
Abstract
In the present study, we isolated three populations of Myxobolus ampullicapsulatus from the gills of crucian carp, Carassius auratus auratus, two from Yongchuan, Chongqing area and one from Poyang Lake, Jiangxi area, China, sequenced their complete small subunit ribosome RNA gene, analyzed their genetic distance and gene similarity, and explored their relationship based on Bayesian inference and maximum likelihood analyses of their small subunit ribosomal DNA. The results combined with their morphological characteristics suggest that M. ampullicapsulatus infecting the gills and pharynx of allogynogenetic gibel carp, Carassius auratus gibelio, should be Myxobolus honghuensis. This study highlights the importance of DNA sequence comparisons for distinguishing Myxobolus species and indicates that the intra-species identification for the two Myxobolus species mentioned in the present research should be less than ten variation sites. In morphology, M. honghuensis Liu et al. (2012) parasitic on the gills of C. auratus auratus (goldfish) was collected from Chongqing area, and its mature spore was 16.5–19.5 × 8.5–10.0 μm in size, polar capsule was 7.0–10.0 × 2.5–4.0 μm in size, and polar filament had 9–10 coils. M. honghuensis Liu et al. (2012) isolated from the pharynx of C. auratus gibelio was sampled in Hubei area, and its mature spore was 15.1–19.5 × 9.0–11.3 μm in size, polar capsule was 7.9–8.1 × 3.0–4.5 μm in size, and polar filament had 7–8 coils.
Similar content being viewed by others


Introduction
Myxobolus Bütschli, 1882 is a genus with the richest species in myxozoan. It has been reported to have more than 850 species (Eiras et al. 2005; Lom and Dyková 2006), most of which produce morphologically similar or identical spores from genetically related hosts. Therefore, its species identification merely based on their morphological characteristics is difficult and much more confusions will be caused. Fortunately, with the development of molecular biology, more and more new technologies have been widely applied in the classification and identification of Myxobolus species harbored from genetically closely related hosts and infection sites (Bartošová et al. 2009; Fiala 2006; Kent et al. 2001; Khlifa et al. 2012; Morsy et al. 2012; Whipps et al. 2004a, b; Zhao et al. 2008). To avoid mis-identification of myxosporean species, researches have recently suggested that morphology, host or organ specificity, tissue tropism, and molecular data should all be taken into consideration (Bartošová et al. 2009; Cone and Overstreet 1998; Dyková and Lom 2007; Lom and Dyková 2006; Molnár 1994; Morsy et al. 2012; Zhao et al. 2008).
Since Zhao et al. (2008) originally found Myxobolus ampullicapsulatus without causing any disease from the gills of Carassius auratus in Chongqing, China, described it in detail, and obtained its small subunit ribosomal DNA sequence, some researchers have studied this species and the related taxa from goldfish, crucian carp, and allogynogenetic gibel carp (Liu et al. 2012; Xi et al. 2011; Zhang et al. 2009, 2010). Among them, Myxobolus wulii (Wu and Li 1986), M. ampullicapsulatus Zhao et al. (2008) and Myxobolus honghuensis Liu et al. (2012) were found in Carassius auratus gibelio in China and M. wulii from the gills and hepatopancreas of goldfish in Japan (Liu et al. 2012; Xi et al. 2011; Zhang et al. 2010). Phylogenetic analyses of small subunit ribosomal RNA gene sequences showed that M. wulii was closely related to M. ampullicapsulatus (Zhang et al. 2010). Meanwhile, Xi et al. (2011) isolated one Myxobolus species from the gills and pharynx of C. auratus gibelio (Bloch) reared in heavily infected pond in Jiangsu area, China and found that it could cause serious disease for host fish. After sequence comparison among the related Myxobolus species, they considered it as M. ampullicapsulatus based on the high similarity of SSU rDNA sequences. M. honghuensis from C. auratus gibelio with serious disease in Hubei area could be distinguished from M. ampulicapsulatus by its equal polar capsules and more polar filament turns and regarded as an independent species (Liu et al. 2012). In this study, we researched the differences and clarified the confusions among M. wulii, M. ampullicapsulatus, and M. honghuensis based on the genetic and molecular information.
