
Abstract
Eimeria tenella is a coccidian parasite of great economical importance for poultry industry. The surface of Eimeria invasive agents, sporozoites and merozoites, is coated with a family of developmentally regulated glycosylphosphatidylinositol (GPI)-linked surface antigens (SAGs), some of them involved in the initiation of the infection process. Using 2D gel electrophoresis followed by mass spectrometry, an antigenic surface protein EtSAG1 (TA4) of E. tenella sporozoites has been identified as a target of neutralizing monoclonal antibody 2H10E3. To clarify the mechanism of invasion inhibition caused by the EtSAG1-specific antibodies, a structural model of EtSAG1 was generated. It appears that “EtSAG fold” does not bear an evolutionary relationship to any known protein structure. The intra- and interchain disulfide bonds could be assigned to certain pairs of six conserved cysteines found in members of the EtSAG protein family. The outward-facing surface of the antigen was found to comprise an expanded positively charged patch, thus suggesting that the parasite invasion process may be initiated by sporozoite attachment to negatively charged sulfated proteoglycans on the surface of the host cell.
Similar content being viewed by others


Introduction
Eimeria species, causative agents of avian coccidiosis, are the most important parasites in poultry. They impair bird growth and feed utilization, cause increased mortality rates, and lead worldwide to tremendous economic losses in the poultry industry (Shirley et al. 2005; Williams 1999). The complex life cycle of Eimeria comprises an exogenous phase in the environment during which excreted oocysts undergo sporulation. After infection via ingestion of sporulated oocysts, an endogenous phase in the chicken intestine consisting of asexual reproduction (schizogony) and sexual differentiation (gamogony) takes place, followed by fertilization and shedding of unsporulated oocysts (Fernando 1990). The key step in the disease process in the chicken is the invasion of gut epithelial cells by the parasite. Gut epithelium invasion is accomplished by sporozoites and merozoites as the extracellular invasive stages and represents an attractive target for orally applied inhibitory antibodies.
Conventional control strategies for coccidiosis depend on vaccination and prophylactic use of anti-coccidial drugs. However, resistance against anti-coccidial compounds has already spread. Vaccination strategies with virulent or attenuated Eimeria strains have been routinely used for 50 years, but the large-scale production of parasites is relatively laborious and expensive. Limited progress has been achieved towards the development of subunit or recombinant vaccines, the major hurdle being the identification of protective antigens (Allen and Fetterer 2002; Dalloul and Lillehoj 2005; Shirley et al. 2005, 2007). During the last years, a number of monoclonal antibodies specific for different Eimeria species have been generated; for several of them, neutralization of sporozoites in an in vitro assay has been shown (Augustine 2001b; Danforth 1983; Sasai et al. 1996; Uchida et al. 1997; Whitmire et al. 1988). Furthermore, application of some monoclonal antibodies, administered intravenously or intraperitoneally for passive immunization, led to a significant reduction of the oocyst output and/or the lesion scores in the chicken gut (Crane et al. 1988; Karim et al. 1996; Wallach et al. 1990). The neutralizing antibodies provide a very useful tool for the identification of parasite antigens, which could potentially be applicable for the generation of recombinant subunit vaccines against coccidia.
In the present work, we analyzed the antigen specificity of a murine monoclonal antibody (mAb) 2H10E3 (Zgrzebski 1994) which was generated by immunizing mice with a microneme fraction of sporozoites of Eimeria tenella, one of the most economically important Eimeria species (Shirley et al. 2005). In this report, we demonstrate the sporozoite-neutralizing activity of mAb 2H10E3 in vitro and describe the determination of its species specificity, identification of the recognized sporozoite surface antigen, as well as generation and characterization of the corresponding recombinant counterpart.
