
Abstract
RNA extraction and construction of complementary DNA (cDNA) library for mites have been quite challenging due to difficulties in acquiring tiny living mites and breaking their hard chitin. The present study is to explore a better method to construct cDNA library for mites that will lay the foundation on transcriptome and molecular pathogenesis research. We selected Psoroptes cuniculi as an experimental subject and took the following steps to construct and verify cDNA library. First, we combined liquid nitrogen grinding with TRIzol for total RNA extraction. Then, switching mechanism at 5′ end of the RNA transcript (SMART) technique was used to construct full-length cDNA library. To evaluate the quality of cDNA library, the library titer and recombination rate were calculated. The reliability of cDNA library was detected by sequencing and analyzing positive clones and genes amplified by specific primers. The results showed that the RNA concentration was 836 ng/μl and the absorbance ratio at 260/280 nm was 1.82. The library titer was 5.31 × 105 plaque-forming unit (PFU)/ml and the recombination rate was 98.21 %, indicating that the library was of good quality. In the 33 expressed sequence tags (ESTs) of P. cuniculi, two clones of 1656 and 1658 bp were almost identical with only three variable sites detected, which had an identity of 99.63 % with that of Psoroptes ovis, indicating that the cDNA library was reliable. Further detection by specific primers demonstrated that the 553-bp Pso c II gene sequences of P. cuniculi had an identity of 98.56 % with those of P. ovis, confirming that the cDNA library was not only reliable but also feasible.
Similar content being viewed by others


Introduction
Mites, belonging to Arachnid, Acari, are distributed widely in the natural world with 50,000 known species. They may affect human and animal health and hence have become an important public health problem. However, different mite species can cause different diseases through different pathogenic mechanisms. Constructing a complementary DNA (cDNA) library is an effective approach to study the function genes that are pathogenic related. Nevertheless, the study of pathogenic mechanism of mites has not been well established due to the following difficulties in cDNA library construction. First, obtaining enough mites for RNA extraction is challenging as mites are tiny and culturing them in vitro has not succeeded for most mites, especially for parasitic mites. The second difficulty is to extract RNA from mites, due to the mites’ hard chitin. Last but not the least, obtaining enough RNA satisfying molecular biology research is not easy as RNase (an enzyme degrading the RNA) is very common in the environment.
Up to now, although there are some English-language papers on cDNA library construction and function genes in Acari, most are about ticks with a larger body (Oberlander et al. 1995; Gao et al. 2008; Heekin et al. 2013; Lees et al. 2014; Radulović et al. 2014; Kotsyfakis et al. 2015), and only a few are about mites with a tiny body. In mites, the only four English-language papers we found had several drawbacks (Lee et al. 1999; Fischer et al. 2003; Kenyon et al. 2003; Ljunggren et al. 2003). First, only Psoroptes ovis and Sarcoptes scabiei mites were studied. Second, the mites used for RNA extraction were up to 250–700 mg, which makes it difficult to replicate the study for other species of mites due to the large extraction amount. Last, there is room for improvement on the recombination rate of cDNA library. Among Chinese-language papers on cDNA library construction of mites, only PCR amplification and agarose gel electrophoresis were used for primary identification of cDNA library. As a result, the reliability of the cDNA library was impaired due to the lack of sequencing of positive clones (Liu et al. 2004; Liu et al. 2008; Xiao et al. 2012; Zhang et al. 2013; Bian et al. 2014; Yu et al. 2014). Therefore, it is necessary to explore a method to construct and detect a full-length cDNA library of mites. This will lay the foundation for future studies of function genes that are pathogenic and control related.
In this study, we constructed a full-length cDNA library of Psoroptes cuniculi as a representative using switching mechanism at 5′ end of the RNA transcript (SMART) technique. The quality of the library was primarily assessed by the library titer, the recombination rate, and the lengths of expressed sequence tags (ESTs) in electrophoresis. Then, the reliability of the cDNA library was detected by dual methods to confirm whether the inserted cDNA fragment belonged to P. cuniculi. One is to sequence the ESTs longer than 500 bp in electrophoresis and conduct the homology searches using the BLAST programs on the NCBI network. Further, a function gene was amplified by specific primers from the P. cuniculi cDNA library and then was sequenced and analyzed. This study is aimed to provide a reference for constructing and detecting a cDNA library for other mite species.
