (Br) viewed through the frequency distribution of relative K s modes. Distributions of K s values were obtained from orthologous gene sets (a) between the genomes and paralogous genes sets (b, c) in the A. thaliana and B. rapa genomes. The vertical axes indicate the frequency of paired sequences and the horizontal axes denote the K s values at 0.05 intervals. The black bars depict the positions of the modes of the K s distribution obtained from orthologous or paralogous gene pairs
To characterize the sequence divergence of the genes, the spatial distribution of repetitive sequences with respect to the genomic position of BrARF genes was examined. We investigated the distribution of five types of repetitive sequences frequently found in the promoter and coding regions of the B. rapa genome (TR, IR, TE, LCR, and SSR). To correlate repetitive sequences with specific genomic fractions, we assigned five categories of sequences, namely, (1) promoter, (2) 5′-UTR, (3) coding exon, (4) intron, and (5) 3′-UTR. As shown in Fig. 6, all the BrARF genes had repetitive sequence insertions and the repetitive sequences were frequent both in the promoter and 5′-UTR regions (1.7 and 2.1 repeats/kb, respectively) compared with the exons (0.1 repeats/kb), introns (1.2 repeats/kb), and 3′-UTR regions (1.0 repeats/kb), due primarily to a higher frequency of LCR. In addition, repeat frequency varied between the different transcribed fractions. Most of the repetitive sequences in the transcribed regions were detected in 5′-UTRs, introns, and 3′-UTRs, with the highest repeat frequency in the 5′-UTRs, which were characterized by elevated levels of LCR and SSR. Exons were significantly underestimated in all repetitive sequences with the exception of TR. Interestingly, even in the duplicate or triplicate paralogs, the degree of repetitive sequence insertion is quite different between paralog members, suggesting that sequence divergence occurred through the insertion of repetitive sequences, mainly in the promoter or noncoding regions of the transcribed fraction. The frequent occurrence of repetitive sequence insertion, both in the promoter and transcribed regions of the genes, may promote the divergence of triplicated genes, resulting in more variants. This finding suggests that the alteration of sequences in the promoter and noncoding regions of the triplicated genes leads to the divergence of expression patterns and function or gene silencing to the pseudogene.

Frequency distribution of five repetitive sequences in the genomic fraction of BrARF genes. The number of repetitive sequences was counted per every 1 kb of promoter, 5′-UTR, exon, intron, and 3′-UTR of BrARFs. TR tandem repeat, IR inverted repeat, TE transposable element, LCR low complexity repeat, SSR simple sequence repeat
RNA-Seq analysis of BrARF gene expression
To determine the expressional characteristics of each BrARF gene member according to auxin treatment and developmental process of the flower, we investigated the expression pattern of the genes using next-generation RNA-Seq technology. The recent development of the RNA-Seq method provides not only information on genome-wide gene expression, but also has the advantage of higher sensitivity and a greater dynamic range of gene expression than array-based technologies (Severin et al. 2010). The sequencing of seedlings, anther, and pistil transcriptomes generated 100 bp paired-end reads for each library covering at least 17× of the genome. The percentage of reads aligned to the genome averaged around 74–75 %, which represents both the quality of the libraries and the relative completeness of the B. rapa draft genome. The current draft genome sequence of B. rapa (284 Mb) covers only 54 % of the whole estimated genome (529 Mb) (Johnston et al. 2005). Through the comparative analysis of sequence datasets against the B. rapa draft genome, a total of 35,220 genes in seedlings, 37,565 genes in anthers, and 35,477 genes in pistils were expressed (data not shown). From these results, we extracted the data for the BrARF gene family and conducted further analysis.