Materials and methods
Production of the monoclonal antibody 2H10E3 and its Fab fragment
The hybridoma line 2H10E3 was grown in Dulbecco's modified Eagle medium (DMEM; Invitrogen GmbH, Karlsruhe, Germany) supplemented with 10% fetal calf serum (FCS, Invitrogen), 1% sodium pyruvate (Sigma-Aldrich Chemie GmbH, Munich, Germany), 1% HEPES (Sigma-Aldrich), 2% l-glutamine (Invitrogen), and 2% penicillin/streptomycin (Invitrogen) at 37°C and 5% CO2. The mAb was isolated from the cell culture supernatant via Protein G-Agarose (Sigma-Aldrich) according to the manufacturer’s instructions. The column-bound antibody was eluted with 10 mM glycine–HCl, pH 3.0, followed by immediate neutralization with 0.5 M Tris–HCl, pH 8.0. Eluted fractions were pooled and thoroughly dialyzed against phosphate-buffered saline (PBS; 137 mM NaCl, 3 mM KCl, 8 mM Na2PO4, 1.5 mM KH2PO4, pH 7.4) or DMEM-light (1.8 mM CaCl2⋅2H2O, 5.4 mM KCl, 0.8 mM MgSO4⋅7H2O, 110 mM NaCl, 44 mM NaHCO3, 1 mM NaH2PO4⋅2H2O, pH 7.4). Fab 2H10E3 was obtained by digesting the mAb with papain followed by separation from the non-cleaved antibody on Protein A-Agarose. For digestion, 5 mg mAb in 5 ml PBS was mixed with 10 mg papain–agarose (Sigma-Aldrich) suspended in 5-ml digestion buffer (22 mM cysteine–HCl in PBS, pH 7.0) and incubated for 1 h at 37°C on a rocking platform. Insoluble enzyme–agarose was removed by centrifugation and the cleared supernatant was filtered, dialyzed against 100 mM Tris–HCl (pH 8.5), and used for loading on a Protein A-Agarose column (Sigma-Aldrich). Flow-through fractions were collected and the protein composition was analyzed by sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS-PAGE) followed by Coomassie staining. Fab purity was determined by enzyme-linked immunosorbent assay (ELISA) with peroxidase (POD)-conjugated goat anti-mouse, Fc- and Fab-specific antibodies (Jackson ImmunoResearch Laboratories, USA). To reach 90–95% purity of the Fab fragment, the chromatography on a Protein A-Agarose column was repeated five times. The resulting fractions of Fab fragments were dialyzed against PBS (or DMEM-light for invasion inhibition assay) and concentrated using Vivaspin-6 or -20 (5,000 MW polyether sulfone, Sartorius AG, Göttingen, Germany). The concentrations of protein preparations were determined according to Bradford (1976) using the Bio-Rad Protein Assay (Bio-Rad Laboratories GmbH, Munich, Germany).
Isolation of parasite material
For indirect fluorescence antibody test (IFAT), invasion inhibition assays, and flow cytometry, viable sporozoites were isolated from freshly passaged and sporulated Eimeria oocysts essentially as described by Raether et al. (1995). In brief, the oocysts were mechanically disrupted by vortexing with glass beads in the medium for excystation Hank's balanced salt solution (HBSS) with 1% Taurodeoxycholate (Invitrogen), 0.25% trypsin. After 2-h excystation at 41°C, the sporozoites were purified by Percoll density gradient centrifugation (Invitrogen; Tomley 1997). Cryopreservation of sporozoites was carried out as described (Shirley 1995). For ELISA, the protein extracts from sporulated oocysts, sporozoites and merozoites, isolated as described (Stotish and Wang 1975), were prepared by vortexing the parasite stages with glass beads in PBS containing 1 mM phenylmethylsulphonyl fluoride, followed by six freeze–thaw cycles and sonication on ice for parasite disruption. Soluble extracts were obtained by additional centrifugation for 10 min at 13,000×g. For ELISA, 0.4 or 0.8 μg total oocyst extract was coated per well of a microtiter plate, and 5 or 10 μg was used per lane in Western blot analyses.