Materials and methods
Sample collection and species identification
The host fish, Carassius auratus auratus were collected during July 2010 and July 2011 from Yongchuan, Chongqing area and Poyang Lake, Jiangxi area, China, necropsied, and examined under the binocular dissecting microscope at ×400 to detect myxosporeans. Plasmodia of three populations of M. ampullicapsulatus were collected from the gills of the host fish, transferred on microscope slides, and ruptured to release spores. After rinsing three times with sterile distilled water, fresh spores were pelleted by centrifugation at 2,000×g, prepared as specimens as previously reported (Zhao et al. 2001), and observed and measured at ×1,000 magnification using a Nikon E600 microscope. The illustrations based on fresh materials were drawn with the aid of the camera lucica and computer software. Photomicrographs were taken using a Nikon DXM1200 microscope at ×1,000 magnification, and measurements based on 25 spores were given in microns (μm) as the arithmetic mean and standard deviation, followed by the range in parentheses. Species identification was conducted as previously reported (Zhao et al. 2001).
DNA extraction, cloning, and sequencing
Genomic DNA of myxosporean isolates from the fish gills were extracted using DNeasy Tissue Kit (QIAGEN). 18S rDNA was amplified using primers ERIB1: 5′-ACC TGG TTG ATC CTG CCA G-3′ and ERIB10: 5′-CTT CCG CAG GTT CAC CTA CGG-3′ at the following conditions: initial denaturation at 94 °C for 5 min followed by 35 cycles of 1 min at 94 °C, 1 min at 56 °C, and 2 min at 72 °C as well as a final elongation at 72 °C for 10 min. Products were purified using a Gel Extraction Kit, measured with the NanoDrop ND-1000 spectrophotometer, inserted into pMD19-T vector, and sequenced in both directions using the ABI PRISM ® 3730 DNA sequencer.
Phylogenetic, sequence, and P-distance analyses
All sequences except those of the newly obtained SSU rRNA genes were obtained using the basic local alignment search tool (BLAST) from GenBank. Sequences selected include those of Myxobolus species which is morphologically similar to M. ampullicapsulatus. Tetracapsuloides bryosalmonae was used as an outgroup taxon. Twenty-two sequences were aligned using Clustal W. Maximum likelihood (ML) analyses were performed using PAUP*4.0b10 (Swofford 2003). Bootstrap confidence values were calculated with a heuristic search using simple sequence addition and 100 replicates. Bayesian analyses were conducted by MrBayes (Ronquist and Huelsenbeck 2003) with parameters setting to 1,000,000 generations and 10,000 trees. Sequences were assembled and manually edited in BioEdit (Hall 1999). In addition, the similarities of seven sequences were calculated by GenBank BLAST, and the P-distance was calculated using MEGA5 (Tamura et al. 2011).
Results
Morphology remarks
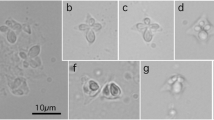
Infection sites and plasmodia morphology for three populations of M. ampullicapsulatus collected from Chongqing area and Poyang Lake, Jiangxi area, China show resemblance to the original population of M. ampullicapsulatus (Zhao et al. 2008). Mature spores were pyriform with bluntly pointed apex and rounded posterior in valvular view (Fig. 1 and Table 1). Generally, the population of M. ampullicapsulatus from Poyang Lake possesses narrower and slightly elongated spores compared with that of M. ampullicapsulatus from Chongqing. In addition, the population of M. ampullicapsulatus isolated from goldfish and crucian carp can be distinguished from M. ampullicapsulatus isolated from the pharynx and gills of C. auratus gibelio from Jiangsu by having a lower ratio of length to width and more bluntly pointed apex (Xi et al. 2011), but displays obvious similarity in both spore morphology and host type to M. honghuensis from the pharynx and gills of C. auratus gibelio in Honghu Lake (Liu et al. 2012). Based on spore morphology and host specificity, M. ampullicapsulatus was mainly parasitic on the gills of goldfish and crucian carp. Thus, M. ampullicapsulatus collected from the pharynx and gills of C. auratus gibelio should be classified as M. honghuensis.

