
Abstract
Mycobacterium tuberculosis represents a severe threat to human health worldwide. Therefore, it is important to expand our knowledge of vital mycobacterial processes, such as that effected by fatty acid synthase type 2 (FASII), as well as to uncover novel ones. Mycobacterial FASII undertakes mycolic acid biosynthesis, which relies on a set of essential enzymes, including 3-oxoacyl-AcpM reductase FabG1/Rv1483. However, the M. tuberculosis genome encodes four additional FabG homologs, designated FabG2–FabG5, whose functions have hitherto not been characterized in detail. Of the four candidates, FabG4/Rv0242c was recently shown to be essential for the survival of M. bovis BCG. The present work was initiated by assessing the suitability of yeast oar1Δ mutant cells lacking mitochondrial 3-oxoacyl-ACP reductase activity to act as a surrogate system for expressing FabG1/MabA directed to the mitochondria. Mutant yeast cells producing this targeted FabG1 variant were essentially wild type for all of the chronicled phenotype characteristics, including respiratory growth on glycerol medium, cytochrome assembly and lipoid acid production. This indicated that within the framework of de novo fatty acid biosynthesis in yeast mitochondria, FabG1 was able to act on shorter (C4) acyl substrates than was previously proposed (C8–20) during mycolic acid biosynthesis in M. tuberculosis. Thereafter, FabG2–FabG5 were expressed as mitochondrial proteins in the oar1Δ strain, and FabG4 was found to complement the mutant phenotype and contain high levels of 3-oxoacyl-thioester reductase activity. Hence, like FabG1, FabG4 is also an essential, physiologically functional 3-oxoacyl-thioester reductase, albeit the latter’s involvement in mycobacterial FASII remains to be explored.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Mycobacterium tuberculosis is the main cause of death due to a single infectious agent (World Health Organisation 1997) among humans, and as many as 2 billion people worldwide are infected with tuberculosis (for a review, see Takayama et al. 2005 and references cited therein). In light of the rise in mycobacterial resistance toward isoniazid, which represents one of less than a handful of first-line antituberculous drugs, there is an urgent need to widen the study of vital M. tuberculosis processes so as to develop novel drugs.
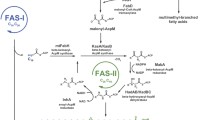
Isoniazid targets the production of mycolic acids (Banerjee et al. 1994), which are very long-chain α-branched β-hydroxylated fatty acids (C54–63) that help form the protective layer around this pathogen, thereby adding to its persistence and virulence (Takayama et al. 2005). Mycolic acid biosynthesis involves a combination of an associative type 1 fatty acid synthase (FASI) similar to the system found in the eukaryotic cytosol, in that it comprises several enzymatic activities located on a single multifunctional synthase (Bloch 1977) and a dissociative type 2 process (FASII) resembling the plant–plastid system that consists of separate enzymes. FASII also occurs in yeast mitochondria (reviewed in Hiltunen et al. 2005).
In M. tuberculosis FASII, FabG1 (MabA) undertakes the reduction of 3-oxoacyl-acyl carrier protein (AcpM) to generate 3-hydroxyacyl-AcpM (Banerjee et al. 1998), whereas in the equivalent mitochondrial process in Saccharomyces cerevisiae, this step is catalyzed by Oar1p (Schneider et al. 1997). In reference to substrate specificities, FabG1 has been demonstrated previously to catalyze the NADP(H)-specific reduction of long-chain (C8–20) 3-oxoacyl-thioester species. Molecular modeling of FabG1 revealed a large substrate-binding pocket capable of accommodating long-chain substrates (Marrakchi et al. 2002), while yeast Oar1p is presumed to act on only short-chain ones. Analysis of the complete sequence of the M. tuberculosis genome (Cole et al. 1998) revealed FabG1 as having four homologs, termed FabG2–FabG5 (Fig. 1), but, so far, not one of them has been fully characterized.

Homology study of M. tuberculosis FabG proteins. Multalin- and Genedoc-based comparison of the deduced amino acid sequence of FabG1 (Rv1483) with those of its homologs FabG2 (Rv1350), FabG3 (Rv2002), FabG4 (Rv0242c) and FabG5 (Rv2766c). Dashes indicate the arrangement of the sequences for best fit, and arrows point to the amino acids forming the catalytic triad (Marrakchi et al. 2002). The first 150 amino acid residues of FabG4 do not match any of those of the other homologs, and were removed from the figure. Black shadings refer to strictly conserved amino acid residues among all the sequences, whereas the darker and lighter gray shadings denote regions with more relaxed residue similarities not necessarily shared by the full set of sequences
The essential requirement for FabG activity has been addressed before in a genome-wide survey using transposon-site hybridization, which revealed just over 600 M. tuberculosis genes that were required for optimal growth (Sassetti et al. 2003), among them fabG2/Rv1350. However, a subsequent study identifying almost 200 M. tuberculosis genes that were required for mycobacterial survival during infection did not pick up a single fabG locus (Sassetti and Rubin 2003). The dispute surrounding the requirement for 3-oxoacyl-thioester activity in mycobacteria was finally settled with the finding that FabG1 is in fact essential for M. tuberculosis survival (Parish et al. 2007), whereas in M. bovis BCG, FabG1 and the hitherto uncharacterized FabG4 have both been exposed as being vital (Beste et al. 2009).