Invasion inhibition assay
The invasion inhibition assay was based on the observation that Eimeria sporozoites are able to invade cultured Madin–Darby bovine kidney (MDBK) cells (Madin and Darby 1958). The MDBK cells were grown in DMEM (Invitrogen) with 10% FCS (Invitrogen), 1% sodium pyruvate (Sigma-Aldrich), 1% HEPES (Sigma-Aldrich), 2% l-glutamine (Invitrogen), and 2% penicillin/streptomycin (Invitrogen) at 37°C and 5% CO2. The assay was performed essentially as previously described (Labbe et al. 2005; Schubert et al. 2005). The washed E. tenella sporozoites (freshly isolated or thawed from cryopreservation) were labeled in HBSS (Invitrogen) with 1 μM 5,6-carboxy-succinimidyl-fluoresceine-ester (Invitrogen) for 30–60 min and washed twice with DMEM medium supplemented with 2.5% FCS (Invitrogen). The labeled sporozoites (30,000–100,000/well) were preincubated with different dilutions of mAb or Fab (1–50 μg/well) or with the buffer control for 2 h at RT and thereafter used for infection of MDBK cells. One day prior the infection, the wells of 48-well Multidishes (Nunc, Wiesbaden, Germany) were seeded with 50,000 MDBK cells in 400 μl medium. The cells were allowed to grow for 24 h to 60–80% confluence in DMEM, 10% FCS. After incubation with the antibody-coated sporozoites at 41°C for 4–16 h, the cells were washed, detached with Accutase (PAA Laboratories GmbH, Pasching, Austria), and analyzed by flow cytometry using a flow cytometer CyFlow SL (Partec GmbH, Münster, Germany). Infected cells containing the labeled sporozoites were differentiated from the fluorescence-free, non-infected MDBK cells at 530 nm. The infected cells, non-infected cells, and free sporozoites were gated using the software FloMax (Partec) for subsequent counting of the infected and non-infected cells. The deduced percentages of infected cells in the presence/absence of inhibitory antibody (Ab) were used for the calculation of the inhibition rates as follows: \( = 100 \times \left( {1 - \left[ {{{\% \,{\text{infected}}\,{\text{cells}}^{\text{Ab}} } \mathord{\left/ {\vphantom {{\% \,{\text{infected}}\,{\text{cells}}^{\text{Ab}} } {\% \,{\text{infected}}\,{\text{cells}}^{{{\text{neg}}.\,{\text{control}}}} }}} \right. } {\% \,{\text{infected}}\,{\text{cells}}^{{{\text{neg}}.\,{\text{control}}}} }}} \right]} \right) \).
For analysis of secreted proteins, the supernatants of infected cells were collected and prepared as described (Brown et al. 2000).
Enzyme-linked immunosorbent assay
For ELISA, the antigen diluted in PBS (100 μl/well) was coated on 96-well microtiter plates (MaxiSorp, Nunc) overnight at 4°C. The extract from sporulated oocysts was used at concentration of 4 or 8 μg/ml. After blocking with PBS containing 3% bovine serum albumin (BSA; PBS–BSA) and washing, the primary antibody diluted in PBS–BSA (1/3,000 in PBS–BSA, Sigma-Aldrich) was added and incubation was continued for 1 h at RT, followed by the POD-conjugated secondary antibody (1/8,000 in PBS–BSA, Sigma-Aldrich) for 1 h at RT. Washing was performed using an ELISA washer (Tecan Deutschland GmbH, Crailsheim, Germany) once with PBS-0.05% Tween-20 and three times with PBS. Bound antibody conjugate was detected via TMB/H2O2 substrate reaction, absorption at 450 nm was measured with an ELISA reader (Tecan), and the data were evaluated with the software Magellan (Tecan). The apparent affinity constants were deduced from the saturation binding experiments. For K D calculation, “One site-specific binding” model of the software Prism (GraphPad, San Diego, CA, USA) was used.
Indirect fluorescence antibody test
For IFAT, the isolated sporozoites of E. tenella were air-dried on Lab-Tek™ Chamber Slides™ (Nunc, 178599). For fluorescent staining of intracellular sporozoites, HepG2 cells (Aden et al. 1979) were grown in Lab-Tek™ II Chamber Slides™ (Nunc, 154534) in DMEM supplemented with 10% FCS, 1% sodium pyruvate, 1% non-essential amino acids (Invitrogen), 2% l-glutamine (Invitrogen), 2% sodium bicarbonate (Invitrogen), and 2% penicillin/streptomycin at 37°C and 5% CO2 overnight followed by infection with 30,000 sporozoites per chamber for at least 2 h. Sporozoites and HepG2 cells, respectively, were fixed and permeabilized by methanol treatment (90% methanol in 100 mM MES, 1 mM EGTA, 1 mM MgCl2, pH 6.9) for 5 min. After blocking for 1 h with 5% BSA–PBS, the slides were incubated with the mAb (10 μg/ml in 5% BSA–PBS, 0.05% Tween-20) for 1 h at RT. The bound antibody was detected by the Alexa488-conjugated anti-mouse antibody (Invitrogen), 1/500 dilution in 5% BSA–PBS, 0.05% Tween-20. After each antibody incubation, the slides were thoroughly washed twice with PBS, 0.05% Tween-20, and once with PBS. In addition, the nuclei of HepG2 cells were stained with DAPI (4′,6-diamidin-2′-phenylindoldihydrochlorid; Invitrogen; 3 μl 0.02% stock solution per 1 ml PBS) for 15 min during the final washing step. After removal from the chambers, the glass slides were mounted with Aqua-Poly/Mount, water-soluble non-fluorescing mounting medium (Polysciences Europe GmbH, Eppelheim Germany). The specimens were examined either by phase contrast light microscopy or by fluorescence microscopy with blue light (470–490 nm) using an Axioplan 2 imaging mot and the AxioCam MR (Carl Zeiss Jena GmbH, Jena, Germany).
