
Abstract
Invasion of Plasmodium falciparum merozoites into host erythrocyte involves a series of highly specific and sequential interaction between merozoite and host erythrocyte surface protein. The key step in the invasion process is the formation of a tight protein–protein interaction between host and parasite called as moving junction. A number of parasite proteins secreted from two organelles, microneme and rhoptry, play a role in initial interaction and junction formation between merozoite with host red blood cells (RBCs) during the invasion process. In the present study, we investigated the role of different domains of a P. falciparum rhoptry neck protein PfRON2. Immunofluorescence assay revealed close association of PfAMA1 and PfRON2 in the merozoites during the invasion process. PfRON2 domains were expressed on COS-7 cell surface, and their interaction was analysed with host RBCs and PfAMA1 protein by rosetting assays. The rosetting assays suggest that the C-terminal cysteine-rich domain of PfRON2 plays a role in binding with host erythrocyte. The C-terminal as well as the central cysteine-rich domain of PfRON2 interact with PfAMA1; this binding can be inhibited by monoclonal antibody (mAb 4 G2) against PfAMA1, suggesting that the hydrophobic groove of PfAMA1 binds to PfRON2. These results suggest that PfRON2 plays a role in merozoite invasion and thus it can be an important vaccine candidate antigen.
Similar content being viewed by others
Introduction
Apicomlexan parasites are important pathogens for humans and animals, which causes diseases including toxoplasmosis, malaria, neosporosis, etc. Many of these pathogens are intracellular parasites and invade a host cell at some point in their lifecycle to complete development. A conserved feature of the invasion process of the apicomlexan parasites is formation of a moving junction (MJ) between the apical tip of the pathogen and the host cell membrane (Aikawa et al. 1978). The MJ is formed by tight interaction of a number of parasite proteins and host surface receptors. During the invasion process, the MJ move along the parasite with the help of gliding motor complex leading to internalization of the parasite in host cell (Alexander et al. 2005; Lebrun et al. 2005). The proteins that are the constituents of the MJ are derived from two different secretory organelles of the parasite, the microneme and the rhoptries. The rhoptries empty their contents apically during the invasion process, after microneme exocytosis. In Toxoplasma gondii, four proteins (RON2, RON4, RON5, and RON8) have been identified to be secreted from the rhoptries and form the RON protein complex, which is targeted to the host cell during formation of MJ at the host parasite interface (Alexander et al. 2005; Lebrun et al. 2005; Straub et al. 2009). In Plasmodium falciparum, three members for the rhoptry neck protein (RON2, RON4, and RON5) have been identified (Alexander et al. 2006; Collins et al. 2009). The subcellular localization studies of RON4, RON5, and RON8 in T. gondii showed that these proteins get localized in the cytosolic face of the host plasma membrane during junction formation and merozoite invasion (Besteiro et al. 2009).
Another important merozoite apical protein is the apical membrane antigen 1 (AMA1) that is secreted from micronemes onto the merozoite surface. A large number of studies has shown that it has an important role during the merozoite invasion. Anti-AMA1 antibodies are shown to block merozoite invasion. In addition, PfAMA1 monoclonal antibodies (mAb 4 G2) are directed to hydrophobic grove, and a small peptide R1 that specially binds with this grove is shown to inhibit merozoite invasion suggesting the role of this region in the invasion process (Collins et al. 2007). However, the exact role of AMA1 during invasion process was not clear earlier. Recently, it is shown that AMA1 directly interacts with RON protein complex in P. falciparum and T. gondii (Tyler and Boothroyd 2011; Lamarque et al. 2011). Therefore, this interaction may be crucial for a direct link of AMA1 and MJ. It was further shown that RON2 is the actual bridging molecule between AMA1 and MJ (Tyler and Boothroyd 2011; Lamarque et al. 2011). However, detail interaction studies of PfRON2 with host red blood cell (RBC) and interaction of different domains of PfRON2 with PfAMA1 are lacking.
In the present study, we have carried out functional analysis of cysteine-rich regions of PfRON2. The study identified the domains of PfRON2 involved in interaction with host RBC and its binding with AMA1. We further show that PfRON2 binds with the hydrophobic grove of AMA1, and thus this interaction may be a key to the MJ formation.
Materials and methods
Parasite culture and indirect immunofluorescence assay
P. falciparum strains 3D7 were cultured on human erythrocytes (4% hematocrit) in RPMI1640 media (Invitrogen) supplemented with 10% O+human serum using standard protocol (Trager and Jensen 1976). Parasite cultures were synchronized by two sorbitol treatments at 4 h apart following Lambros and Vanderberg (1979). Indirect immunofluorescence assays were performed on P. falciparum 3D7 parasite lines as described earlier (Ramasamy et al. 2007; Dasaradhi et al. 2005). Thin smears of P. falciparum infected erythrocytes were made on glass slide and fixed with a mixture of methanol/acetone. Slides were blocked in blocking buffer (1× PBS, 10% FCS) for 2 h at 37°C. After blocking, slides were incubated with primary antibody diluted in blocking buffer (mice anti-PfRON2-M, 1:100; mice anti-PfRON2-C, 1:100; rabbit anti-PfAMA1 1:300) for 1 h at 37°C. Slides were washed with 1× PBS for 1 h and incubated with appropriate secondary antibody conjugated to fluorescent dye (FITC or Cy3; dilution 1:250) for 1 h. The slides were stained with DAPI for 30 min at 37°C at a final concentration of 2 μg/ml, then washed twice in 1× PBS Tween-20 0.05%, once in 1× PBS, and mounted on a cover slip in the presence of anti-fade mounting media (Bio-Rad). The slides were viewed on Nikon TE 2000-U fluorescence microscope.