The latter finding of two non-redundant FabGs in M. bovis BCG raised the pertinent question of whether a similar situation might also occur in M. tuberculosis, whereby FabG1 is not necessarily the sole essential 3-oxoacyl-thioester reductase after all. This prompted the deployment of an alternative strategy for studying the functions of the remaining four FabG homologs. To this end, S. cerevisiae was exploited as a surrogate host for investigating M. tuberculosis proteins (Gerum et al. 2002) with potential FASII activities. Here, mitochondrially targeted versions of FabG1 to FabG5 were expressed in yeast oar1Δ mutant cells (Schneider et al. 1997), which lack 3-oxoacyl-ACP reductase activity and do not respire or grow on glycerol, assemble cytochrome complexes or produce lipoic acid. The effects were compared to those of ectopically expressing native Oar1p. This oar1Δ strain was applied previously to expose the identity of nematode and human 3-oxoacyl-thioesters (Gurvitz 2009; Chen et al. 2009). The outcome of this study is discussed in terms of the identification of the essential mycobacterial fabG4 gene as encoding a novel 3-oxoacyl-thioester reductase, as well as the role of protein–protein interactions in mycobacterial FASII.
Materials and methods
S. cerevisiae strains, plasmids and oligonucleotides
Yeast strains, plasmids and oligonucleotides used are listed in Table 1. The Escherichia coli strain TOP10 F′ was used for all plasmid amplifications and isolations. The wild-type yeast strain BY4741 and its oar1Δ derivative were obtained from EUROSCARF (www.uni-frankfurt.de). To generate strains yPLM37 to yPLM43, plasmids were introduced into BY4741oar1Δ cells using a published protocol (Chen et al. 1992), and transformants were selected on solid synthetic defined glucose (SD-Ura) medium lacking uracil.
Plasmid constructions
DNA manipulations and plasmid constructions were performed according to standard techniques (Ausubel et al. 1989). The five fabG genes were isolated from H37Rv genomic DNA by polymerase chain reaction (PCR) using Phusion high-fidelity DNA polymerase (Finnzymes Oy, Espoo, Finland) and the corresponding oligonucleotides (Table 1), as described previously (Gurvitz et al. 2008a,b, 2009). Electrophoretic resolution of the PCR products, and their excision, purification and subsequent ligation to an EcoRV-digested pBluescript plasmid vector (Stratagene, La Jolla, CA) were performed as done previously (Gurvitz et al. 2008b). The five pBluescript inserts were subcloned following digestion with NcoI and HindIII restriction enzymes, and ligated behind the CTA1 promoter within a similarly digested plasmid pYE352:mitQOR (pPLM62). As a result of this digestion, the QOR open reading frame (ORF) encoding E. coli quinone reductase was removed to leave behind the nucleotides for the yeast Coq3p (Hsu et al. 1996) mitochondrial leader sequence (MLS), as described in the literature (Torkko et al. 2001; Gurvitz et al. 2008b). Following import into the mitochondria, the fused MLS is cleaved off to leave behind a shorter fusion protein preceded by eight amino acids remaining from the MLS fusion (Torkko et al. 2001). Nucleotide sequencing of the fabG inserts verified that no mutations were introduced during the amplification process and that the COQ3-fabG junction remained intact. Construction of plasmid YEp352:OAR1 (pPLM188) expressing S. cerevisiae mitochondrial OAR1 from the CTA1 promoter is described (Chen et al. 2009). Briefly, the OAR1 ORF was amplified from yeast genomic DNA using standard procedure and, following digestion with the appropriate restriction enzymes, the PCR product was ligated to a similarly digested pYE352:CTA1 (Filppula et al. 1995), from which the CTA1 ORF was removed (Chen et al. 2009).
Media and growth conditions
Standard yeast (Rose et al. 1990) and E. coli (Sambrook et al. 1989) media were prepared as described. S. cerevisiae strains were propagated on solid, rich glucose medium YPD consisting of 1% (wt/vol) yeast extract, 2% (wt/vol) peptone (YP), 2% (wt/vol) d-glucose and 2% (wt/vol) agar. YEp352-based episomal plasmids marked with the URA3 gene (Hill et al. 1986) were maintained in transformed strains on solid, synthetic defined (SD-Ura) medium consisting of 0.67% (wt/vol) yeast nitrogen base without amino acids, 2% (wt/vol) d-glucose and 3% (wt/vol) agar, with all necessary supplements added except for uracil (Sigma–Aldrich Inc., MO, USA). Synthetic complete glycerol (SCglycerol) medium was prepared essentially as SD-Ura, except that uracil was added and glucose was replaced with 3% (wt/vol) glycerol. Induction of yeast cells in oleic acid medium has been described previously (Gurvitz et al. 1997).