One-dimensional SDS-PAGE and Western blot analyses
Complex protein extracts from sporulated oocysts and recombinant antigens were boiled either in reducing or non-reducing sample buffer Roti®-Load 1 or Roti®-Load 2 (Roth, Karlsruhe, Germany), respectively, prior to loading on 15% SDS-PAA gel for electrophoresis. The separated proteins were transferred on a Protran® nitrocellulose membrane (Whatman, Dassel, Germany), blocked with 3% BSA–Tris-buffered saline (TBS) for 1 h and incubated either with the mAb 2H10E3 (1/500 in 1% BSA–TBS) or with an mouse anti-StrepII antibody (IBA, Göttingen, Germany, 1/5,000 in 1% BSA–TBS), respectively. Detection of bound antibodies was performed with an AP-conjugated goat anti-mouse antibody (Sigma-Aldrich, 1/30,000 in 1% BSA–TBS) for 1 h followed by adding NBT-BCIP substrate (Sigma-Aldrich). Between incubations with different antibodies, the membranes were washed twice with TBS-0.05% Tween-20 and once with TBS for 5 min each.
2DE and protein staining
Protein extracts were subjected to isoelectric focusing (IEF) and subsequent SDS–PAGE as described (Schlesier and Mock 2006) using buffers without dithiothreitol as reducing agent. For loading by rehydration, 35 μg of the protein extract was applied to immobilized pH gradient (IPG) strips of 7 cm in length with a pH gradient of 3–10. IEF on an IPGphor 2 unit (GE Healthcare) was carried out at 20°C with an active rehydration step of 14 h (50 V), then 30 min gradient to 250 V, 30 min gradient to 500 V, 30 min gradient to 4,000 V and 5.30 h 4,000 V with a total of about 25 kVh. Before the second dimension, the strips were equilibrated for 15 min in buffer A (50 mM Tris–HCl, pH 8.8, 6 M urea, 30%, v/v, glycerol, 2%, w/v, SDS, 0.01% bromphenol blue) and then 15 min in buffer B (50 mM Tris–HCl, pH 8.8, 6 M urea, 30%, v/v, glycerol, 2%, w/v, SDS, 135 mM iodoacetamide, 0.01% bromphenol blue). The second dimension was performed on 11.25% SDS-PAA gels covered with 0.5% agarose using a Hoefer S600 apparatus (GE Healthcare). Afterwards, the gels were washed in water for 5 min and the protein spots were visualized using ruthenium(II)-tris-(bathophenanthroline-disulphonate) staining (RuBP) as described (Lamanda et al. 2004). In short, the two-dimensional electrophoresis (2DE) gels were incubated in 30% EtOH, 10% acetic acid overnight at 4°C and washed four times with 20% EtOH for 30 min. Staining was performed with 1 μM RuBP solution in 20% EtOH for 6 h in the dark with shaking. Stained 2DE gels were washed in water for 10 min and destained in 40% EtOH, 10% acetic acid overnight at 4°C. Prior to scanning, the gels were equilibrated twice in water for 10 min. The image acquisition was performed on Fuji Image Reader FLA-5100 (Fuji Film, Tokyo, Japan) equipped with FLA-5000 v1.0 software. The following scanning parameters were used: resolution 100 μm, 16-bit pictures, excitation wavelength 473 nm, emission filter 580 nm.