It has been reported that auxin treatment induced or repressed the expression of some ARF members in A. thaliana (Okushima et al. 2005), rice (Wang et al. 2007), and maize (Xing et al. 2011). The response of BrARF genes to exogenous auxin stimuli was investigated in seedlings after 1 μM NAA treatment for 2 h. RNA-Seq analysis revealed that a total of 27 out of 31 BrARFs displayed transcriptional activity in seedlings, and no expression was detected for BrARF24–27 under any of the conditions tested in this study (Fig. 7). The most highly expressed genes in seedlings was BrARF2-2 and 2-3, which showed 3- to 85-fold higher expression than other members. Most of the BrARFs were responsive to exogenous auxin treatment but showed diverse expression patterns. As with their Arabidopsis homologs AtARF16 and 19 (Okushima et al. 2005; Wang et al. 2005), auxin treatment gradually enhanced transcription for BrARF16-1, 16-2, and 19-2, while decreased expression was found for BrARF2-1, 5-2, 8-2, and 10. In addition, 8 genes showed up-regulation by auxin treatment followed by down-regulation (BrARF1, 2-2, 2-3, 5-1, 5-3, 6, 7-1, and 11) and 12 genes were down-regulated first but up-regulated later (BrARF3-1, 3-2, 4, 7-2, 8-1, 9-1, 9-2, 17-1, 17-2, 18-1, 18-2, and 19-1). In either cases, transcriptional changes were observed within 0.5–1 h after auxin treatment. Of particular interest, one of the duplicate or triplicate paralog members was more abundantly expressed than the other members, suggesting that there are functional redundancies among the genes. Furthermore, expression patterns between paralogs in response to auxin treatment were different. These transcriptional differences may involve the modification of regulatory elements. A database search of plant promoters (PLACE) detected several auxin response elements in the promoter region of all of BrARFs except BrARF19-1, supporting the auxin responsiveness of BrARF genes (Supplementary Table 4). This finding demonstrates the complexity of the auxin-regulated expression of BrARF genes and thus the relationship between auxin response elements and the change in expression of BrARF genes by auxin treatment need to be further investigated.

Expression of BrARF genes in response to exogenous auxin treatment. Five-day-old seedlings were harvested after 0, 0.5, 1, and 2 h of incubation in 0.5× MS agar media containing 1 μM NAA. The whole mRNA level for each gene per treatment was estimated using mRNA sequencing followed by TMM normalizaton. The average value of three biological replicates was presented with the standard deviation. No expression of BrARF24–27 was detected in this experiment. X axes indicate the NAA incubation time and Y axes denote normalized read count
We also studied the expression profile of BrARF genes during flower development based on the recent functional studies of AtARF genes that showed their important role in flower, embryo, and seed development (Hardtke and Berleth 1998; Schruff et al. 2005; Nishimura et al. 2005; Finet et al. 2010). An investigation of gene expression was performed with anthers and pistils at floral development stages 9–13, at which both reproductive organs rapidly elongate and gametophyte development is completed (Smyth et al. 1990; Yu et al. 2005). It is known that ARF genes are constitutively expressed in plant tissues (Wang et al. 2007). However, RNA-Seq analysis of BrARF genes exhibited dynamic expression patterns in reproductive organs during flower development. Except BrARF11, 24, 25, 26, and 27 in Group I, all the BrARF genes were expressed in both the anther and pistil tissues, and some of them showed relatively higher expression in specific tissues and development stages (Fig. 8). In addition, the expression patterns of duplicate or triplicate paralogs also varied considerably. Similar to auxin treatment, one of the duplicates or triplicates was dominantly expressed over the others. Genes in Group II were more abundantly expressed in the pistil than the anther, whereas Group IV members showed relatively high expression in anther. Genes in Groups I and III, however, presented complicated expression patterns in that some genes exhibited relatively high expression in the pistil, whereas other genes demonstrated significant induction at specific stages of anther and pistil development. For example BrARF2-2 and 2-3 in Group I and BrARF5-1 and 5-2 in Group III were highly induced at stage 10 of anther development (uni-nuclear stage of male gametophyte), whereas BrARF18-2 in Group I and BrARF5-1 and 5-2 in Group III showed up-regulation in pistils at the floral development stage 9–10 (initiation of ovule primodia). Among the genes, the most predominantly expressed gene in both the anther and pistil was BrARF2-2, and this is in good agreement with the presence of abundant ESTs (Table 1) and expression data in seedlings (Fig. 7). It is worthy to note that this gene might be the primary auxin response gene in B. rapa because its mRNA was highly up-regulated when treated with exogenous auxin and significantly induced or sustained at high level at specific developmental stages of the anther or pistil. Transcriptional differentiation of paralog members was also noticed between vegetative and floral tissues. In seedlings, BrARF5-1 and 18-1 were abundantly expressed, whereas BrARF5-2 and 18-2 were dominant forms expressed in floral tissues. In addition, consistent with the results shown in Fig. 7, BrARF24, 25, and 26 were not expressed in anthers or pistils at any development stages. Only BrARF27 showed very low expression levels at the early stage of anther development, suggesting that genes emerging recently by dispersed duplication are more likely to be silenced. Otherwise, their function can be restricted to a specific tissue or development stage including embryogenesis or seed development based on the role of A. thaliana homologs (Okushima et al. 2005).