Miscellaneous
For 3-oxoacyl-thioester reductase activity assays, soluble protein extracts were prepared from yeast cells that were propagated in oleic acid medium and processed as described (Gurvitz et al. 2008a,b, 2009). Protein concentrations were determined according to a published method (Bradford 1976). Reductase activity was assayed spectrophotometrically at 23°C as described (Dommes and Kunau 1984). The reductase activity assay mixture consisted of 50 mM KPi (pH 8.0) and 50 µg/ml bovine serum albumin, 2.0 µl purified protein representing the hydratase 2 domain from Drosophila melanogaster MFE2 (unpublished), 125 µM NADP+ or NAD+, and 60 µM 2-trans-decenoyl-CoA or 2-trans-hexenoyl-CoA that were synthesized via the mixed anhydride system (Goldman and Vagelos 1961). Reduction of the electron carrier was monitored spectrophotometrically at 340–385 nm. The assembly of cytochrome complexes was followed with a spectrophotometer at 480–640 nm, using yeast cells that were applied as a thick paste onto the glass surface of an otherwise aluminum cold-temperature cuvette, which was chilled in liquid nitrogen (Lindenmayer and Estabrook 1958). The lipoic acid content of yeast strains was monitored by a biological assay as described previously (Brody et al. 1997; Hayden et al. 1993). Operon prediction was undertaken using OperonDB (http://operondb.cbcb.umd.edu/cgi-bin/operondb/operons.cgi). Multalin (npsa-pbil.ibcp.fr/cgi-bin/npsa_automat.pl?page =/NPSA/npsa_multalin.html) and Genedoc (www. nrbsc.org/gfx/genedoc/index.html) were used to generate the sequence comparison in Fig. 1.
Results
Mycobacterial FabG1 and FabG4 restore respiratory growth of S. cerevisiae oar1Δ cells
The M. tuberculosis genome (Cole et al. 1998) contains five genes with the potential to encode 3-oxoacyl-AcpM reductases, termed fabG1 to fabG5 (Takayama et al. 2005). FabG1 is chronicled as a 3-oxoacyl-AcpM reductase involved in mycolic acid biosynthesis (Banerjee et al. 1998; Marrakchi et al. 2002); however, the function of the remaining four homologs has hitherto remained unknown. To determine whether these could encode 3-oxoacyl-thioester reductases, FabG1–FabG5 were expressed as fusion constructs that were preceded by the Coq3p mitochondrial leader sequence (Hsu et al. 1996) so as to generate mitochondrially targeted versions. The five fusions were tethered behind the promoter of the CTA1 gene encoding peroxisomal catalase A (Cta1p), which is only moderately de-repressed on non-fermentable carbon sources such the glycerol is used here for complementation assays (Filipits et al. 1993; Rottensteiner et al. 1996). These tests were performed on yeast cells lacking Oar1p that have been exploited previously to demonstrate the physiological function of ectopically expressed 3-oxoacyl-ACP reductases (Gurvitz 2009; Chen et al. 2009).
Mutant oar1Δ yeast cells were transformed with multicopy episomal plasmids loaded with the fusion constructs. Following selection on synthetic defined glucose medium lacking uracil (SD-Ura), survivors were streaked on synthetic complete medium containing glycerol (SCglycerol) as the sole carbon source. Only those mutants expressing FabG1 or FabG4 were capable of forming single colonies on this medium type. A dedicated analysis of the reasons for the failure of FabG2, FabG3 or FabG5 to achieve phenotype rescue was beyond the scope of the present study, these causes being all the more enigmatic since no obvious differences among the homologs emerged from the data shown in Fig. 1.
To demonstrate more accurately that expression of mitochondrial versions of FabG1 or FabG4 in the oar1Δ mutant cells could compensate for the missing activity attributed to native Oar1p, the four strains (each expressing FabG1, FabG4, Oar1p or Cta1p) were propagated overnight in liquid SD-Ura medium. Following tenfold serial dilution, cultures were spotted onto solid SD-Ura (glucose) or SCglycerol (glycerol) media, and the plates were incubated at 30°C until single colonies were detectable (Fig. 2). As anticipated, mutant oar1Δ cells expressing Oar1p could form single colonies on glycerol, whereas those cells expressing Cta1p were not able to proliferate on this medium, vindicating the experimental design. Importantly, the results demonstrated that the respiratory deficient phenotype of the oar1Δ mutant could be rescued by FabG1 and FabG4, indicating functional complementation.