Cloning and expression recombinant protein, peptides, and generation of polyclonal anti-sera
A fragment of PfAMA1 gene (97–543 aa) corresponding to the complete ectodomain, was amplified by polymerase chain reaction (PCR) from P. falciparum 3D7 cDNA using primers 824A (5′CGC CCG GGA TCC ATT GAA ATA GTA GAA AGA AG 3′) and 825A (5′ C GGT CGA CTC GAG ATC ATA AGT TGG TTT ATG TTC 3′). The amplified PCR products were cloned into the BamHI and XhoI sites of pET28a expression vector (Novagen). The resultant plasmid pET28a-PfAMA1 was transformed into expression cells BLR (DE3) for expression of the recombinant protein. These Escherichia coli BLR(DE3) cells were grown in Luria broth containing tetracycline (25 μg/ml) and kanamycin (100 μg/ml) at 37°C with shaking to an OD600 of 0.6–0.7, and expression of recombinant protein was induced with isopropyl-β-thiogalactopyranoside (IPTG) at a final concentration of 1 mM. The cultures were further grown at 37°C for 3–4 h, and the E. coli cells were harvested by centrifugation. The cell pellet was suspended in lysis buffer (20 mM Tris pH 8.0, 500 mM NaCl, 1 mM benzamidine hydrochloride, and 1% Tween 20), and the bacterial cells were lysed by sonication (Torebeo Ultrasonic Processor 36800, Cole Parmer). The lysate was centrifuged at 15,000 × g for 30 min at 4°C, and the supernatant was discarded. The pellet was dissolved in 6 M GuHCl (20 mM Tris, 300 mM NaCl) pH 8.0 and incubated with Ni-nitrilotriacetic acid (Ni2+-NTA) agarose resin (Qiagen), pre-equilibrated with the 6 M GuHCl buffer pH 8.0, at 4°C for 1 h. The suspension was applied to a column and washed with 10 bed volumes of the wash buffer (6 M GuHCl pH 6.3, 20 mM Tris, 300 mM NaCl). The bound protein was eluted with 10 bed volume of elution buffer (6 M GuHCl pH 4.3 in 20 mM Tris, 300 mM NaCl). The eluates were analyzed on SDS-PAGE and the fractions containing the recombinant protein with a clear single band were pooled and the protein concentration was determined using the bicinchoninic acid assay (BCA method) using a standard curve of bovine serum albumin. Refolding of the Ni2+ eluates was carried out using the rapid dilution (1:40, v/v) method using buffer containing 50 mM sodium phosphate pH 7.4, 1 M urea, 1 mM EDTA, 1 mM reduced glutathione (GSH), 0.25 mM oxidized glutathione(GSSG), at 10 ° C for 24 h. The refolded protein was dialyzed in 50 mM sodium phosphate buffer pH 6.5 with 1 M urea.
The pooled protein was further purified by anion-exchange chromatography on a column of SP-Sepharose resin (GE Healthcare) equilibrated with equilibration buffer (50 mM sodium phosphate buffer pH 6.5, 10 mM NaCl). The bound proteins were eluted with a linear gradient of NaCl (10 mM–1 M) in phosphate buffer (pH 6.5). Eluates were analyzed by SDS-PAGE, fractions containing a single protein band of PfAMA1 were pooled, and the protein concentration was determined by BCA.
Mice antisera were raised against the middle and the C-terminal cysteine-rich regions of PfRON2 (labeled as PfRON2-M and PfRON2-C, respectively). Priorly, three polypeptides overlapping with the PfRON2-M, PLERLYYNSLALGELVEPC (1,073–1,090 aa), TGTKSFYSLPTILTANSC (1,139–1,155 aa), and DNMFYANLFVLTALSRC (1,473–1,488 aa), and two polypeptides corresponding to PfRON2-C, KIDDKDFVHNFKMILGC (1,907–1,922 aa) and MTLGSLSAYTLFSAC (2,006–2,019 aa), were linked with KLH and purified using KLH conjugation kit (Pierce) as manufacturer’s instruction. Mixtures of polypeptides (50 μg/mice), formulated in complete Freund’s adjuvant (Sigma, USA), were immunized into separate set of female BALB/c mice. Subsequently, the mice were administered three booster doses (days 14, 28, and 42) of the polypeptides formulated in Freund’s incomplete adjuvant. The mice serum was collected 10 days after the second boost and third boost.
Yeast two-hybrid assay for PfAMA1 and PfRON2 fragments
The gene fragment corresponding to mature ectodomain of PfAMA1 protein (289–1,629 bp; 96–543 aa) was amplified from total cDNA of the parasite by PCR using primers 824 and 825 A. The amplified PCR product was digested with SmaI and SalI and cloned into SmaI and SalI sites of pGBKT7 vector (BD Biosciences) to give pGBK-PfAMA1 construct.
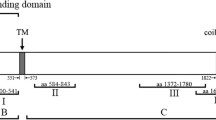
Four gene fragments spanning the whole PfRON2 protein, except the variable asparagine-rich region at the N-terminus, were amplified and cloned in pGADT7 vector (BD Biosciences). These include the middle cysteine-rich region (2,848–3,816 bp, 949–1,272 aa; PfRON2-M), C-terminal cysteine-rich region (5,572–6,186 bp, 1,857–2,062 aa; PfRON2-C) and two fragment corresponding to the region between these two cysteine-rich domains (3,688–4,602 bp, 1,229–1,534 aa; PfRON2-M1; and 4,555–5,613 bp, 1,518–1,871 aa; PfRON2-M2). Location of these fragments with respect to the full protein is indicated in Fig. 1a. The primer combination used to amplify each of the fragments is given in Table S1.