Expression profiling of BrARF genes during anther and pistil development. Anther and pistil at floral development stages 9–13 were harvested from growth chamber-grown plants. Whole mRNA levels for each gene in the anther and pistil tissues were estimated using RNA sequencing followed by TMM normalization. The average value of three biological replicates was presented with the standard deviation. No expression of BrARF24–26 was detected in the anther or pistil tissues at any stage. X axes indicate flower development stage and Y axes denote normalized read count
Discussion
Polyploidy is ubiquitous in the plant genomes (Cui et al. 2006; De Bodt et al. 2005; Jiao et al. 2011). All the plant genomes characterized so far experienced whole genome duplication events during their evolution. Polyploidy facilitates the accumulation of duplicated genes in the genome, leading to the increased complexity and diversity of the gene network. Duplication at the whole genome level led to gene redundancy, and resulting redundant copies of amplified genes might be pseudogenes (nonfunction) or gain additional (subfunction) or novel functions (neofunction). Otherwise, the duplicated genes may play a role in preventing potential harmful mutations (buffering). In any of these cases, the ultimate fate of duplicated genes varied according to the individual plant and species (Sémon and Wolfe 2007; Taylor and Raes 2004). In this study, we investigated the organization and expression of 31 BrARF genes. Phylogenetic analysis and comparative mapping of genes on the AK genome blocks revealed that the BrARF gene family consists of four sister groups. The tree topology of the current analysis is very similar to the previously reported trees based on A. thaliana and rice ARF proteins (Okushima et al. 2005; Wang et al. 2007). Furthermore, most of the BrARF genes shared same ancestral origin with the AtARF homologs mapped to the same AK blocks. Thus, the expansion of the BrARF gene family could be explained by the recent WGT (Mun et al. 2009a; The Brassica rapa Genome Sequencing Project Consortium 2011). Considering that B. rapa underwent WGT, the BrARF gene family ought to have approximately three times as many members as that of A. thaliana. However, only 31 members were identified in the B. rapa genome, suggesting a substantial loss of genes following hexaploidy formation by WGT.
The collapse of the BrARF gene complement might be a result of genome-level gene loss, termed paralog reduction or fractionation, which is typical of the diploidization of eukaryotes. It is noteworthy that, following polyploidy events, loss events of gene duplicates through processes such as epigenetic silencing, pseudogenization, and deletion of chromosomal segments containing one or more genes following polyploidy randomly occurred in the genome (Sankoff et al. 2010). The current analysis of ARF gene distribution in B. rapa agrees with this hypothesis, where loss of paralog members had occurred in the genome. Although it is uncertain as to how the redundant genes were deleted, it is possible that the insertion of repetitive sequences might lead to the pseudogenization of paralogs. Analysis of repetitive sequences in the promoter and non-coding regions in the genic region of BrARF genes revealed that all the BrARF genes contain repetitive sequence insertions in the sequences. Moreover, the higher frequency of repetitive sequences, especially LCR, which is the most abundant repetitive sequence in the B. rapa genome, in the promoter, 5′-UTR, and intronic regions was identified, suggesting that sequence divergence may possibly be preceded by the suppression of transcription and/or a change to the exonic splicing pattern. Repetitive sequence insertion possibly changes the structure of the original gene. For example, transposon activation can induce the alteration of cis-regulatory elements or sequence loss. In addition, LCR and SSR in duplicate genes may become active to increase the mutation rate when the species suffer under harsh environments. TR and IR sequences together promote the variation and divergence of the introns of paralogs. This finding is consistent with recent reports that the splicing pattern and cis-regulatory region of the duplicates genes changed rapidly after duplication and led to changes in the protein constitutions (Chen 2010; Paterson et al. 2009; Zhang et al. 2009). Thus, the variation in the promoter and genic regions may possibly promote sequence divergence and gene inactivation and eventually result in paralog reduction in the BrARF gene family. We note that the BrARF24, 25, and 26 genes, which may have originated from the dispersed duplication of BrARF27 after WGT, showed no expression in the tissues investigated. This finding indicates that the genes recently emerging after WGT were silenced or gained different expression characteristics compared with other members, suggesting the birth and death of duplicated paralogs is an ongoing process. Therefore, internal sequence divergence along with natural selection can serve at the ultimate factors that influence the fate of duplicated genes.