Growth of S. cerevisiae oar1Δ mutants expressing M. tuberculosis FabG1 or FabG4. Yeast oar1Δ cells expressing native mitochondrial 3-oxoacyl-ACP reductase (Oar1p positive control), native peroxisomal catalase A (Cta1p negative control), mitochondrially targeted FabG1 or mitochondrially targeted FabG4 were grown in liquid SD-Ura medium that selected for plasmid presence. Following serial dilution, cells were applied to solid (SD-Ura) glucose or (SCglycerol) glycerol media. The plates were incubated at 30°C until single colonies appeared and recorded photographically. The yPLM strains used were 37, 38, 41 and 43
Yeast oar1Δ cells expressing FabG4 contain 3-oxoacyl-thioester reductase activity
Soluble protein extracts from BY4741oar1Δ cells expressing M. tuberculosis FabG1 or FabG4 were examined for 3-oxoacyl-thioester reductase activity using 2-trans-hexenoyl-CoA (trans-C6:1(2)) or 2-trans-decenoyl-CoA (trans-C10:1(2)) as substrates. The reactions additionally included purified protein consisting of the hydratase 2 domain from Drosophila melanogaster peroxisomal type 2 multifunctional enzyme (MFE2), which hydrated the substrates to generate the 3-hydroxyacyl-CoA species that could be metabolized, in the reverse reaction, by the 3-oxoacyl-thioester reductases examined. The reactions were monitored spectrophotometrically at wavelengths of 340–385 nm, by following the reduction of NADP+ or NAD+ to their corresponding protonated species (Gurvitz 2009). Both electron carrier types were considered in this assay, as recent work revealed that 3-oxoacyl-thioester reductase activity might actually depend on NAD(H), poignantly evidenced by the nematode and human 3-oxoacyl-ACP reductases of mitochondrial FASII (Gurvitz 2009; Chen et al. 2009).
To obtain high levels of protein expression from the fatty acid-inducible CTA1 promoter (Filipits et al. 1993; Rottensteiner et al. 1996), cells were grown overnight on oleic acid medium. The results of the enzyme assays with 2-trans-hexenoyl-CoA as substrate and NADP+ as cofactor (in the presence of hydratase 2 activity) performed on oar1Δ cells over-expressing native Oar1p or Cta1p indicated that any reductase activity present was below the detection limit of the assay used. Recent work on Oar1p by others revealed that its 3-oxoacyl-thioester reductase activity could only be detected in assays performed on a recombinant protein (Chen et al. 2009). Similarly, it was impossible to detect any activity in soluble protein extracts obtained from oar1Δ cells over-expressing FabG1 when NADP(H) was supplied as cofactor in the reaction, but this was anticipated since previous measurements were also performed solely on a recombinant protein (Marrakchi et al. 2002). Surprisingly, a volume of 0.1 µl of soluble protein extract containing approximately 5 mg/ml protein from mutant cells over-expressing FabG4 gave rise to an NAD(H)-dependent oxidation of the C6 substrate at the rate of 0.21 ± 0.02 (SD, n = 3) µmol/mg protein × min−1 (Table 2). When the C10 substrate was used under the same reaction conditions, the soluble extracts containing FabG4 yielded a rate of 0.25 ± 0.08 (SD, n = 3) µmol/mg protein × min−1 (Table 2). Further examination of FabG4 extracts with NADP+ and C6 resulted in a rate of 3.5 ± 0.3 (SD, n = 3) nmol/mg protein × min−1 (Table 2), indicating that FabG4 might also utilize NADP(H), albeit inefficiently. Hence, heterologous expression of FabG4 in mutant oar1Δ cells resulted in protein extracts containing readily detectable levels of 3-oxoacyl-thioester reductase activity.
FabG1 and FabG4 restore the assembly of cytochrome complexes in the oar1Δ mutant
Yeast cells carrying a deletion at the OAR1 gene have previously been shown to lack assembled cytochrome complexes (Schneider et al. 1997). Hence, rescue of oar1Δ cells from their fermentative phenotype should be associated with re-assembly of these structures. To verify this recovered function by means of a qualitative assay, the yeast strains described in “Yeast oar1Δ cells expressing FabG4 contain 3-oxoacyl-thioester reductase activity” were monitored for cytochrome complex assembly using the cold-temperature cuvette method (Lindenmayer and Estabrook 1958). Cell pastes were collected and applied to cuvettes that were then immersed in liquid nitrogen prior to being placed in a spectrophotometer. The result demonstrated that oar1Δ cells expressing peroxisomal Cta1p were not efficient at assembling the complete retinue of the cytochrome complexes (e.g., complexes a and a3; Fig. 3). On the other hand, the signals representing these complexes were more pronounced in mutant cells expressing native Oar1p or mycobacterial FabG1 and FabG4. Hence, this indicated that the ability of oar1Δ mutants to grow on glycerol was coincidental with the recuperation of cytochrome complex assembly in their mitochondria.