a Schematic diagram of PfRON2 protein, showing signal sequence (SS) and predicted TM domain (TM). The two cysteine-rich regions are also marked. Different fragments of PfRON2 cloned in present study for RBC-binding assays and PfAMA1-binding assays are also indicated. b Schematic diagram of T. gondii RON-2 (TgRON2) showing signal sequence (SS) and three predicted transmembrane domains (TM1, TM2, TM3). c Immunofluorescence assay showing co-localisation of PfRON2 with PfAMA1 at apical end at the time of meozoite attachment during merozoite invasion
The pGBKT7-PfAMA1 construct was co-transformed with each of the PfRON2 construct into the AH109 yeast cells, and potential interactions were assessed by growth of the co-transformants on selective media.
COS-7 cell expression plasmid constructs, COS-7 cell culture, transfection, and immunofluorescence assays
To generate COS-7 cell expression plasmid constructs, two fragments of PfRON2, one corresponding to middle cysteine-rich region PfRON2-M (950–1,272 aa) of gene and the other representing the C-terminal cysteine-rich region PfRON2-C (1,858–2,062 aa), were amplified from the genomic DNA using primers 679A (5′ CAG CTG GGA GCG AAC TTT TCA AAT GAA G 3′) and 680A (5′ GGG CCC AGG ATT ATA TAA ACT TGA TTC TTC 3′) for PfRON2-M and primers 681A (5′ CAG CTG CGA CTT AGA GAA TTA CAA AAT TC 3′) and 682A (5′ GGG CCC AGT GGA AGT AGA TAA TGC CG 3′) for PfRON2-C. The amplified PCR products were cloned into PvuII and ApaI sites of pRE4 vector (Cohen et al. 1988) in frame with the signal sequence and transmembrane segment of HSVgD. The resultant plasmids were labeled as pRE4-PfRON2-M and pRE4-PfRON2-C, respectively. The COS-7 cells (American Type Culture Collection [ATCC] CRL 1651; Rockville, MD) were cultured in Dulbecco-modified Eagle medium (DMEM; Invitrogen) with 10% heat-inactivated fetal calf serum (FCS) in a humidified CO2 (5%) incubator at 37°C. Fresh monolayer of 40–60% confluent COS-7 cells growing in 35-mm-diameter wells were transfected with 2–4 μg plasmid DNA using lipofectamine plus reagent (Invitrogen) following manufacturer’s instructions. To detect expression of the fusion proteins on the surface of transfected COS-7 cells and to calculate the transfection efficiency, immunofluorescence assays were performed, 36–40 h after transfection, following Chitnis and Miller (1994), using mouse polyclonal antibody against middle or C-terminal cysteine-rich region of PfRON2 (PfRON2-M and PfRON2-C).
Erythrocyte-binding assay with COS-7 cells expressing PfRON2 fragments
Human erythrocytes were washed three times in incomplete RPMI1640 media (Life Technologies) and resuspended in RPMI1640. Erythrocytes were treated with neuraminidase, chymotrypsin, and trypsin as described earlier (Chitnis and Miller 1994). Briefly, for neuraminidase treatment, 5.5 ml of a 5% (vol/vol) suspension of the washed human erythrocytes in incomplete medium was incubated twice with 3 milliunits of neuraminidase (Vibrio cholerae; Sigma) for 1 h at 37°C each time. For trypsin treatment, washed human erythrocytes were incubated with 1 mg/ml tosyl-phenylalanine-chloromethyl-ketone-treated trypsin (Sigma) for 1 h at 37°C. For chymotrypsin treatment, erythrocytes were incubated with 1 mg/ml chymotrypsin (Sigma) for 1 h at 37°C. COS-7 cells transfected with constructs pRE4-PfRON2-M and pRE4-PfRON2-C were tested for binding to normal, neuraminidase-treated, chymotrypsin-treated, and trypsin-treated human erythrocytes, 36–40 h after transfection, following Chitnis and Miller (1994). COS-7 cells transfected with plasmid pHVDR22 (Chitnis and Miller 1994) were used as positive control. The number of rosettes of erythrocytes bound to transfected COS-7 cells was scored in 20 fields at 200× magnification for each set. A cluster of eight or more erythrocytes bound to a COS-7 cell was scored as a rosette. The number of rosettes observed was normalized for transfection efficiency of 5% for all the sets. The statistical significance of the results were determined by using unpaired Student’s t-test.
Magnetic beads coating and COS-7 cell-binding assay and inhibition of binding with mAb
The recombinant PfAMA1 protein was coated onto magnetic beads through the anti-PfAMA1 antibody. Briefly, Dynal magnetic beads coated with sheep anti-rabbit IgG (Dynal Biotech) were incubated with antiPfAMA1 antibody (100 μg/ml) and then cross-linked with DMP (20 mM) in triethanolamine buffer (0.2 M, pH 8.2) following manufacturer’s instructions. Magnetic beads cross-linked anti-PfAMA1 antibody was incubated with recombinant PfAMA1 (20 μg/ml) for 1 h at 4°C with intermittent shaking. These PfAMA1-coated beads were washed and used for binding assays. To asses binding inhibition by mAb, the PfAMA1-coated beads were incubated with mAb 4 G2 or purified antibody from preimmune sera. These antibody-treated beads were washed and used in the binding assays. COS-7 cells transfected with constructs pRE4-PfRON2-M and pRE4-PfRON2-C were tested for binding to magnetic beads, 36–40 h after transfection, following Chitnis and Miller (1994). The number of rosettes of beads bound to transfected COS-7 cells was scored in 20 fields at 200× magnification for each set. A cluster of eight or more beads bound to a COS-7 cell was scored as a rosette. The number of rosettes observed was normalized for transfection efficiency of 5% for all the sets.