Materials and methods
Mite collection and RNA extraction
The pre-experiment of cDNA library construction using Demodex caprae being preserved at −80 °C for more than 1 year failed; hence, living P. cuniculi mites were used. P. cuniculi were collected from naturally infected rabbits from Xi’an, China. Crusts were taken out from the external auditory canal of the rabbits by a sterile forceps and live mites were isolated under 37 °C in thermostat. After being identified as P. cuniculi under a microscope, 68 mg mites were collected into a 1.5-ml EP tube. Mites were then washed with 5 % phosphate-buffered saline (PBS; pH = 7.4), 1 % SDS, and 5 % PBS (pH = 7.4) once, respectively, and assembled by centrifugation. The mites had been ground into powder by liquid nitrogen before total RNA was extracted by TRIzol (Roche, Swiss). The concentration and purity of RNA were measured by an ultraviolet spectrophotometer. The completeness of RNA was estimated by 2 % agarose gel electrophoresis. The RNA sample was then digested by DNase I (Takara, Japan) to eliminate potential genomic DNA (gDNA).
Single-strand and double-strand cDNA synthesis
First, RNA was reverse transcribed to single-strand cDNA (ss cDNA) using reverse transcriptase M-MLV (Takara, Japan) with primers P1 (SMART IV) and P2 (CDS/3′ PCR). Second, double-strand cDNA (ds cDNA) was synthesized by LD-PCR (Takara, Japan) with primers P3 (5′ PCR anchor) and P2 (CDS/3′ PCR) (Table 1). Finally, the PCR products of ds cDNA were separated by 2 % agarose gel electrophoresis, and those longer than 500 bp were isolated and purified by a gel extraction kit (Omega, USA).
Double-strand cDNA cloning
The purified ds cDNA was linked with pMD-19T vector (Takara, Japan), and 2 μl recombination plasmid was transformed into 20 μl Escherichia coli DH-5α. Seventy-eight microliters of LB liquid medium (Amp +) was added to make the total volume of 100 μl, which was then smeared evenly on the blue and white Luria broth (LB) medium Amp (+) (10 cm in diameter). After the medium was incubated at 37 °C overnight for 16–18 h, the colonies were observed.
cDNA library quality assessment
The library titer and recombination rate were calculated as follows: library titer = (number of colonies × dilution factor × 103 μl/ml) / (μl of diluted volume plated), recombination rate = number of white colonies / (number of white colonies + number of blue colonies). Positive clones (white colonies) were selected randomly by a sterile tip for PCR amplification using the universal primers for pMD-19T vector (Table 1). The reaction volume was 12.5 μl and the reaction parameters were as follows: initial denaturation at 94 °C for 5 min, followed by 35 cycles of denaturation at 94 °C for 30 s, annealing at 63 °C for 30 s, extension at 72 °C for 1 min, and final extension at 72 °C for 10 min. The lengths of inserted fragments were estimated by electrophoretic separation using 2 % agarose gel.
Sequencing and function prediction of positive clones
Positive clones longer than 500 bp in electrophoresis were sequenced by Suzhou GENEWIZ Biotechnology Co. Ltd., China. The length of expressed sequence tag (EST) was calculated as the length of the band—216 bp (59 bp SMART IV + 157 bp pMD-19T vector). To verify whether the ESTs belonged to P. cuniculi, homology searches were conducted using the BLASTn and BLASTx programs on the NCBI network. The open reading frames (ORFs) were found by ORF finder. The sequences of Psoroptes were aligned by Clustal X1.8 and the sequence identity was computed. Neighbor-joining (NJ) tree across Psoroptidia based on known ESTs was reconstructed by MEGA 5.0. The tree confidence was assessed by 1000 bootstraps.