Comparison of spores among the populations of M. ampullicapsulatus and M. honghuensis morphologically. a M. ampullicapsulatus CQLg (from Zhao et al. 2008). b M. ampullicapsulatus CQ collected in the present study. c M. ampullicapsulatus JXa.a collected in the present study. d M. ampullicapsulatus JSa.g (from Xi et al. 2011). e M. honghuensis HBa.g (from Liu et al. 2012). Bar = 10 μm
Sequence analyses
The three new sequences of M. ampullicapsulatus were obtained and submitted to GenBank (Fig. 2), and their lengths are shown in Table 1. Based on GenBank BLAST searches, the three new sequences were most similar to that of M. ampullicapsulatus CQLg (DQ339482) with similarities of 99, 100, and 100 %, respectively, and to that of M. ampullicapsulatus HBa.a (JQ690373) with similarity of 99 % for all (Table 2). Furthermore, Clustal W multiple alignment shows that the coverage length of seven sequences was 1,554 nucleotides, and sequence analyses show that 18S rDNA sequences had little variability among the former five populations of M. ampullicapsulatus (Fig. 3). A total of ten variation sites was scattered in the five sequences. However, 24 variation sites were found in both populations of M. ampullicapsulatus JS and M. ampullicapsulatus CQLg (Zhao et al. 2008; Xi et al. 2011) and only two variation sites in both populations of M. ampullicapsulatus JS and M. honghuensis (Liu et al. 2012). The similarities of the seven sequences are listed in Table 2 and Fig. 3.

Regions of alignment of SSU rRNA gene sequences of populations of M. ampullicapsulatus and M. honghuensis. Regions are separated by vertical lines and the reference sequence. Insertions and deletions were compensated by introducing alignment gaps (dash). Matched sites are represented by dots. Distinct sequence signatures of each clade are shaded

Phylogenetic tree generated by maximum likelihood analysis of the aligned small subunit ribosomal RNA gene sequences. Nodal supports are indicated for bootstrap confidence values resulting from ML and posterior probability of Bayesian analysis. “-” reflects minor differences between maximum likelihood and other methods. GenBank accession numbers for each taxon are listed. The distance scale is shown under the tree. Schematic of (a) M. ampullicapsulatus and (b) M. honghuensis. Bar = 10 m
P-distance analyses
P-distance inferred from 18S rDNA data for the seven sequences are shown in Table 2. Their P-distances to the former five M. ampullicapsulatus (<0.005) are far smaller than their distances to M. ampullicapsulatus JS and M. honghuensis (0.013–0.016). In addition, the P-distances between the latter two is only 0.001.
Phylogenetic analysis
Since the topology of ML tree is similar to that of Bayesian inference (BI) tree, they were combined into one (Fig. 3). Twenty-one myxosporeans were grouped into two main phylogenetic lineages: freshwater lineage and marine lineage. The result of topologies of the two trees strongly supports that M. ampullicapsulatus and M. honghuensis should be placed into two clades. However, M. ampullicapsulatus JSa.g from the pharynx and gills of C. auratus gibelio in Yancheng, Jiangsu area (Xi et al. 2011) is clustered with M. honghuensis from the same host and infection site. The sister clade of M. honghuensis contains five populations of M. ampullicapsulatus from infected gills of C. auratus auratus. Bayesian analyses strongly support a large clade including a sister group formed by M. wulii with another one consisting of Myxobolus koi and Myxobolus longisporus.