Sequencing of pfron-2 genes from different P. falciparum isolates
The region containing the middle and C-terminal domains in the pfron-2 (PF14_0495) gene were PCR-amplified using high-fidelity PCR enzyme mix (MBI Fermentas) and total genomic DNAs of six different P. falciparum field isolates (Table S1) using the 816A/817A-primer pair for middle and 818A/819A for C-terminal cysteine-rich region. The resulting PCR products (968 and 614 bp, respectively) were purified using a PCR purification kit (Qiagen) and sequenced directly. For each DNA sample, amplified products from two separate PCRs were sequenced in both the forward and reverse direction using big dye termination chemistry. The sequence data were aligned using the ClustalW algorithm in Bio-Edit software (Tom Hall, Ibis Biosciences, Carlsbad, CA, USA).
Results
PfRON2 colocalizes with AMA1 at the MJ during merozoite invasion
The PfRON2 protein is localized at the apical end of the P. falciparum merozoite. We studied its localization with respect to AMA1 in the merozoite during the invasion into the host RBCs. Anti-sera were raised using small peptide corresponding to the middle and C-terminal cysteine-rich regions. Immunofluorescence studies were carried out using antibodies against apical membrane antigen-1 (PfAMA1) and anti-PfRON2 antisera. At the time of attachment of the merozoite with the host RBCs, the PfRON2 staining was at the apical tip, the PfAMA1 staining was also mainly at the apical tip and with some distribution at the merozotie surface. During parasite invasion, the PfRON2 and PfAMA1 showed colocalization at the apical end of merozoite (Fig. 1b).
PfRON2 binds with human RBCs through its C-terminal cysteine-rich region
To assess the functional role of PfRON2, the middle and C-terminal cysteine-rich domains (PfRON2-M, 949–1,272 aa; PfRON2-C, 1,857–2,062 aa) were expressed on the surface of COS-7 cells in order to test their binding with the human erythrocytes. The secretory signal sequence and transmembrane segment of Herpes simplex virus glycoprotein D (HSV gD) gene in the pRE4 mammalian expression vector were used (Cohen et al. 1988) to target these protein to the surface of transfected COS cells. A construct pHVDR22, designed to express P. vivax Duffy-binding protein region II (PvRII) in the same way (Chitnis and Miller 1994), was used as the positive control in these assays. Immunofluorescence assay of the unpermeabilized transfected cells using DL6 antibodies and antisera against PfRON2-M and PfRON2-C confirmed expression of these protein fragments on COS cell surfaces (Fig. 2a). The untransfected control cells showed no staining with these antibodies. Transfection efficiency was also calculated from the immunostaining of transfected cells and was observed to be 4–7%, and therefore, the transfection efficiency of 5% was used to normalized the rosetting data following Chitnis and Miller (1994).

Erythrocyte-binding assay of PfRON2 protein fragments. a Immunofluorescence assay to confirm the expression of recombinant proteins corresponding to PfRON2 fragments on transfected COS cells, as revealed by the anti-PfRON2-M and anti-PfRON2-C antibodies. b Rosetting of human RBCs on COS-7 cells expressing PfRON2 cysteine-rich regions (PfRON2-M and PfRON2-C) on their surfaces. Untransfected/mock transfected COS-7 cells were used as negative controls. c Rosetting of enzyme-treated human RBCs on COS-7 cells expressing PfRON2-C on their surfaces
The C-terminal cysteine-rich region of PfRON2 (construct pRE4-PfRON2-C) showed binding with human erythrocytes although its binding efficiency were lower than that of PvRII construct (Fig. 2b, Table 1). The middle cysteine-rich region of PfRON2 (construct pRE4-PfRON2-M) did not show efficient binding with human RBCs (Fig. 2b, Table 1), suggesting that PfRON2 binds with the host RBCs only through the C-terminal region. Treatment of RBCs with chymotrypsin did not affect binding with the PfRON2-C-expressing COS cells, whereas trypsin and neuraminidase treatment significantly (P < 0.001 and P < 0.001, respectively) reduced the binding (Fig. 2c, Table 1). These results suggest that PfRON2-C binds with a receptor on RBC that is resistant to chymotrypsin treatment but is sensitive to trypsin and neuraminidase.
PfRON2 cysteine-rich domains interact with PfAMA1
To assess the PfRON2 and PfAMA1 interaction and to identify the PfRON2 region involved in this interaction, we first utilized the yeast two-hybrid system. The ectodomain of PfAMA1 and different PfRON2 fragments (PfRON2-M, PfRON2-M1, PfRON2-M2, and PfRON2-C; Fig. 1a) were cloned in yeast expression vectors in fusion with Gal4 DNA-binding domain (pGBKT7) and Gal4 activation domain (pGAD), respectively. The yeast AH109 cells co-transformed with pGBKT7-PfAMA1 and pGAD-PfRON2-M or pGAD-PfRON2-C constructs were able to form new colonies after 3 days of growth on triple drop-out media (L-,T-, H-) containing up to 10 mM of 3-amino-triazole (Table S2). However, cells co-transformed with pGBKT7-PfAMA1 with other two PfRON2 constructs, pGAD-PfRON2-M1 and pGAD-PfRON2-M2, were not able to grow on the triple drop-out media. These results show that PfRON2 interacts directly with PfAMA1, and both the cysteine-rich domains are involved in this interaction.