PCR amplification and detection by specific primers
In order to further verify the reliability of the library, specific primers were designed according to the two allergen II sequences of P. ovis from GenBank (two forward primers were designed as there were several different bases at the 5′ terminal of the two sequences) (Table 1) for PCR amplification using the bacteria liquid of P. cuniculi cDNA library as template. The sequences of P. cuniculi and P. ovis were aligned by Clustal X1.8, and the sequence identity was computed. NJ tree across Psoroptidia was reconstructed by MEGA 5.0. The tree confidence was assessed by 1000 bootstraps.
Results
RNA quality
The quality of the obtained RNA is good. The concentration of total RNA was 836 ng/μl. The OD260/280 and OD260/OD230 were 1.82 and 2.08, respectively, indicating high purification. Figure 1 shows three bands of 28S, 18S, and 5S with the 28S significantly lighter than 18S, indicating good integrity of RNA.

Electrophoretogram of total RNA of P. cuniculi. Lane M DL5000 DNA marker, lane 1 total RNA
cDNA library quality
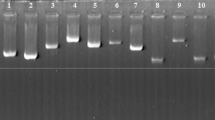
There were 1062 bacterial colonies in the plate of cDNA library with a recombination rate of 98.21 % (1043/1062). The library titer was 5.31 × 105 plaque-forming unit (PFU)/ml (1062/2 × 10−3). Thirty-eight randomly selected clones were identified to be longer than 500 bp in the electrophoresis, including 4 clones of 1500–2000 bp, 8 clones of 1000–1500 bp, and 26 clones of 500–1000 bp (Fig. 2). The cDNA library was preliminarily proved to be successfully constructed.

PCR products of positive clones of the P. cuniculi cDNA library. Lane M DL5000 DNA marker, lanes 1–38 positive clones, PC positive control of 500 bp, NC negative control
Sequencing and function prediction
The 38 clones identified above were sequenced. Two sequences were identified as ribosomal genes of mite and three were identified as genome of bacteria. The other 33 clones were identified as ESTs of P. cuniculi which was made up of 6.06 % (2/33) of known genes, 18.18 % (6/33) of putative genes, and 75.76 % (25/33) of novel genes. The lengths of ESTs ranged from 319 to 1658 bp (average 711 bp).
Table 2 presented the 13 sequences with known characteristics, of which 8 sequences (clones 1–8) were ESTs of P. cuniculi. The protein function predicted by BLASTn and BLASTx was the same, demonstrating that the blast results were reliable. The clones 1 and 4 (1656 and 1658 bp, respectively) were almost identical with only three variable sites detected. The total score of BLASTn for clones 1 and 4 was high with SRP54 of P. ovis (2471 and 2453, respectively), and the nucleotide identified was as high as 99.63 % (5/1350) (Fig. 3), indicating that both clones 1 and 4 should be SRP54, and P. cuniculi and P. ovis should be of the same species. The phylogenetic tree based on SRP54 sequences also showed that P. cuniculi clustered with P. ovis first, and then gathered with Chorioptes and Otodectes as a clade of Psoroptidae, and finally gathered with Turbinoptidae, Pterolichidae, and Pyroglyphidae as Psoroptidia (Fig. 4). This was in accordance with morphological classification. Therefore, the constructed cDNA library of P. cuniculi was reliable.

Alignments of three SRP54 sequences of Psoroptes

Radiative NJ phylogenetic trees across Psoroptidia inferred from SRP54. P Psoroptes, Ch Chorioptes, O Otodectes, A Aniacarus, Co Congocoptes, D Dermatophagoides, H Hirstia, S Sturnophagoides, E Euroglyphus, G Gymnoglyphus
Detection by specific primers
The Pso c II gene fragments of P. cuniculi were successfully amplified by the two pairs of primers (Fig. 5) and sequenced to be 553 bp (JZ875784, JZ875785). The sequence identified of P. cuniculi and P. ovis (BQ834604, AF187083) was 98.56 % (8/554) (Fig. 6), indicating that Psoroptes mites parasitizing in rabbit and sheep were of the same species. The phylogenetic tree also showed that P. cuniculi and P. ovis gathered as a clade between Pyroglyphidae and Acaridae (Fig. 7). Hence, the cDNA library can satisfy the need for amplification.