The dynamic and differential distribution of auxin hormone in the plant controls the diverse morphological differentiation, development, and growth. Under domestication and selection, Brassica species have a notable propensity to develop new morphological variants rapidly (Paterson et al. 2001). The current draft genome showed that the gene families involved in auxin signal transduction (transport inhibitor response 1, TIR1; topless, TPL; ARF; AUX/IAA) have been expanded and amplification by tandem duplication beyond that of A. thaliana can be observed for GH3 and SAUR in the B. rapa genome (The Brassica rapa Genome Sequencing Project Consortium 2011). ARFs are transcription factors that are upstream regulators of the auxin-related signaling process. Although several members were deleted and silenced after WGT, the expression of duplicated ARF genes in B. rapa would amplify expression of downstream auxin-related genes in the signaling pathway. Moreover, the divergence of paralogs may result in the alteration of the function and expression pattern of an ancestral gene. Of practical importance, functional changes to BrARF genes may have crucial effect on gametophyte and seed development in B. rapa, considering the role of AtARF genes during flower development. Since a limited number of ESTs was reported, we studied the expression of all BrARF genes in seedlings and flowers by whole transcriptome sequencing to investigate their global expression pattern and putative function. The RNA-Seq showed a great dynamic range of gene expression, with low variation between technical replicates.
In this study, we found that 27 BrARF genes were expressed in seedlings and that they responded to exogenous auxin treatment. Identification of various auxin responsive elements in the promoter region seems to be related with a transcriptional change to the BrARF genes. We found that the expression of BrARF16-1, 16-2, and 19-2 was increased, whereas that of BrARF2-1, 5-2, 8-2, and 10 was decreased by auxin treatment, although they contained the same auxin responsive transcriptional activation element in the promoter region. Posttranscriptional down-regulation of the transcript by miRNA is possible for these genes, based on action of miRNAs on AtARF transcripts (Wang et al. 2005; Wu et al. 2006; Yang et al. 2006). BrARF2-1 and 5-2 have the target sites for miRNA396 and miRNA156, respectively. BrARF10 contains those for miRNA156 and miRNA160; however, the target sites of miRNA160 were also detected in BrARF16-1 and 16-2. The differential insertion of repetitive sequences in the promoter region or unidentified novel miRNA of additional epigenetic markings may be involved in the regulation of gene expression. Therefore, we anticipate that the regulation of BrARF gene expression in auxin signaling is more complicated. Expression changes between gene duplicates in polyploidal B. rapa may reflect the retention of redundant copies with functional divergence. The expression patterns of BrARF genes in the anther and pistil also indicate that several members are transcriptionally induced at specific stages of anther or pistil development. High expression levels of BrARF2-2, 2-3, 5-1, and 5-2 at stage 9–10 in both anther and pistil development suggest that these genes play a more general role during early flower development. In particular, the differential expression of BrARF10, 16-1, 16-2, 17-1, and 17-2 in the anther and BrARF3-1, 6, 8-1, 8-2, and 18-1 in the pistil implies that these genes might be involved in a distinct floral development process. This result indicates that more diverse functions can be played by BrARF in floral tissues. Although the role of BrARF genes needs to be further investigated, the information on the structure and expression of BrARF generated here will accelerate their functional analysis in the future.