To further confirm that the PfRON2 interacts with PfAMA1 through the cysteine-rich domains, we again used the heterologous expression system. The middle and C-terminal cysteine-rich regions of PfRON2 expressed on the COS cell surfaces were assessed for their interaction with PfAMA1 protein. The ectodomain region of PfAMA1 (96–543 aa) was expressed in E. coli, purified and refolded by rapid dilution method. The refolded protein was coated onto the magnetic beads with the help of anti-PfAMA1 antibodies. The PfAMA1-coated beads were then used for the binding assays with the COS cell expressing PfRON2-M or PfRON2-C. Binding of >8 beads on a COS cell were treated as single rosette. The PfRON2-M and PfRON2-C domains showed binding with the PfAMA1 (Fig. 3, Table 2). These results suggest that PfRON2 binds with PfAMA1 through its cysteine-rich regions.

Interaction of PfRON-2 fragments with AMA1. Rosetting assay of AMA1-coated magnetic beads to assess the binding between PfAMA1 and PfRON2 fragments. COS-7 cells expressing PfRON2 cysteine-rich regions (PfRON2-M and PfRON2-C) on their surfaces were incubated with magnetic beads coated with PfAMA1 and rossetting was analysed. Untransfected COS-7 cells were used as negative controls. To assess inhibition of binding, the beads were preincubated with mAb 4 G2 (1:100; 1:250) before the binding assays. Purified preimmune mice serum antibodies were used as control
PfAMA1 hydrophobic groove is involved in its binding with PfRON2
The interaction of PfAMA1 with PfRON2 was further analyzed with respect to its hydrophobic binding pocket in the PfAMA1 ectodomain. The monoclonal antibodies (mAb 4 G2) against the hydrophobic pocket were used in the binding assays of PfAMA1-coated beads and COS cell surface-expressed PfRON2 protein. Presence of mAb 4 G2 in the binding assay (at 89 μg/ml final concentration) showed 50–60% reduction in the binding of PfRON2-M and PfRON2-C with PfAMA1 (P < 0.0001 and P < 0.01, respectively; Fig. 3, Table 2). This suggests that binding of mAb 4 G2 in the hydrophobic groove of PfAMA1 interfere with its binding with PfRON2. These results show that PfRON2 binds with the hydrophobic groove of PfAMA1.
The middle and C-terminal cysteine-rich functional regions are highly conserved
After confirming that the middle and C-terminal cysteine-rich regions are the functional regions of the protein, we assessed if these regions contain any polymorphic residues. The gene fragments corresponding to these regions were amplified from total DNAs of six different field isolates and sequenced. Both the fragments were found to be highly conserved among all the isolates, there was no change in the amino acid sequence, and these regions also do not have any non-synonymous changes in the nucleotide sequence. The SNP data available at PlasmoDB was also analyzed for any change in the amino acid sequence of the protein for ten different laboratory strains of P. falciparum. There is only one SNP (2,914 bp) among these sequences lying in the middle cysteine-rich region of the protein in strains HB3 and Santalucia, D(972)H.

Model of Plasmodium falciparum moving junction and interaction of RON protein complex with AMA1 and host erythrocyte. The PfRON2 protein interacts with a host surface receptor as well as with PfAMA1 to form a bridge between host and the parasite. Other proteins of RON complex also interact with PfRON2, and during the invasion process these proteins are positioned to anchor with host cytoskeleton. Rh Rhoptries, M microneme, PPM parasite plasma membrane
Discussion
Invasion of the host erythrocyte by P. falciparum merozoites is a critical step during blood stage infection, which involves a cascade of protein–protein interactions between the parasite and the host. A number of these interactions are mediated by parasite proteins in the apical secretory organelles of the merozoites, microneme and rhoptries (Cowman and Crabb 2006). After the release of merozoites from schizont and during the invasion process, contents of both rhoptries and micronemes get excreted through the ductules at the neck of the rhoptry (Healer et al. 2002; Bannister et al. 2003). Micronemes secretes a number of proteins belonging to ebl family, including EBA-175, EBA-140 and EBA-180, which binds with the host RBC in a sialic acid-dependent manner (Cowman and Crabb 2006). Another important microneme protein is AMA1; several lines of experiments have shown that it plays a key role in merozoite invasion. The anti-AMA1 antibodies inhibit merozoite invasion, and an AMA1-binding R1 peptide block critical invasion steps after reorientation of the merozoite and apical end attachment (Harris et al. 2005, 2009; Treeck et al. 2009). Recent discovery of rhoptry neck protein complex consisting of RON-2, RON-4, and RON-5 helped to understand the functional role of AMA1 during invasion and process of MJ formation. The RON protein complex interacts with AMA1 during the junction formation and that anchors the merozoite to host cell surface and promotes the invasion process (Richard et al. 2010). The RON2 homolog in T. gondii is shown to be the key component of RON protein complex that forms a bridge between the host and the parasite by interacting with AMA1 toward parasite site and with other RON proteins toward the host cell side (Lamarque et al. 2011). In the present study, we have analyzed different fragments of PfRON2 to assess its interaction with the host cell and with the PfAMA1 protein in the parasite.