Discussion
Morphological comparison for four populations of M. ampullicapsulatus and M. honghuensis indicates that the former three populations of M. ampullicapsulatus have longer pointed apex and ampullaceous polar capsules with longer neck region (Fig. 1), while M. ampullicapsulatus from Jiangsu area and M. honghuensis possess ampullaceous polar capsules with shorter neck region. Moreover, the spores of M. ampullicapsulatus from Jiangsu area can be distinguished from those of the former M. ampullicapsulatus by having a lower ratio of length to width and more bluntly pointed apex and displaying a similarity to M. honghuensis. Liu et al. (2012) have mentioned that M. honghuensis is differed from M. ampullicapsulatus by subtle morphological differences. As for the host range and tissue tropism, M. ampullicapsulatus from Jiangsu and M. honghuensis were both from the pharynx and gills of C. auratus gibelio (Liu et al. 2012; Xi et al. 2011), whereas M. ampullicapsulatus from Chongqing, Jiangxi, and Hubei were all obtained from the gills of C. auratus auratus (Zhao et al. 2008). All the abovementioned morphological characteristics, host specificity, and tissue tropism play important roles in the classification of the complicated myxosporean species (Li et al. 2012; U-taynapun et al. 2011). Therefore, the different host and parasite locations can be used as good references for taxonomy prior to recognizing the importance of the minor differences in the spore shape and size.
Analysis of variation sites based on the alignment of SSU rRNA gene sequences also revealed that M. ampullicapsulatus obtained previously from Jiangsu area is distinct from the five populations of M. ampullicapsulatus in the study. The sequences of the five M. ampullicapsulatus isolated from C. auratus auratus have high identity (99–100 %) to each other but slightly lower identity (98–99 %) to the sequences of two M. ampullicapsulatus isolated from C. auratus gibelio (Table 2). Moreover, the sequence of M. ampullicapsulatus from C. auratus gibelio is nearly identical to that of M. honghuensis from C. auratus gibelio (99 % over 1,996 nt), with only two variation sites. Genetically, these two sequences are intra-specific variations among those reported species of myxozoans (Ferguson et al. 2008; Molnár et al. 2006; Whipps et al. 2004a, b; Whipps and Diggles 2006; Whipps and Kent 2006; Suo et al. 2010). Therefore, defining the population of M. ampullicapsulatus from C. auratus gibelio as M. ampullicapsulatus is debatable, and M. ampullicapsulatus and M. honghuensis from C. auratus gibelio should be regarded as the same species. Blast analyses indicate that M. ampullicapsulatus JSa.a is distinguishable from the other M. ampullicapsulatus sequenced to date. In conclusion, the result indicates that the intra-species identification for the two Myxobolus species mentioned in the present research should be less than ten variation sites.
Depending on estimates of evolutionary divergence among the seven sequences (Table 2) except M. ampullicapsulatus JSa.a, the genetic distances inferred from 18S rDNA sequences for five populations of M. ampullicapsulatus are very close (<0.005). However, their genetic distances to M. ampullicapsulatus JSa.a and M. honghuensis are greater (>0.013). The population of M. ampullicapsulatus JSa.a is genetically closer to M. honghuensis (0.001), indicating that they are in the same species. The study suggests that M. ampullicapsulatus from C. auratus gibelio was mis-identified and should be M. honghuensis.
ML and BI phylogenetic trees strongly suggest that M. ampullicapsulatus and M. honghuensis should be grouped into two different clades: one contains all the populations for M. ampullicapsulatus except M. ampullicapsulatus JSa.a isolated from the pharynx and gills of C. auratus gibelio in Yancheng, Jiangsu area and the other only includes both M. ampullicapsulatus JSa.a and M. honghuensis Liu et al. (2012) from the pharynx of C. auratus gibelio in Honghu Lake, Hubei area. M. ampullicapsulatus was originally described from the gills of goldfish. The two populations of M. ampullicapsulatus in the present research were also from the gills of goldfish or crucian carp, indicating that tissue specificity is an important factor in the evolution of myxozoans (Blaylock et al. 2004; Burger et al. 2007; Cone et al. 2005; Easy et al. 2005; Eszterbauer 2004; Fiala 2006; Kent et al. 2001; Molnár et al. 2006; Whipps et al. 2004a, b; Zhao et al. 2008).