Spectrophotometric assay for mitochondrial cytochrome complexes. Cell pastes from the indicated strains were collected from liquid cultures that were cultivated overnight in oleic acid medium and applied to the glass face of otherwise aluminum cold-temperature cuvettes that were immersed in liquid nitrogen prior to being placed in a spectrophotometer for analysis. The pastes were scanned at wavelengths between 480 and 640 nm. Numbers to the left indicate units of absorbance (ABS), those at the bottom refer to wavelength in nm. Peaks corresponding to known cytochromes are indicated (letters with arrows). The yPLM strains used were 37, 38, 41 and 43
Expression of FabG1 or FabG4 renews lipoic acid production in the oar1Δ mutant
Previous work demonstrated that the C8 octanoic acid product of mitochondrial FASII acts as the starting point for the synthesis of lipoic acid, and that a mutation in any one of the yeast genes encoding mitochondrial FASII enzyme causes a dramatic drop in the levels of lipoic acid (Hiltunen et al. 2005). However, this deficiency can be reversed when such mutants are supplied with known or presumed FASII enzyme activities of the corresponding type. To link FabG1 and FabG4 expression with a rehabilitated FASII system in yeast mitochondria, lipoic acid levels were examined in the four strains using an in vivo growth assay that exploits a lipoic acid-deficient E. coli strain (Brody et al. 1997; Hayden et al. 1993). Extracts prepared from the negative control strain expressing Cta1p supported only a nominal growth level of these E. coli cells, commensurate with 7 ng lipoic acid present per gram wet weight of yeast cells (Table 3). In contrast, yeast mutant cells expressing native Oar1p produced sufficient amounts of lipoic acid to support levels of bacterial growth equivalent to 193 ng of lipoic acid per gram wet weight. Importantly, expression of FabG1 or FabG4 in the yeast mutant resulted in extracts containing the respective lipoic acid levels of 174 and 164 ng of lipoic acid per gram wet weight, which were over 20-fold higher than the levels of the negative control, thereby underscoring further the function of FabG4 as a putative FASII-like 3-oxoacyl-thioester reductase.
Discussion
The identification of a novel 3-oxoacyl-thioester reductase encoded by the essential mycobacterial gene fabG4 is described herein. The report takes into account the findings whereby FabG4 (1) restored the respiratory growth of an otherwise obligatory fermentative yeast mutant devoid of Oar1p; (2) contained high levels of 3-oxoacyl-thioester reductase activity; and (3) replaced Oar1p during lipoic acid synthesis. Future experimentation dedicated to knocking out fabG4 would likely help to elucidate whether FabG4 is in fact vital for M. tuberculosis persistence and virulence. Nevertheless, since FabG4 was practically indistinguishable from the essential mycobacterial 3-oxoacyl-AcpM reductase FabG1 within the parameters of phenotype rescue in yeast, it is attractive to speculate that FabG4 might also be involved in some hitherto elusive aspect of fatty acid biosynthesis in M. tuberculosis. Additional important results to emerge here regarding FabG1 include the refinement of its in vivo substrate specificities (down to C4), as well as exposure of its ability to function in yeast without resorting to protein–protein interactions with other mycobacterial FASII enzymes.
The role of FabG1 has been described previously within the context of mycobacterial FASII, which is outlined in great detail in Takayama et al. (2005). FASII begins with a C20-CoA intermediate emerging from FASI, with additional rounds of C2 extensions being undertaken by FASII so as to arrive at the final length (up to C63) required for incorporation into mature mycolic acids. In a FASII pre-step, 3-oxoacyl-AcpM synthase mtFabH (see below) converts the incoming C20-CoA to a (C22) 3-oxoacyl-AcpM based on the availability of C3 malonyl-AcpM. Further FASII extensions are effected by two additional 3-oxoacyl-AcpM synthases, represented by KasA and KasB, giving rise to a 3-oxoacyl-AcpM intermediate. This metabolite acts as substrate for the aforementioned FabG1, which reduces the latter intermediary to 3-hydroxyacyl-AcpM, with the concomitant oxidation of NADPH. The subsequent two FASII steps include dehydration and reduction (Takayama et al. 2005).
With reference to the non-rescuing FabG homologs, the published data for FabG2, FabG3 and FabG5 remain rather scant. The crystal structure of FabG3/Rv2002 has been determined, but there are no data available on its physiological function or enzyme activity (Yang et al. 2002). Another homolog, FabG5/Rv2766c, was picked up with additional proteins (including InhA and the isoniazid-sensitive dihydrofolate reductase DfrA) in a screen for proteins capable of interacting with a resin mimicking the isoniazid–NAD/P adduct formed with the active species of isoniazid (Argyrou et al. 2006). Interestingly, isoniazid-sensitive FabG1 itself (Banerjee et al. 1998; Ducasse-Cabanot et al. 2004) was not identified in this search. In the aforementioned scan for genes needed for optimal growth (Sassetti et al. 2003), of the five fabG genes only fabG2/Rv1350 was found to be required for this parameter. Moreover, none of them came up in a transposon-based screen for altered virulence (McAdam et al. 2002), or was upregulated in M. tuberculosis treated with isoniazid and other antimicrobials (Betts et al. 2003). Hence, further work will be required to underpin the functions of the remaining FabG homologs. This notwithstanding, it should be noted that since the present study was conducted entirely in a yeast background, the observations presented here may not necessarily apply to mycobacterial biochemistry. In other words, because phenotype rescue could not be achieved with the remaining three homologs, it would be premature to proclaim that they do not in fact play a role in fatty acid biosynthesis.