PCR products of Pso c II of P. cuniculi. Lane M 100 bp DNA ladder marker, lane 1 Pso c II (F1), lane 2 Pso c II (F2), NC negative control

Alignments of four allergen II sequences of Psoroptes

Radiative NJ phylogenetic trees across Psoroptidia inferred from allergen II. P Psoroptes, G Gymnoglyphus, T Tyrophagus, L Lepidoglyphus, E Euroglyphus, D Dermatophagoides
Discussion
At present, the traditional cDNA library and the full-length cDNA library are the two main types of cDNA library. The full-length cDNA library could satisfy the requirement for large-scale, high throughput, and highly efficient studies of functional genomics due to its advantages over traditional cDNA library, including long-fragment cloning, high proportion of full-length cDNAs, less invalid cloning, etc. The construction of cDNA library becomes more appealing after the introduction of a new and more efficient method, namely SMART (Chenchik et al. 1996). It has been widely used in microbial pathogens (anisopliae, monascus, etc.) (Zhang et al. 2007; Zhu et al. 2009) and parasites (roundworm, tapeworms, Giardia lamblia, and others) (Wei et al. 2007; Zhang et al. 2009; Guo et al. 2012; Zhou et al. 2011). However, it is rarely used in medical mites because of the difficulties in mite collection and RNA extraction.
In this study, we successfully constructed the full-length cDNA library of P. cuniculi by optimizing experimental conditions, as well as improving experimental design and operation. P. cuniculi was selected as the study object to facilitate comparison with the same species, P. ovis, whose cDNA library has been constructed. The mites were ground into powder by liquid nitrogen and RNA was extracted with TRIzol to ensure complete lysis of tissue and cells. SMART technique was selected to construct a full-length library. Instead of phage vector TriplEx2 and expression vector ZAP, plasmid vector pMD-19T was used to increase the recombination rate and sequencing convenience. As a result, the quality of RNA satisfied the requirement for cDNA library construction. The concentration of RNA was 836 ng/μl, and the absorbance ratios of the RNA at 260/280 and 230/260 nm were 1.82 and 2.08, respectively. The mites needed for RNA extraction significantly reduced from 250 to 700 mg to 68 mg. The library quality was significantly improved: the library titer increased from 2.80 × 105 PFU/ml (Lee et al. 1999) to 5.31 × 105 PFU/ml, and the recombination rate increased from 76 to 89 % (Lee et al. 1999; Kenyon et al. 2003) to 98.21 %, consistent with the quality standard proposed by Clontech company (USA) where primary library titer ≥5 × 105 PFU/ml and recombination rate ≥90 %. In this way, the mites used for RNA extraction were decreased and the quality of cDNA library was improved significantly.
It is important to detect the reliability of the library with two independent methods conducted. In this study, first, the clone 1 and 4 sequences of P. cuniculi showed 99.63 % identity with the SRP54 gene sequence of P. ovis, which not only confirmed that both clones were SRP54 gene but also indicated that P. cuniculi and P. ovis were of the same species, in line with the result obtained by Pegler based on ribosomal ITS2 (Pegler et al. 2005). Second, the bacteria liquid of P. cuniculi cDNA library was used as a template for PCR amplification using specific primers designed according to allergen II gene sequences of P. ovis. These sequences of P. cuniculi and P. ovis showed 98.56 % identity, further indicating that the constructed cDNA library was reliable.
It is a common practice among Chinese scholars to assess the cDNA library only by PCR amplification and electrophoresis of positive clones (Liu et al. 2004; Liu et al. 2008; Tao et al. 2011; Xiao et al. 2012; Zhang et al. 2013; Bian et al. 2014; Hu et al. 2014; Yu et al. 2014). However, that simple assessment is hardly satisfactory as the electrophoretic bands could suggest only the length of the inserted fragments, but not their nature. The inserted fragments might be the cDNA/gDNA of the studied mites, the gDNA of bacteria carried by the studied mites, or even the gDNA/cDNA of the host. In this study, according to the blast results of the 38 clones which were longer than 500 bp identified by electrophoresis (Table 2), clones 9–10 were highly homologous with ribosomal ITS1-5.8S gene of P. cuniculi and ribosomal 5.8S-ITS2-28S gene of Dermatophagoides pteronyssinus, respectively, while clones 11–13 showed 74, 99, and 90 % identities with Burkholderia pseudomallei, E. coli, and Serratia marcescens, respectively. Therefore, we suggested that sequencing long fragments (usually ≥500 bp) and/or PCR amplification with specific primers should be applied to confirm if the cDNA library was successfully constructed.