Based on the discussion above, M. ampullicapsulatus seems to be a gill-specific parasite of C. auratus auratus, whereas M. honghuensis is a pharynx or gill parasite, which has a shift in tissue microhabitat. M. ampullicapsulatus and M. honghuensis can be distinguished with careful scrutiny, as the latter has shorter bottleneck, a lower ratio of length to width, and less polar filament turns. Furthermore, the study infers that M. ampullicapsulatus and M. honghuensis are two different species, and the population of M. ampullicapsulatus from Jiangsu area should be regarded as the same species as M. honghuensis. Taken together, this study highlights the importance of DNA sequence comparisons for distinguishing Myxobolus species and also indicates that morphologically similar or identical spores collected from genetically close hosts are important for species classification. The intra-species identification for the two Myxobolus species mentioned in the present research should be less than ten variation sites and more than ten variation sites for inter-species identification of Myxobolus can be inferred. However, some molecular criterion for the intra-species or inter-species identification based on the comprehensive factors, including sequence length, coverage, similarity, variation sites and genetic distances, should be extracted and proved by much more research data.
References
Bartošová P, Fiala I, Hypša V (2009) Concatenated SSU and LSU rDNA data confirm the main evolutionary trends within myxosporeans (Myxozoa: Myxosporea) and provide an effective tool for their molecular phylogenetics. Mol Phylogenet Evol 53:81–93. doi:10.1016/j.ympev.2009.05.018
Blaylock RB, Bullard SA, Whipps CM (2004) Kudoa hypoepicardialis n. sp. (Myxozoa: Kudoidae) and associated lesions from the heart of seven perciform fishes in the northern Gulf of Mexico. J Parasitol 90:584–593. doi:10.1645/GE-161R
Burger MAA, Cribb TH, Adlard RD (2007) Patterns of relatedness in the Kudoidae with descriptions of Kudoa chaetodoni n. sp. and K. lethrini n. sp. (Myxosporea: Multivalvulida). Parasitology 134:669–681. doi:10.1017/S0031182006001995
Cone DK, Overstreet RM (1998) Species of Myxobolus (Myxozoa) from the bulbus arteriosus of centrarchid fishes in North America, with a description of two new species. J Parasitol 84:371–374. doi:10.2307/3284499
Cone DK, Yang J, Sun G, Easy R (2005) Taxonomy and molecular phylogeny of Myxobolus bilobus n. sp. (Myxozoa) parasitizing Notemigonus crysoleucas (Cyprinidae) in Algonquin Park, Ontario, Canada. Dis Aquat Org 66:227–232. doi:10.3354/dao066227
Dyková I, Tyml T, Fiala I, Lom J (2007) New data on Soricimyxum fegati (Myxozoa) including analysis of its phylogenetic position inferred from the SSU rRNA gene sequences. Folia Parasitol 54:272–276. doi:10.1016/j.protis.2008.09.004
Easy RH, Johnson SC, Cone DK (2005) Morphological and molecular comparison of Myxobolus procerus (Kudo, 1934) and M. intramusculi n. sp. (Myxozoa) parasitising muscles of the trout-perch Percopsis omiscomaycus. Syst Parasitol 61:115–122. doi:10.1007/s11230-005-3135-9
Eiras JC, Molnár K, Lu YS (2005) Synopsis of the species of Myxobolus Bűtschli, 1882 (Myxozoa: Myxosporea: Myxobolidae). Syst Parasitol 61:1–46. doi:10.1007/s11230-004-6343-9