The PfRON2 protein is a large protein with a signal sequence and a large low complexity region at the N-terminus. In addition, two cysteine-rich regions are identified in the protein. The first is in the middle followed by the low complexity region, and the second is close to the C-terminus just before the C-terminal transmembrane domain. In many of the parasite proteins, the globular cysteine-rich regions are shown to be the functional domains; the cysteine-rich domains in merozoite surface/apical proteins are shown to be involved in receptor–ligand interaction between host and the parasite during invasion (Goel et al. 2003; Li et al. 2004; Sim et al. 1994). Since a number of merozoite rhoptry proteins play a role in binding and invasion of RBC by the merozoites (Cowman and Crabb 2006), we tried to assess a possible role of PfRON2 in RBC binding. The two cysteine-rich domains of PfRON2 were assessed for their RBC-binding properties. Our results of binding assays using COS cell surface expression system suggest that PfRON2 is involved in binding of the merozoite with the host RBCs, and this interaction is mediated through its C-terminal cysteine-rich region. During the merozoite invasion into the host erythrocyte, a number of surface proteins are suggested to be involved in initial binding of the merozoite with the RBC, whereas proteins secreted from apical organelles, microneme and rhoptries, are involved in binding after reorientation of the merozoite (Cowman and Crabb 2006). A number of receptors on the erythrocyte surface interact with these parasite ligands. These receptor–ligand interactions between the merozoite surface/apical proteins and erythrocyte surface receptors are characterized by their enzyme sensitivities to neuraminidase, trypsin, and chymotrypsin (Cowman and Crabb 2006, Sim et al. 1994; Doolan et al. 1994; Triglia et al. 2001; Rayner et al. 2001; Duraisingh et al. 2003; Wickramarachchi et al. 2008). We found that PfRON2 binds with RBC in a chymotrypsin-resistant manner, and this binding can be inhibited by trypsin and neuraminidase treatments, suggesting that PfRON2 utilizes a receptor which is sensitive to trypsin and neuraminidase treatments. Taken together, the results of localization and binding assays suggested that the PfRON2 is involved in apical interaction of merozoite with the host RBC and formation of MJ complex during the process of invasion (Fig. 4).
Interaction of AMA-1 with RON protein complex is clearly shown in T. gondii as well as in P. falciparum; it is further evident that this interaction is crucial for the invasion process (Tyler and Boothroyd 2011; Lamarque et al. 2011). Furthermore, a small region at the C-terminus of TgRON2 was shown to be involved in binding with TgAMA1 (Tyler and Boothroyd 2011; Tonkin et al. 2011). Based upon these results, interaction of C-terminal region of PfRON-2 and PfAMA-1 was predicted (Tonkin et al. 2011). We evaluated a different region of PfRON2 for binding with the ecto-domain of PfAMA1 protein. We carried out yeast two-hybrid assays as well as PfAMA1 recombinant protein-binding assays with the COS cell surface-expressed PfRON2 fragments. In both assays, the PfRON2 cysteine-rich region present in the middle of the protein, PfRON2-M, and the cysteine-rich region present at the C-terminus, PfRON2-C, showed binding affinity with PfAMA1. Our data suggest that PfRON2 binds with PfAMA1 mainly through its cysteine-rich regions; the two cysteine-rich region may act in cooperation to form a protein complex with PfAMA1. Malaria antigens are known to exhibit extensive polymorphism which may be one of the strategies of the parasites to evade host immune mechanisms; therefore, the antigens that are under natural immune pressure tend to have higher levels of polymorphism (Hisaeda et al. 2005; Rich et al. 2000). On the other hand, it is also shown that the polymorphic regions in an antigen are usually different than the functionally critical domains which are relatively conserved and are more suitable target for vaccine development (Singh et al. 2006; Del Portillo et al. 1991).We found that the two cysteine-rich regions of PfRON2 are highly conserved among different field isolates and laboratory strains, which further support the functional importance of these regions.
The interaction of PfAMA1 with RON protein complex is suggested to be through a domain located within a hydrophobic groove in the ectodomain of PfAMA1 protein (Collins et al. 2009). The hydrophobic groove has residues conserved across Plasmodia and similar hydrophobic resides are present in AMA1 from other apicomplexa. A monoclonal antibody, mAb 4 G2, directed against an epitope in the hydrophobic groove is shown to inhibit merozoite invasion (Collins et al. 2007). It is later shown that binding of mAb 4 G2 with the hydrophobic groove prevented binding of AMA1 with rhoptry neck proteins (Collins et al. 2007). To find out if the hydrophobic groove in PfAMA1 is directly involved in binding with PfRON2, we assessed efficacies of mAb 4 G2 to inhibit the interaction of PfAMA1 and PfRON2 in the COS cell surface-binding assays. Our data showed that mAb 4 G2 is able to specifically inhibit the interaction of PfAMA1 with PfRON2, suggesting that PfAMA1 binds with PfRON2 through its hydrophobic groove. It has been suggested that, during the merozoite invasion process, the RON proteins are discharged from the rhoptries and associate with the AMA1 for the junction formation, whereas inhibition of this association by mAb 4 G2 prevents junction formation and thus inhibits invasion (Collins et al. 2007). Together with these studies, our results suggest that the interaction of PfRON2 with PfAMA1 through its hydrophobic groove may be a key step in junction formation and merozoite invasion; these results have important implication on rational design of a subunit-based malaria vaccine.