The current work has contributed to an increased knowledge of the ARF gene family in B. rapa. Based on the genomic organization and expression characteristics, we can hypothesize that overrepresentation of BrARF genes, together with other auxin-related genes in the signaling pathway, may enrich the auxin-related gene networks in the plant. Considering that the possible role of multiple auxin-related gene networks in environmental adaptation is complex, systematic studies on the target genes of ARF in auxin signaling pathway, such as AUX/IAA, GH3, and SAUR, will increase our understanding of the development and adaptation of B. rapa.
References
Altschul SF, Madden TL, Schäffer AA, Zhang J, Zhang Z, Miller W, Lipman DJ (1997) Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acid Res 25:3389–3402
Benson G (1999) Tandem repeats finder: a program to analyze DNA sequences. Nucleic Acid Res 27:573–580
Cannon SB, Kozik A, Chan B, Michelmore R, Young ND (2003) DiagHunter and GenoPix2D: programs for genomic comparisons, large-scale homology discovery and visualization. Genome Biol 4:R68
Chen ZJ (2010) Molecular mechanisms of polyploidy and hybrid vigor. Trends Plant Sci 15:57–71
Cui L, Wall PK, Leebens-Mack JH, Lindsay BG, Soltis DE, Doyle JJ, Soltis PS, Carlson JE, Arumuganathan K, Barakat A, Albert VA, Ma H, dePamphilis CW (2006) Widespread genome duplications throughout the history of flowering plants. Genome Res 16:738–749
De Bodt S, Maere S, Van de Peer Y (2005) Genome duplication and the origin of angiosperm. Trends Ecol Evol 20:591–597
Ellis CM, Negpal P, Young JC, Hagen G, Guilfoyle TJ, Reed JW (2005) AUXIN RESPONSE FACTOR1 and AUXIN RESPONSE FACTOR2 regulate senescence and floral organ abscission in Arabidopsis thaliana. Development 132:4563–4574
Fahlgren N, Montgomery TA, Howell MD, Allen E, Dvorak SK, Alexander AL, Carrington JC (2006) Regulation of AUXIN RESPONSE FACTOR3 by TAS3 ta-siRNA affects developmental timing and patterning in Arabidopsis. Curr Biol 16:939–944
Finet C, Fourquin C, Vinauger M, Berne-Dedieu A, Chambrier P, Paindavoine S, Scutt SP (2010) Parallel structural evolution of auxin response factors in the angiosperms. Plant J 63:952–959
Finn RD, Tate J, Mistry J, Coggill PC, Sammut JS, Hotz HR, Ceric G, Forslund K, Eddy SR, Sonnhammer EL, Bateman A (2008) The Pfam protein families database. Nucleic Acid Res 36:D281–D288
Fukai H, Taniguchi N, Tasaka M (2006) PICKLE is required for SOLITARY-ROOT/IAA14-mediated repression of ARF7 and ARF19 activity during Arabidopsis lateral root initiation. Plant J 48:380–389
Goetz M, Vivian-Smith A, Johnson SD, Koltunow AM (2006) AUXIN RESPONSE FACTOR8 is a negative regulator of fruit initiation in Arabidopsis. Plant Cell 18:1873–1886
Goldman N, Yang Z (1994) A codon-based model of nucleotide substitution for protein-coding DNA sequences. Mol Biol Evol 11:725–736
Guilfoyle TJ, Hagen G (2007) Auxin response factors. Curr Opin Plant Biol 10:453–460
Hagen G, Guilfoyle TJ (2002) Auxin-responsive gene expression: genes, promoters and regulatory factors. Plant Mol Biol 49:373–385
Hardtke CS, Berleth T (1998) The Arabidopsis gene MONOPTEROUS encodes a transcription factor mediating embryo axis formation and vascular development. EMBO J 17:1405–1411
Hayashi K (2012) The interaction and integration of auxin signaling components. Plant Cell Physiol 53:965–975