Returning to the essential requirement of FabG4 for mycobacterial existence, although an M. tuberculosis mutant knockout at fabG4 has been reported previously to be symptomless in the context of survival during infection (Sassetti and Rubin 2003), very important recent studies revealed fabG4 (as well as fabG1, fabD and acpS, all affiliated with FASII) to be essential, at least in terms of growth of M. bovis BCG on Roisin’s medium (Beste et al. 2009). Interestingly, the fabG4 locus (Rv0242c) occurs in close proximity to that of htdX (Rv0241c), whose latter product has been shown recently to act in yeast hdt2Δ cells as a physiological 3-hydroxyacyl-thioester dehydratase (Gurvitz et al. 2009). This represents the enzyme activity following FabG in the fatty acid biosynthesis scheme, and the two genes are predicted to occur in a single operon (operonDB; confidence = 84; n = 57). Since similar pair arrangements have been observed in M. tuberculosis with other genes of mycolic acid biosynthesis, including Rv1484/inhA and Rv1483/fabG1 (Takayama et al. 2005), it is at least plausible that HtdX and FabG4 might also function within a shared FASII-like pathway.
Regarding the issue of whether FabG1 is essential for pathogen survival, this has only recently been resolved. A study dedicated to addressing this specific issue revealed that knocking out the gene for FabG1 in the pathogen was only possible if an additional plasmid-borne gene copy was made available, a procedure formally accepted as genetic proof for the essentiality of this gene (Parish et al. 2007). Although the requirement for FabG1 in M. tuberculosis could be compensated by using the homologous protein from M. smegmatis, functional complementation of the mycobacterial mutant devoid of its native FabG1 could not be achieved using an ectopically expressed E. coli FabG. It was argued that failure to complement the mutant phenotype using the bacterial protein was likely due to structural differences between the bacterial and mycobacterial proteins (Parish et al. 2007), in that the former is not able to accommodate long-chain fatty acids or undertake the suggested specific protein–protein interactions necessary for FASII (Veyron-Churlet et al. 2004, 2005).
Hence, the finding here that FabG1 could alleviate the growth phenotype of oar1Δ mutant cells was remarkable in light of the conclusions drawn regarding the central position of FabG1 in the complex presumed to exist within the structure of mycobacterial FASII (Veyron-Churlet et al. 2004). Using the yeast two-hybrid approach, native FabG1 was shown to interact only very weakly with the aforementioned FabH, albeit subsequent co-immunoprecipitation failed to confirm this putative interaction altogether. Nevertheless, mutant versions of FabG1 were constructed so that they failed to form the native FabG1 multimer; these displayed more extensive protein–protein interactions, namely with the previously mentioned KasA, KasB, and FabH. Based on the results with these mutant FabG1 monomers, the authors concluded that the wild-type FabG1 monomer must also interact with each of the aforementioned condensing enzymes in the native state, and that the high propensity of these monomers to multimerize helps maintain the macromolecular organization of FASII. Since expression of these FabG1 mutants in M. tuberculosis appeared to block FASII, presumably by forming unproductive FabG1 multimers, this led to the suggestion that the quaternary structure of FabG1 is critical for a functional FASII complex (Veyron-Churlet et al. 2004). The conclusion that FabG1 represents a pivotal interacting protein that is vital for maintaining FASII complexes in M. tuberculosis is difficult to reconcile with the observations made here, whereby FabG1 could function in yeast FASII to 90% efficiency (Table 3) in an environment within which it has clearly not evolved to form highly specific protein–protein interactions.
A further point of contention regarding FabG1 is its physiological substrate specificity. Previous work on recombinant FabG1 showed that it was active in vitro using C4 acetoacetyl-CoA as substrate (Banerjee et al. 1998; Ducasse-Cabanot et al. 2004). However, other studies found that FabG1 preferentially acts on long-chain C8–20 CoA substrates, and that it metabolizes the C4 substrate only poorly (Marrakchi et al. 2002). Unfortunately, even following extensive over-expression in cells grown on oleic acid medium, the enzyme assay used here failed to detect any reductase activity for FabG1 using C6 or C10 substrates. Nevertheless, it can still be concluded that, at least within an in vivo yeast context, FabG1 was almost as efficient as native Oar1p in metabolizing short-chain C4–8 substrates, despite fabG1 being expressed at only modest levels commensurate with glucose derepression. Herein lays one of the major advantages of using yeast for examining the in vivo efficacy of enzymes for substrates that are otherwise not metabolized facilely in vitro. It is predicted that yeast FASII mutants will continue to serve as a convenient tool for identifying novel pathogen proteins.