References
Aikawa M, Miller LH, Johnson J, Rabbege J (1978) Erythrocyte entry by malarial parasites. A moving junction between erythrocyte and parasite. J Cell Biol 77(1):72–82
Alexander DL, Mital J, Ward GE, Bradley P, Boothroyd JC (2005) Identification of the moving junction complex of Toxoplasma gondii: a collaboration between distinct secretory organelles. PLoS Pathog 1(2):e17
Alexander DL, Arastu-Kapur S, Dubremetz JF, Boothroyd JC (2006) Plasmodium falciparum AMA1 binds a rhoptry neck protein homologous to TgRON4, a component of the moving junction in Toxoplasma gondii. Eukaryot Cell 5(7):1169–1173
Bannister LH, Hopkins JM, Dluzewski AR, Margos G, Williams IT, Blackman MJ, Kocken CH, Thomas AW, Mitchell GH (2003) Plasmodium falciparum apical membrane antigen 1 (PfAMA-1) is translocated within micronemes along subpellicular microtubules during merozoite development. J Cell Sci 116(Pt 18):3825–3834
Besteiro S, Michelin A, Poncet J, Dubremetz JF, Lebrun M (2009) Export of a Toxoplasma gondii rhoptry neck protein complex at the host cell membrane to form the moving junction during invasion. PLoS Pathog 5(2):e1000309
Chitnis CE, Miller LH (1994) Identification of the erythrocyte binding domains of Plasmodium vivax and Plasmodium knowlesi proteins involved in erythrocyte invasion. J Exp Med 180(2):497–506
Cohen GH, Wilcox WC, Sodora DL, Long D, Levin JZ, Eisenberg RJ (1988) Expression of herpes simplex virus type 1 glycoprotein D deletion mutants in mammalian cells. J Virol 62(6):1932–1940
Collins CR, Withers-Martinez C, Bentley GA, Batchelor AH, Thomas AW, Blackman MJ (2007) Fine mapping of an epitope recognized by an invasion-inhibitory monoclonal antibody on the malaria vaccine candidate apical membrane antigen 1. J Biol Chem 282(10):7431–7441
Collins CR, Withers-Martinez C, Hackett F, Blackman MJ (2009) An inhibitory antibody blocks interactions between components of the malarial invasion machinery. PLoS Pathog 5(1):e1000273
Cowman AF, Crabb BS (2006) Invasion of red blood cells by malaria parasites. Cell 124(4):755–766, Review
Dasaradhi PV, Mohmmed A, Kumar A, Hossain MJ, Bhatnagar RK, Chauhan VS, Malhotra P (2005) A role of falcipain-2, principal cysteine proteases of Plasmodium falciparum in merozoite egression. Biochem Biophys Res Commun 336(4):1062–1068
Del Portillo H, Longacre S, Khouri E, David PH (1991) Primary structure of the Merozoite surface antigen 1 of Plasmodium vivax reveals sequences conserved between different Plasmodium species. Proc Natl Acad Sci U S A 88:4030–4034
Doolan DL, Beck HP, Good MF (1994) Evidence for limited activation of distinct CD4+ T cell subsets in response to the Plasmodium falciparum circumsporozoite protein in Papua New Guinea. Parasite Immunol 16(3):129–136
Duraisingh MT, Maier AG, Triglia T, Cowman AF (2003) Erythrocyte-binding antigen 175 mediates invasion in Plasmodium falciparum utilizing sialic acid-dependent and -independent pathways. Proc Natl Acad Sci U S A 100(8):4796–4801
Goel VK, Li X, Chen H, Liu SC, Chishti AH, Oh SS (2003) Band 3 is a host receptor binding merozoite surface protein 1 during the Plasmodium falciparum invasion of erythrocytes. Proc Natl Acad Sci U S A 100(9):5164–5169
Harris KS, Casey JL, Coley AM, Masciantonio R, Sabo JK, Keizer DW, Lee EF, McMahon A, Norton RS, Anders RF, Foley M (2005) Binding hot spot for invasion inhibitory molecules on Plasmodium falciparum apical membrane antigen 1. Infect Immun 73(10):6981–6989
Harris KS, Casey JL, Coley AM, Karas JA, Sabo JK, Tan YY, Dolezal O, Norton RS, Hughes AB, Scanlon D, Foley M (2009) Rapid optimization of a peptide inhibitor of malaria parasite invasion by comprehensive N-methyl scanning. J Biol Chem 284(14):9361–9371
Healer J, Crawford S, Ralph S, McFadden G, Cowman AF (2002) Independent translocation of two micronemal proteins in developing Plasmodium falciparum merozoites. Infect Immun 70(10):5751–5758
Hisaeda H, Yasutomo K, Himeno K (2005) Malaria: immune evasion by parasites. Int J Biochem Cell Biol 37:700–706
Lamarque M, Besteiro S, Papoin J, Roques M, Vulliez-Le Normand B, Morlon-Guyot J, Dubremetz JF, Fauquenoy S, Tomavo S, Faber BW, Kocken CH, Thomas AW, Boulanger MJ, Bentley GA, Lebrun M (2011) The RON2-AMA1 interaction is a critical step in moving junction-dependent invasion by apicomplexan parasites. PLoS Pathog 7(2):e1001276
Lambros C, Vanderberg JP (1979) Synchronization of Plasmodium falciparum erythrocytic stages in culture. J Parasitol 65(3):418–420
Lebrun M, Michelin A, El Hajj H, Poncet J, Bradley PJ, Vial H, Dubremetz JF (2005) The rhoptry neck protein RON4 re-localizes at the moving junction during Toxoplasma gondii invasion. Cell Microbiol 7(12):1823–1833