Jiao Y, Wickett NJ, Ayyampalayam S, Chanderbali AS, Landherr L, Ralph PE, Tomsho LP, Hu Y, Liang H, Soltis PS, Soltis DE, Clifton SW, Schlarbaum SE, Schuster SC, Ma H, Leebens-Mack J, dePamphilis CW (2011) Ancestral polyploidy in seed plants and angiosperms. Nature 473:97–100
Johnston JS, Pepper AE, Hall AE, Chen ZJ, Hodnett G, Drabek J, Lopez R, Price HJ (2005) Evolution of genome size in Brassicaceae. Ann Bot 95:229–235
Kalluri UC, Difazio SP, Brunner AM, Tuskan GA (2007) Genome-wide analysis of Aux/IAA and ARF gene families in Populus trichocarpa. BMC Plant Biol 7:59
Koch MA, Haubold B, Mitchell-Olds T (2000) Comparative evolutionary analysis of chalcone synthase and alcohol dehydrogenase loci in Arabidopsis, Arabis, and related genera (Brassicaceae). Mol Biol Evol 17:1483–1498
Krzywinski MI, Schein JE, Birol I, Connors J, Gascoyne R, Horsman D, Jones SJ, Marra MA (2009) Circos: an information aesthetic for comparative genomics. Genome Res 19:1639–1645
Kumar R, Tyagi AK, Sharma AK (2011) Genome-wide analysis of auxin response factor (ARF) gene family from tomato and analysis of their role in flower and fruit development. Mol Genet Genomics 285:245–260
Li H, Johnson P, Stepanova A, Alonso JM, Ecker JR (2004) Convergence of signaling pathways in the control of differential cell growth in Arabidopsis. Dev Cell 7:193–204
Liu PP, Montgomery TA, Fahlgren N, Kasschau KD, Nonogaki H, Carrington JC (2007) Repression of AUXIN RESPONSE FACTOR 10 by microRNA160 is critical for seed germination and ost-germination stages. Plant J 52:133–146
Lukens LN, Quijada PA, Udall J, Pires JC, Schranz ME, Osborn TC (2004) Genome redundancy and plasticity within ancient and recent Brassica crop species. Biol J Linn Soc Lond 82:665–674
Lysak MA, Berr A, Pecinka A, Schmidt R, McBreen K, Schubert I (2006) Mechanisms of chromosome number reduction in Arabidopsis thaliana and related Brassicaceae species. Proc Natl Acad Sci USA 103:5224–5229
Mun JH, Kwon SJ, Yang TJ, Seol YJ, Jin M, Kim JA, Lim MH, Kim J, S., Lee SI, Baek S, Choi BS, Kim DS, Kim N, Yu HJ, Lim KB, Lim YP, Bancroft I, Hahn JH, Park BS (2009a) Genome-wide comparative analysis of the Brassica rapa gene space reveals genome shrinkage and differential loss of duplicated genes after whole genome triplication. Genome Biol 10:R111
Mun JH, Yu HJ, Park S, Park BS (2009b) Genome-wide identification of NBS-encoding resistance genes in Brassica rapa. Mol Genet Genomics 282:617–631
Mun JH, Kwon SJ, Seol YJ, Kim JA, Jin M, Kim JS, Lim MH, Lee SI, Hong JK, Park TH, Lee SC, Kim BJ, Seo MS, Baek S, Lee MJ, Shin JY, Hahn JH, Hwang YJ, Lim KB, Park JY, Lee J, Yang TJ, Yu HJ, Choi IY, Choi BS, Choi SR, Ramchiary N, Lim YP, Fraser F, Drou N, Soumpourou E, Trick M, Bancroft I, Sharpe AG, Parkin IA, Batley J, Edwards D, Park BS (2010) Sequence and structure of Brassica rapa chromosome A3. Genome Biol 11:R94
Nishimura T, Wada T, Yamamoto KT, Okada K (2005) The Arabidopsis STV1 protein, responsible for translation reinitiation, is required for auxin-mediated gynoecium patterning. Plant Cell 17:2940–2953
Okushima Y, Overvoorde PJ, Arima K, Alonso JM, Chan A, Chang C, Ecker JR, Hughes B, Lui A, Nguyen D, Onodera C, Quach H, Smith A, Yu G, Theologis A (2005) Functional genomic analysis of the AUXIN RESPONSE FACTOR gene family members in Arabidopsis thaliana: unique and overlapping functions of ARF7 and ARF19. Plant Cell 17:444–463