It should be noted that the actual length of ESTs is usually shorter than the bands in electrophoresis (≥500 bp). In this study, the length of ETSs is equal to the length of the band minus 216 bp (SMART IV + pMD-19T vector). Therefore, the length of the shortest EST is 319 bp.
High-quality RNA is an important prerequisite of cDNA library construction. To extract high-quality RNA, you should make sure of the following issues: First, to guarantee the completeness of RNA, use living mites for RNA extraction because RNA would degrade rapidly in dead mites. Second, to ensure the RNA releases completely, use liquid nitrogen grinding for complete homogenate. Third, to avoid RNase contamination, execute the operation skillfully and quickly under a clean, unpolluted environment. Last, to avoid DNA or protein residual, suck up RNA from the upper water phase accurately and then treat RNA with DNase. In our pre-experiment of cDNA library construction using D. caprae, the reason for failure was probably that the mites had been dead and RNA was degraded as they were cryopreserved for more than a year.
In the study, the successful attempt to construct a full-length cDNA library using P. cuniculi will provide a valuable experience for cDNA library construction of other mite species. The six function genes obtained play important roles in protein synthesis, processing and metabolism, immune evasion, as well as growth and development. Those genes are essential for protein expression, pathogenic mechanism study, and preventive vaccine development.
References
Bian YH, Zhou Y, Yu LL, Sun JX, Yang L, Teng FX, Cui YB (2014) Construction and primary identification of a full-length cDNA library for Dermatophagoides farinae. J Pathog Biol 9:546–551
Chenchik A, Moqadam F, Siebert P (1996) RNA: isolation, analysis and synthesis. Wiley-Liss, New York, pp 273–321
Fischer K, Holt DC, Haruma P, Currie BJ, Walton SF, Kem DJ (2003) Generation and characterization of cDNA clones from Sarcoptes scabiei var. hominis for an expressed sequence tag library: identification of homologues of house dust mite allergens. Am J Trop Med Hyg 68:61–64
Gao JL, Luo JX, Fan RQ, Fingerle V, Guan GQ, Liu ZJ, Li YQ, Zhao HP, Ma ML, Liu JL, Liu AH, Ren QY, Dang ZS, Sugimoto C, Yin H (2008) Cloning and characterization of a cDNA clone encoding calreticulin from Haemaphysalis qinghaiensis (Acari: Ixodidae). Parasitol Res 102:737–746
Guo JL, Jiang J, Zheng WY, Li ML, Tian XF, Feng XM, Wang YH, Ju XH, Kong YQ (2012) Construction and characterization of a full-length cDNA library from non-fresh Giardia lamblia. Asian Pac J Tropl Med 5:931–934
Heekin AM, Guerrero FD, Bendele KG, Saldivar L, Scoles GA, Dowd SE, Gondro C, Nene V, Djikeng A, Brayton KA (2013) Gut transcriptome of replete adult female cattle ticks, Rhipicephalus (Boophilus) microplus, feeding upon a Babesia bovis-infected bovine host. Parasitol Res 112:3075–3090
Hu PF, Li XC, Liu HK, Guan WJ, Ma YH (2014) Construction and characterization of a cDNA expression library from the endangered Hu sheep. Genet Mol Res 13:9019–9023
Kenyon F, Welsh M, Parkinson J, Whitton C, Blaxter ML, Knox DP (2003) Expressed sequence tag survey of gene expression in the scab mite Psoroptes ovis—allergens, proteases and free-radical scavengers. Parasitology 126:451–460
Kotsyfakis M, Kopáček Petr, Franta Zdeněk, Pedra JHF, Ribeiro JMC (2015) Deep sequencing analysis of the Ixodes ricinus haemocytome. PLOS Neglect Trop D. doi:10.1371/journal.pntd.0003754