Li X, Chen H, Oo TH, Daly TM, Bergman LW, Liu SC, Chishti AH, Oh SS (2004) A co-ligand complex anchors Plasmodium falciparum merozoites to the erythrocyte invasion receptor band 3. J Biol Chem 279(7):5765–5771
Okoyeh JN, Pillai CR, Chitnis CE (1999) Plasmodium falciparum field isolates commonly use erythrocyte invasion pathways that are independent of sialic acid residues of glycophorin A. Infect Immun 67:5784–5791
Ramasamy G, Gupta D, Mohmmed A, Chauhan VS (2007) Characterization and localization of Plasmodium falciparum homolog of prokaryotic ClpQ/HslV protease. Mol Biochem Parasitol 152(2):139–148
Rayner JC, Vargas-Serrato E, Huber CS, Galinski MR, Barnwell JW (2001) A Plasmodium falciparum homologue of Plasmodium vivax reticulocyte binding protein (PvRBP1) defines a trypsin-resistant erythrocyte invasion pathway. J Exp Med 194(11):1571–1581
Rich SM, Ferreira MU, Ayala FJ (2000) The origin of antigenic diversity in Plasmodium falciparum. Parasitol Today 16:390–396
Richard D, MacRaild CA, Riglar DT, Chan JA, Foley M, Baum J, Ralph SA, Norton RS, Cowman AF (2010) Interaction between Plasmodium falciparum apical membrane antigen 1 and the rhoptry neck protein complex defines a key step in the erythrocyte invasion process of malaria parasites. J Biol Chem 285(19):14815–14822
Sim BK, Chitnis CE, Wasniowska K, Hadley TJ, Miller LH (1994) Receptor and ligand domains for invasion of erythrocytes by Plasmodium falciparum. Science 264(5167):1941–1944
Singh SK, Hora R, Belrhali H, Chitnis CE, Sharma A (2006) Structural basis for Duffy recognition by the malaria parasite Duffy-binding-like domain. Nature 439:741–744
Straub KW, Cheng SJ, Sohn CS, Bradley PJ (2009) Novel components of the Apicomplexan moving junction reveal conserved and coccidia-restricted elements. Cell Microbiol 11(4):590–603
Tonkin ML, Roques M, Lamarque MH, Pugnière M, Douguet D, Crawford J, Lebrun M, Boulanger MJ (2011) Host cell invasion by apicomplexan parasites: insights from the co-structure of AMA1 with a RON2 peptide. Science 333(6041):463–467
Trager W, Jensen JB (1976) Human malaria parasites in continuous culture. Science 193(4254):673–675
Treeck M, Zacherl S, Herrmann S, Cabrera A, Kono M, Struck NS, Engelberg K, Haase S, Frischknecht F, Miura K, Spielmann T, Gilberger TW (2009) Functional analysis of the leading malaria vaccine candidate AMA-1 reveals an essential role for the cytoplasmic domain in the invasion process. PLoS Pathog 5(3):e1000322
Triglia T, Thompson JK, Cowman AF (2001) An EBA175 homologue which is transcribed but not translated in erythrocytic stages of Plasmodium falciparum. Mol Biochem Parasitol 116(1):55–63
Tyler JS, Boothroyd JC (2011) The C-terminus of Toxoplasma RON2 provides the crucial link between AMA1 and the host-associated invasion complex. PLoS Pathog 7(2):e1001282
Wickramarachchi T, Devi YS, Mohmmed A, Chauhan VS (2008) Identification and characterization of a novel Plasmodium falciparum merozoite apical protein involved in erythrocyte binding and invasion. PLoS One 3(3):e1732
Acknowledgments
We are grateful to Roselyn Eisenberg and Gary Cohen for plasmid pRE4 and DL6 antibodies. We thank Michael Blackman for providing mAb 4 G2 and Chetan Chitnis for providing anti-PfAMA-1 antibodies and pHVDR22 vector. We also thank the Rotary blood bank, New Delhi for providing human RBCs, Rakesh and Ashok for assisting in animal handling, and Asad for helping. MEH is supported by predoctoral research fellowship by ICGEB.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Table S1
List of primers used in the study to PCR amplify different fragments of PfRON2 and PfAMA1. The restriction sites are given in lower case (DOC 33 kb)
Table S2
Growth of yeast AH109 cells on different selection media (with amino acid dropout) after cotransformation with pGBK-PfAMA-1 plasmid construct and a pGAD construct harboring PfRON-2 fragment. (DOC 38.5 kb)
Table S3
Table showing details of P. falciparum strains and field isolates used for sequencing of pfdblmsp genes (DOC 42 kb)
Fig. S1
Expression, purification and refolding of PfAMA1. A SDS-PAGE of total Escherichia coli lysate before (U) and after induction (I) of recombinant protein expression; the gel shows expression of recombinant protein corresponding to ecto-domain of PfAMA1. B SDS-PAGE showing Ni-NTA eluate after purification of recombinant protein from inclusion bodies. C SDS-PAGE of purified and refolded PfAMA1 in eluate of ion-exchange chromatography (DOC 230 kb)
Rights and permissions
About this article
Cite this article
Hossain, M.E., Dhawan, S. & Mohmmed, A. The cysteine-rich regions of Plasmodium falciparum RON2 bind with host erythrocyte and AMA1 during merozoite invasion. Parasitol Res 110, 1711–1721 (2012). https://doi.org/10.1007/s00436-011-2690-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00436-011-2690-z