Eszterbauer E (2004) Genetic relationship among gill-infecting Myxobolus species (Myxosporea) of cyprinids: molecular evidence of importance of tissue-specificity. Dis Aquat Org 58:35–40. doi:10.3354/dao058035
Ferguson JA, Atkinson SD, Whipps CM, Kent ML (2008) Molecularand morphological analysis of Myxobolus spp. of salmonid fishes with the description of Myxobolus fryeri n. sp. J Parasitol 94:1322–1334. doi:10.1645/GE-1606.1
Fiala I (2006) The phylogeny of Myxosporea (Myxozoa) based on small subunit ribosomal RNA gene analysis. Int J Parasitol 36:1521–1534. doi:10.1016/j.ijpara.2006.06.016
Hall TA (1999) BioEdit: a user-friendly biological sequence alignment editor and analysis program for windows 95/98/NT. Nucleic Acids Symp Ser 41:95–98
Kent ML, Andree KB, Bartholomew JL, El-Matbouli M, Desser SS, Devlin RH, Feist SW, Hedrick RP, Hoffmann RW, Khattra J, Hallett SL, Lester RJG, Longshaw M, Palenzeula O, Siddall ME, Xiao C (2001) Recent advances in our knowledge of the Myxozoa. J Eukaryot Microbiol 48:395–413
Khlifa S, Miller TL, Adlard RD, Faye N, Sasal P (2012) Henneguya mauritaniensis n. sp. (Myxozoa) from the arterial bulb of Pagrus caeruleostictus (Valenciennes, 1830) off Mauritania. Parasitol Res 111:1287–1294. doi:10.1007/s00436-012-2963-1
Li YC, Sato H, Kamata Y, Ohnishi T, Sugita-Konishi Y (2012) Three novel myxobolid species of genera Henneguya and Myxobolus (Myxosporea: Bivalvulida) from marine fish in Japan. Parasitol Res 111:819–826. doi:10.1007/s00436-012-2904-z
Liu Y, Whipps CM, Gu ZM, Zeng C, Huang MJ (2012) Myxobolus honghuensis n. sp. (Myxosporea: Bivalvulida) parasitizing the pharynx of allogynogenetic gibel carp Carassius auratus gibelio (Bloch) from Honghu Lake, China. Parasitol Res 110:1331–1336. doi:10.1007/s00436-011-2629-4
Lom J, Dyková I (2006) Myxozoan genera: definition and notes on taxonomy, life-cycle terminology and pathogenic species. Folia Parasitol 53:1–36
Molnár K (1994) Comments on the host, organ and tissue specificity of fish in myxosporeans and on the types of their intrapiscine development. Parasitol Hung 27:5–20
Molnár K, Marton S, Eszterbauer E, Székely C (2006) Comparative morphological and molecular studies on Myxobolus spp. infecting chub from the river Danube, Hungary, and description of M. muellericus sp. n. Dis Aquat Org 73:49–61. doi:10.3354/dao073049
Morsy K, Abdel-Ghaffar F, Bashtar AR (2012) Morphology and small subunit ribosomal DNA sequence of Henneguya suprabranchiae (Myxozoa), a parasite of the catfish Clarias gariepinus (Clariidae) from the River Nile, Egypt. Parasitol Res 111:1423–1435. doi:10.1007/s00436-012-2976-9
Ronquist F, Huelsenbeck JP (2003) Bayesian phylogenetic inference under mixed models. Biogeosciences 19:1572–1574. doi:10.1093/bioinformatics/btg180
Suo D, Zhao YJ (2010) Morphological redescription of Thelohanellus nikolskii Achmerov, 1995 (Myxozoa, Bivalvulida) and phylogenic analysis of Thelohanellus nikolskii inferred from 18S rDNA. Acta Zoot Sin 35:90–95 (in Chinese with English summary)
Swofford DL (2003) PAUP*: phylogenetic analysis using parsimony (* and other methods), version 4.0b10. Sinauer Associates, Sunderland, Massachusetts
Tamura K, Peterson D, Peterson N, Stecher G, Nei M, Kumar S (2011) MEGA5: molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol Biol Evol 28:2731–2739. doi:10.1093/molbev/msr121