Lee AJ, Elwyn Isaac R, Coates D (1999) The construction of a cDNA expression library for the sheep scab mite Psoroptes ovis. Vet Parasitol 83:241–252
Lees K, Jones AK, Matsuda K, Akamatsu M, Sattelle DB, Woods DJ, Bowman AS (2014) Functional characterisation of a nicotinic acetylcholine receptor α subunit from the brown dog tick, Rhipicephalus sanguineus. Int J Parasitol 44:75–81
Liu L, Peng JL, Zhou Y, Cui YB (2008) Construction and primary identification of cDNA expression library for Dermatophagoides pteronyssinus. J Fourth Mil Med Univ 29:143–146
Liu ZG, Zhou ZW, Gao B, Luo SW, Chen SZ, Zhang ZS (2004) DNA library construction for Dermatophagoides farinae. Chinese J Zoonoses 20:923–925
Ljunggren E, Nilsson D, Mattsson JG (2003) Expressed sequence tag analysis of Sarcoptes scabiei. Parasitology 127:139–145
Oberlander U, Adam R, Berg K, Seeber F, Lucius R (1995) Molecular cloning and characterization of the filarial LIM domain proteins AvL3-1 and OvL3-1. Exp Parasitol 81:592–599
Pegler KR, Evans L, Stevens JR, Wall R (2005) Morphological and molecular comparison of host-derived populations of parasitic Psoroptes mites. Med Vet Entomol 19:392–403
Radulović ŽM, Kim TK, Porter LM, Sze SH, Lewis L, Mulenga A (2014) A 24–48 h fed Amblyomma americanum tick saliva immuno-proteome. BMC Genomics 15:518
Tao KE, Dong CH, Mao H, Zhao YZ, Liu HY, Liu SY (2011) Construction of a normalized full-length cDNA library of sesame developing seed by DSN and SMART™. Hort Environ Biotechnol 10:1004–1009
Wei DX, Deng Y, Wu SQ, Lin RQ, Song HQ, Zhu XQ (2007) Construction of a cDNA library for female Ascaris suum using SMART technique Chinese. J Prev Vet Med 29:180–184
Xiao KY, Guo SL, Liu YR, Su L, Feng YX, Yuan FS (2012) Construction and identification of the cDNA library for Demodex. J Shandong Univ (Health Sci) 50:15–19
Yu LL, Zhou Y, Sun JX, Yang L, Bian YH, Teng FX, Cui YB (2014) Construction and primary identification of the cDNA expression library for Tyrophagus putrescentiae. Chinese J Immunol 30:1227–1229
Zhang DW, Zeng YL, Wei FL, Zhou DG (2013) Construction and identification of the cDNA library for Tetranychus urticae. Guangdong Agr Sci 40:149–151
Zhang H, Zhao WJ, Han ML, Wang XH, Bo XW (2009) Construction of a full-length cDNA library for adult Moniezia expansa using SMART technique. China Anim Husb Vet Med 36:51–55
Zhang SZ, Wang ZK, Peng GX, Cao YQ, Yin YP, Xie L, Liu J, Xia YX (2007) Construction of a normalized full-length cDNA library of Metarhizium anisopliae var. acridum in the sporulation stage by DSN and SMART™. J Agr Biotechnol 15:884–887
Zhou RQ, Xia QY, Huang HC, Lai M, Wang ZX (2011) Construction of a cDNA library from female adult of Toxocara canis, and analysis of EST and immune-related genes expressions. Exp Parasitol 129:120–126
Zhu BY, Gao L, Huang X, Li HM (2009) Construction of a full-length cDNA library for monascus using SMART technique. J Guangdong Pharm Coll 25:310–313
Acknowledgments
This work was supported by the National Natural Science Foundation of China (No. 81271856; No. 81471972).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Hu, L., Zhao, Y., Cheng, J. et al. Constructing and detecting a cDNA library for mites. Parasitol Res 114, 3893–3901 (2015). https://doi.org/10.1007/s00436-015-4621-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00436-015-4621-x