Paterson AH, Lan TH, Amasino R, Osborn TC, Quiros C (2001) Brassica genomics: a complement to, and early beneficiary of the Arabidopsis sequence. Genome Biol 2:Reviews1011
Paterson AH, Bowers JE, Bruggmann R, Dubchak I, Grimwood J, Gundlach H, Haberer G, Hellsten U, Mitros T, Poliakov A, Schmutz J, Spannagl M, Tang H, Wang X, Wicker T, Bharti AK, Chapman J, Feltus FA, Gowik U, Grigoriev IV, Lyons E, Maher CA, Martis M, Narechania A, Otillar RP, Penning BW, Salamov AA, Wang Y, Zhang L, Carpita NC, Freeling M, Gingle AR, Hash CT, Keller B, Klein P, Kresovich S, McCann MC, Ming R, Peterson DG, Mehboob-ur-Rahman, Ware D, Westhoff P, Mayer KF, Messing J, Rokhsar DS (2009) The Sorghum bicolor genome and the diversification of grasses. Nature 457:551–556
Plant Biosafety Office (1999) The Biology of Brassica rapa L. In: Agency CFI (ed) Biology Document BIO 1922-02 edn
Robinson MD, Oshlack A (2010) A scaling normalization method for differential expression analysis of RNA-seq data. Genome Biol 11:R25
Sankoff D, Zheng C, Zhu Q (2010) The collapse of gene complement following whole genome duplication. BMC Genomics 11:313
Schranz ME, Lysak MA, Mitchell-Olds T (2006) The ABC’s of comparative genomics in the Brassicaceae: building blocks of crucifer genomes. Trends Plant Sci 11:535–542
Schruff MC, Spielman M, Tiwari S, Adams S, Fenby N, Scott RJ (2005) The AUXIN RESPONSE FACTOR 2 gene of Arabidopsis links auxin signaling, cell division, and size of seeds and other organs. Development 133:251–261
Sémon M, Wolfe KH (2007) Consequences of genome duplication. Curr Opin Genet Dev 17:505–516
Severin AJ, Woody JL, Bolon YT, Joseph B, Diers BW, Farmer AD, Muehlbauer GJ, Nelson RT, Grant D, Specht JE, Graham MA, Cannon SB, May GD, Vance CP, Shoemaker RC (2010) RNA-Seq Atlas of Glycine max: a guide to the soybean transcriptome. BMC Plant Biol 10:160
Smyth DR, Bowman JL, Meyerowitz EM (1990) Early flower development in Arabidopsis. Plant Cell 2:755–767
Sorin C, Bussell JD, Camus I, Ljung K, Kowalczqk M, Geiss G, McKhann H, Garcion C, Vaucheret H, Sandberg G, Bellini C (2005) Auxin and light control of adventitious rooting in Arabidopsis require ARGONAUTE1. Plant Cell 17:1343–1359
Tamura K, Peterson D, Peterson N, Stecher G, Nei M, Kumar S (2011) MEGA5: molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol Biol Evol 28:2731–2739
Taylor JS, Raes J (2004) Duplication and divergence: the evolution of new genes and old ideas. Annu Rev Genet 38:15–43
Temnykh S, Clerck GD, Lukashova A, Lipovich L, Cartinhour S, McCouch S (2001) Computational and experimental analysis of microsatellites in rice (Oryza sativa L.): frequency, length variation, transposon associations, and genetic marker potential. Genome Res 11:1441–1452
The Brassica rapa Genome Sequencing Project Consortium (2011) The genome of the mesopolyploid crop species Brassica rapa. Nat Genet 43:1035–1040
Thompson JD, Higgins DG, Gibson TJ (1994) CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acid Res 22:4673–4680
Ulmasov T, Liu ZB, Hagen G, Guilfoyle TJ (1995) Composite structure of auxin response elements. Plant Cell 7:1611–1623
Ulmasov T, Hagen G, Guilfoyle TJ (1997) ARF1, a transcription factor that binds to auxin response elements. Science 276:1865–1868