U-taynapun K, Penprapai N, Bangrak P, Metaka T, Itami T, Tantikitti C (2011) Myxobolus supamattayai n. sp. (Myxosporea: Myxobolidae) from Thailand parasitizing the scale pellicle of wild mullet (Valamugilseheli). Parasitol Res 109:81–91. doi:10.1007/s00436-010-2223-1
Whipps CM, Kent ML (2006) Phylogeography of the cosmopolitan marine parasite Kudoa thyrsites (Myxozoa: Myxosporea). J Eukaryot Microbiol 53:364–373. doi:10.1111/j.1550-7408.2006.00114.x
Whipps CM, Grossel G, Adlard RD, Yokoyama H, Bryant MS, Munday BL, Kent ML (2004a) Phylogeny of the multivalvulidae (Myxozoa: Myxosporea) based on comparative ribosomal DNA sequence analysis. J Parasitol 90:618–622. doi:10.1645/GE-153R
Whipps CM, El-Matbouli M, Hedrick RP, Blazer V, Kent ML (2004b) Myxobolus cerebralis internal transcribed spacer 1 (ITS-1) sequences support recent spread of the parasite to North America and within Europe. Dis Aquat Org 60:105–108. doi:10.3354/dao060105
Whipps CM, Diggles BK (2006) Kudoa alliaria in flesh of Argentinian Hoki Macruronus magellanicus (Gadiformes; Merlucciidae). Dis Aquat Org 69:259–263. doi:10.3354/dao069259
Xi BW, Xie J, Zhou QL et al (2011) Mass mortality of pond-reared Carassius gibelio caused by Myxobolus ampullicapsulatus in China. Dis Aquat Org 93:257–260. doi:10.3354/dao02297
Zhang JY, Yokoyama H, Wang JG, Li AH, Gong XN, Ryu-Hasegawa A, Iwashita M, Ogawa K (2010) Utilization of tissue habitats by Myxobolus wulii Landsberg & Lom, 1991 in different carp hosts and disease resistance in allogynogenetic gibel carp: redescription of M. wulii from China and Japan. J Fish Dis 33:57–68. doi:10.1111/j.1365-2761.2009.01102.x
Zhang QZ, Zhao YJ, He GL (2009) The microscopic structure and ultrastructue observations of the mature spore for Myxobolus ampullicapsulatus Zhao et al., 2008 (Myxosporea, Myxobolidae). Acta Zoot Sin 34:531–539
Zhao YJ, Ma CL, Song WB (2001) Illustrated guide to the identification of pathogenetic Protozoa in mariculture—II. Diagnostic methods for the Myxosporea J Ocean Uni Qingdao 31:681–688
Zhao YJ, Sun CY, Kent ML, Deng JL, Whipps CM (2008) Description of a new species of Myxobolus (Myxozoa: Myxobolidae) based on morphological and molecular data. J Parasitol 94:737–742. doi:10.1645/GE-1429.1
Acknowledgments
This work was supported by grants from the National Natural Science Foundation of China (nos. 31101637, 31172068), project of Chongqing Science & Technology Commission (no. CSTC, 2010CA1010), the Science Research Foundation of the Education Committee of Chongqing (no. KJ090814), and Science Founding of Chongqing Normal University (no. 11XLB025). The authors also thank Mr. Zhigang XIE for sampling myxosporean from Jiangxi area.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution License which permits any use, distribution, and reproduction in any medium, provided the original author(s) and the source are credited.
About this article
Cite this article
Zhao, Y.J., Li, N.N., Tang, F.H. et al. Remarks on the validity of Myxobolus ampullicapsulatus and Myxobolus honghuensis (Myxozoa: Myxosporea) based on SSU rDNA sequences. Parasitol Res 112, 3817–3823 (2013). https://doi.org/10.1007/s00436-013-3569-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00436-013-3569-y