Abbreviations
- ACP:
-
Acyl carrier protein
- AcpM:
-
Mycobacterial ACP
- BCG:
-
Bacillus Calmette-Guérin
- FAS:
-
Fatty acid synthase
- FASI:
-
Type 1 associative FAS
- FASII:
-
Type 2 dissociative FAS
- MLS:
-
Mitochondrial leader sequence
- ORF:
-
Open reading frame
- PCR:
-
Polymerase chain reaction
- MFE2:
-
Peroxisomal type 2 multifunctional enzyme
References
Argyrou A, Jin L, Siconilfi-Baez L et al (2006) Proteome-wide profiling of isoniazid targets in Mycobacterium tuberculosis. Biochemistry 45:13947–13953
Ausubel FM, Brent R, Kingston RE et al (1989) Current protocols in molecular biology. Wiley, NY
Banerjee A, Dubnau E, Quémard A et al (1994) inhA, a gene encoding a target for isoniazid and ethionamide in Mycobacterium tuberculosis. Science 263:227–230
Banerjee A, Sugantino M, Sacchettini JC et al (1998) The mabA gene from the inhA operon of Mycobacterium tuberculosis encodes a 3-ketoacyl reductase that fails to confer isoniazid resistance. Microbiology 144:2697–2704
Beste DJV, Espasa M, Bonde B et al (2009) The genetic requirements for fast and slow growth in mycobacteria. PLoS One 4:e5349
Betts JC, McLaren A, Lennon MG et al (2003) Signature gene expression profiles discriminate between isoniazid-, thiolactomycin-, and triclosan-treated Mycobacterium tuberculosis. Antimicrob Agents Chemother 47:2903–2913
Bloch K (1977) Control mechanisms for fatty acid synthesis in Mycobacterium smegmatis. Adv Enzymol 45:1–84
Bradford MM (1976) A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein–dye binding. Anal Biochem 72:248–254
Brody S, Oh C, Hoja U et al (1997) Mitochondrial acyl carrier protein is involved in lipoic acid synthesis in Saccharomyces cerevisiae. FEBS Lett 408:217–220
Chen D-C, Yang B-C, Kuo T-T (1992) One-step transformation of yeast in stationary phase. Curr Genet 21:83–84
Chen Z-J, Kastaniotis AJ, Miinalainen IJ et al. (2009) 17β-Hydroxysteroid dehydrogenase type 8 and carbonyl reductase type 4 assemble as a ketoacyl reductase of human mitochondrial FAS. FASEB J. doi:10.1096/fj.09-133587
Cole ST, Brosch R, Parkhill J et al (1998) Deciphering the biology of Mycobacterium tuberculosis from the complete genome sequence. Nature 393:537–544
Dommes V, Kunau W-H (1984) 2,4-Dienoyl coenzyme A reductases from bovine liver and Escherichia coli. Comparison of properties. J Biol Chem 259:1781–1788
Ducasse-Cabanot S, Cohen-Gonsaud M, Marrakchi H et al (2004) In vitro inhibition of the Mycobacterium tuberculosis β-ketoacyl-acyl carrier protein reductase MabA by isoniazid. Antimicrob Agents Chemother 48:242–249
Filipits M, Simon MM, Rapatz W et al (1993) A Saccharomyces cerevisiae upstream activating sequence mediates induction of peroxisome proliferation by fatty acids. Gene 132:49–55
Filppula SA, Sormunen RT, Hartig A et al (1995) Changing stereochemistry for a metabolic pathway in vivo. Experiments with the peroxisomal β-oxidation in yeast. J Biol Chem 270:27453–27457
Gerum AB, Ulmer JE, Jacobus DP et al (2002) Novel Saccharomyces cerevisiae screen identifies WR99210 analogues that inhibit Mycobacterium tuberculosis dihydrofolate reductase. Antimicrob Agents Chemother 46:3362–3369
Goldman P, Vagelos PR (1961) The specificity of triglyceride synthesis from diglycerides in chicken adipose tissue. J Biol Chem 236:2620–2623
Gurvitz A (2009) Caenorhabditis elegans F09E10.3 encodes a 3-oxoacyl-thioester reductase of mitochondrial type 2 fatty acid synthase FASII that is functional in yeast. J Biomed Biotechnol. http://www.hindawi.com/journals/jbb/aip.235868.html
Gurvitz A, Rottensteiner H, Kilpeläinen SH et al (1997) The Saccharomyces cerevisiae peroxisomal 2,4-dienoyl-CoA reductase is encoded by the oleate-inducible gene SPS19. J Biol Chem 272:22140–22147
Gurvitz A, Hiltunen JK, Kastaniotis AJ (2008a) Identification of a novel mycobacterial 3-hydroxyacyl-thioester dehydratase HtdZ (Rv0130) by functional complementation in yeast. J Bacteriol 190:4088–4090
Gurvitz A, Hiltunen JK, Kastaniotis AJ (2008b) Function of heterologous Mycobacterium tuberculosis InhA, a type 2 fatty acid synthase enzyme involved in extending C20 fatty acids to C60-to-C90 mycolic acids, during de novo lipoic acid synthesis in Saccharomyces cerevisiae. Appl Environ Microbiol 74:5078–5085
Gurvitz A, Hiltunen JK, Kastaniotis AJ (2009) Heterologous expression of mycobacterial proteins in yeast reveals two physiologically functional 3-hydroxyacyl-thioester dehydratases, HtdX and HtdY, in addition to HadABC and HtdZ. J Bacteriol 191:2683–2690
Hayden MA, Huang IY, Iliopoulos G et al (1993) Biosynthesis of lipoic acid: characterization of the lipoic acid auxotrophs Escherichia coli W1485-lip2 and JRG33-lip9. Biochemistry 32:3778–3782
Hill JE, Myers AM, Koerner TJ et al (1986) Yeast/E. coli shuttle vectors with multiple unique restriction sites. Yeast 2:163–167