Vaucheret H, Vazquez F, Crete P, Bartel DP (2004) The action of ARGONAUTE1 in the miRNA pathway and its regulation by the miRNA pathway are crucial for plant development. Genes Dev 18:1187–1197
Wang JW, Wang LJ, Mao YB, Cai WJ, Xue HW, Chen XY (2005) Control of root cap formation by microRNA-targeted auxin response factors in Arabidopsis. Plant Cell 17:2204–2216
Wang D, Pei K, Fu Y, Sun Z, Li S, Liu H, Tang K, Han B, Tao Y (2007) Genome-wide analysis of the auxin response factors (ARF) gene family in rice (Oryza sativa). Gene 394:13–24
Warburton PE, Giordano J, Cheung F, Gelfand Y, Benson G (2004) Inverted repeat structure of the human genome: the X-chromosome contains a preponderance of large, highly homologous inverted repeats that contain testes genes. Genome Res 14:1861–1869
Waterhouse AM, Procter JB, Martin DMA, Clamp M, Barton GJ (2009) Jalview Version 2—a multiple sequence alignment editor and analysis workbench. Bioinformatics 25:1189–1191
Wu MF, Tian Q, Reed JW (2006) Arabidopsis microRNA167 controls patterns of ARF6 and ARF8 expression, and regulates both female and male reproduction. Development 133:4211–4218
Xing H, Pudake RN, Guo G, Xing G, Hu Z, Zhang Y, Sun Q, Ni Z (2011) Genome-wide identification and expression profiling of auxin response factor (ARF) gene family in maize. BMC Genomics 12:178
Yang Z (2007) PAML 4: phylogenetic analysis by maximum likelihood. Mol Biol Evol 24:1586–1591
Yang JH, Han SJ, Yoon EK, Lee WS (2006) Evidence of an auxin signal pathway, microRNA167-ARF8-GH3, and its response to exogenous auxin in cultured rice cells. Nucleic Acid Res 34:1892–1899
Yu HJ, Hogan P, Sundaresan V (2005) Analysis of the female gametophyte transcriptome of Arabidopsis by comparative expression profiling. Plant Physiol 139:1853–1859
Yu HJ, Park SG, Oh M, Hwang HJ, Kim N, Chung H, Sohn SH, Park BS, Mun JH (2011) The Brassica rapa tissue-specific EST database. Kor J Hort Sci Technol 29:511–518
Zhang ZG, Zhou L, Wang P, Liu Y, Chen XF, Hu LD, Kong XY (2009) Divergence of exonic splicing elements after gene duplication and the impact on gene structures. Genome Biol 10:R120
Acknowledgments
We thank Dr. Bumjin Kim and Sin-Gi Park of Department of Genomics, the National Academy of Agricultural Science, RDA for bioinformatic supports. This work was supported by grants from the National Academy of Agricultural Science and the Next-Generation Biogreen21 program (PJ006759 and PJ008019), Rural Development Administration, Korea and The Catholic University of Korea (M-2012-B002-00103).
Open Access
This article is distributed under the terms of the Creative Commons Attribution License which permits any use, distribution, and reproduction in any medium, provided the original author(s) and the source are credited.
Author information
Authors and Affiliations
Corresponding authors
Additional information
Communicated by Y. Van de Peer.
J.-H. Mun and H.-J. Yu contributed equally to this work.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 2.0 International License (https://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Mun, JH., Yu, HJ., Shin, J.Y. et al. Auxin response factor gene family in Brassica rapa: genomic organization, divergence, expression, and evolution. Mol Genet Genomics 287, 765–784 (2012). https://doi.org/10.1007/s00438-012-0718-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00438-012-0718-4