Hiltunen JK, Okubo F, Kursu VA et al (2005) Mitochondrial fatty acid synthesis and maintenance of respiratory competent mitochondria in yeast. Biochem Soc Trans 33:1162–1165
Hsu AY, Poon WW, Shepherd JA et al (1996) Complementation of coq3 mutant yeast by mitochondrial targeting of the Escherichia coli UbiG polypeptide: evidence that UbiG catalyzes both o-methylation steps in ubiquinone biosynthesis. Biochemistry 35:9797–9806
Lindenmayer A, Estabrook RW (1958) Low-temperature spectral studies on the biosynthesis of cytochromes in baker’s yeast. Arch Biochem Biophys 78:66–82
Marrakchi H, Ducasse S, Labess G et al (2002) MabA (FabG1), a Mycobacterium tuberculosis protein involved in the long-chain fatty acid elongation system FAS-II. Microbiology 148:951–960
McAdam RA, Quan S, Smith DA et al (2002) Characterization of a Mycobacterium tuberculosis H37Rv transposon library reveals insertions in 351 ORFs and mutants with altered virulence. Microbiology 148:2975–2986
Parish T, Roberts G, Laval F et al (2007) Functional complementation of the essential gene fabG1 of Mycobacterium tuberculosis by Mycobacterium smegmatis fabG but not Escherichia coli fabG. J Bacteriol 189:3721–3728
Rose MD, Winston F, Heiter P (1990) Methods in yeast genetics: a laboratory manual. Cold Spring Harbor Laboratory Press, Cold Spring Harbor
Rottensteiner H, Kal AJ, Filipits M et al (1996) Pip2p: a transcriptional regulator of peroxisome proliferation in the yeast Saccharomyces cerevisiae. EMBO J 15:2924–2934
Sambrook J, Fritsch EF, Maniatis T (1989) Molecular cloning: a laboratory manual. Cold Spring Harbor Laboratory, Cold Spring Harbor
Sassetti CM, Rubin EJ (2003) Genetic requirements for mycobacterial survival during infection. Proc Natl Acad Sci USA 100:12989–12994
Sassetti CM, Boyd DH, Rubin EJ (2003) Genes required for mycobacterial growth defined by high density mutagenesis. Mol Microbiol 48:77–84
Schneider R, Brors B, Burger F et al (1997) Two genes of the putative mitochondrial fatty acid synthase in the genome of Saccharomyces cerevisiae. Curr Genet 32:384–388
Takayama K, Wang C, Besra GS (2005) Pathway to synthesis and processing of mycolic acids in Mycobacterium tuberculosis. Clin Microbiol Rev 18:81–101
Torkko JM, Koivuranta KT, Miinalainen IJ et al (2001) Candida tropicalis Etr1p and Saccharomyces cerevisiae Ybr026p (Mrf1’p), 2-enoyl thioester reductases essential for mitochondrial respiratory competence. Mol Cell Biol 21:6243–6253
Veyron-Churlet R, Guerrini O, Mourey L et al (2004) Protein–protein interactions within the fatty acid synthase-II system of Mycobacterium tuberculosis are essential for mycobacterial viability. Mol Microbiol 54:1161–1172
Veyron-Churlet R, Bigot S, Guerrini O et al (2005) The biosynthesis of mycolic acids in Mycobacterium tuberculosis relies on multiple specialized elongation complexes interconnected by specific protein–protein interactions. J Mol Biol 353:847–858
World Health Organisation (1997) Anti-tuberculosis drug resistance in the world: the WHO/IUATLD global project on anti-tuberculosis drug resistance surveillance. WHO Global Tuberculosis Programme, Geneva
Yang JK, Yoon HJ, Ahn HJ et al (2002) Crystallization and preliminary X-ray crystallographic analysis of the Rv2002 gene product from Mycobacterium tuberculosis, a β-ketoacyl carrier protein reductase homologue. Acta Crystallogr D Biol Crystallogr 58:303–305
Acknowledgments
This work is dedicated to J. Kalervo Hiltunen, on the occasion of his 60th birthday. Johanna Mäkinen from the Mycobacterial Reference Laboratory at the National Public Health Institute in Turku, Finland, is thanked for providing M. tuberculosis H37Rv genomic DNA. J. Kalervo Hiltunen was instrumental for the enzyme assays and supported this work in a multitude of ways, Alexander J. Kastaniotis undertook the lipoic acid measurements and gave invaluable advice throughout this study, Zhi-Jun Chen is thanked for furnishing plasmid pYE352:OAR1, and Tatu Haataja for donating the hydratase 2 domain of D. melanogaster MFE2 (all from the University of Oulu, Finland). This work was funded by the grant P20764-B03 from the Austrian Science Fund (FWF).
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Author information
Authors and Affiliations
Corresponding author
Additional information
Communicated by G. Klug.
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License (https://creativecommons.org/licenses/by-nc/2.0), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
Gurvitz, A. The essential mycobacterial genes, fabG1 and fabG4, encode 3-oxoacyl-thioester reductases that are functional in yeast mitochondrial fatty acid synthase type 2. Mol Genet Genomics 282, 407–416 (2009). https://doi.org/10.1007/s00438-009-0474-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00438-009-0474-2