Mass spectrometry
Spots selected for mass spectrometry (MS) analysis were excised from the gel using a spot picker (Proteineer spII., Bruker Daltonics, Bremen, Germany), washed, and digested with trypsin as previously described (Witzel et al. 2007). Acquisition of peptide mass fingerprint data was performed on a REFLEX III matrix-assisted laser desorption/ionization–time of flight mass spectrometer (Bruker Daltonics) operating in a reflector mode. The spectra were calibrated using external calibration and subsequent internal mass correction. Protein identification was performed with the MASCOT search engine (Matrix Science, London, UK) searching for all entries in the NCBI non-redundant protein sequence database. The following parameters were used for the search: monoisotopic mass accuracy, 100- to 200-ppm tolerance, missed cleavages 1, and the allowed variable modifications: oxidation (Met), propionamide (Cys) and carbamidomethyl (Cys). For further characterization, the samples were subjected to analysis by electrospray ionization quadrupole time-of-flight tandem MS (nanoLC-ESI-Q-ToF MS/MS) and de novo sequencing according to Witzel et al. (2007) with minor modifications. Two microliters of the digest was subjected to nanoscale RP LC analysis on a nanoAcquity UPLC system (Waters Corporation, Milford, MA, USA). A 20-mm × 180-μm Symmetry 5-μm column was used for concentration and desalting of the digest samples. The subsequent peptide elution was performed on a 100-mm × 100-μm BEH130 C18 1.7-μm column (Waters Corporation). The eluate from the analytical column was directed to the NanoLockSpray source of a Q/ToF Premier hybrid orthogonal accelerated time-of-flight (oa-ToF) mass spectrometer (Waters Corporation). MS and MS/MS data were acquired in a continuum mode using MassLynx 4.1 software (Waters Corporation). Data processing and database searching of de novo sequences against a protein index of the non-redundant NCBI database was done using ProteinLynx GlobalSERVER v2.3 software (Waters). A 10 ppm peptide, 0.1-Da fragment tolerance, one missed cleavage and variable oxidation (Met) and propionamide (Cys) were used as search parameters.
Generation of recombinant Eimeria antigens
As a source of genetic material, an E. tenella sporozoite-specific cDNA library (λ ZAP II; Refega et al. 2003) was used, which was kindly provided by Dr. Marie Labbé (Laboratoire de Virologie et Immunologie Moléculaires INRA F 78352, Jouy-en-Josas, France). A gene encoding a 25-kDa precursor of a GPI-anchored surface antigen (EtSAG1; original name TA4 antigen; EMBL accession AJ586531.2) of E. tenella sporozoites (Brothers et al. 1988) devoid of GPI signal and anchor sequence was amplified by polymerase chain reaction (PCR) using a forward (sense) primer, 5′-ATG GTA GGT CTC AGG CCA TGC AGG ATT ACC CAA CAG CAG T, and a reverse (anti-sense) primer, 5′-ATG GTA GGT CTC AGC GCT GAC TGG AGA AAC TCC GCC CTT C and cloned into BsaI-digested pASK-IBA2 expression vector (IBA). In the generated plasmid, the EtSAG1 gene was placed under transcriptional control of the tetracycline promoter/operator and was preceded by the OmpA signal sequence for secretion of the recombinant protein into the bacterial periplasm. In addition, the expression product contained a C-terminal Strep-Tactin affinity tag (Strep-tag II) for purification. For similar cloning into BsaI-digested pASK-IBA2 plasmid, a gene encoding a 19-kDa sporozoite antigen of E. tenella antigen was amplified by PCR using the following primer combination: 5′-ATG GTA GGT CTC AGG CCG GAG AAG CAG ACA CCC AGG CC and 5′-ATG GTA GGT CTC AGC GCT GAA GCC GGA CTG GTG AAG GTA C. The expression of recombinant antigens was induced in Escherichia coli (BL21 DE3) shaking cultures by adding anhydrotetracycline to a final concentration of 200 µg/l. Purification of the recombinant proteins was carried out by affinity chromatography on a Strep-Tactin Sepharose column according to the manufacturer's instructions.
Molecular modeling of EtSAG1
BLAST (Altschul et al. 1997) search for 3D templates found no close homolog of EtSAG1 in the pdb databases. Therefore, secondary structure prediction and fold recognition were performed using GeneSilico Metaserver (Kurowski and Bujnicki 2003), which provides common interface to several protein structure prediction methods. The majority of the fold recognition methods [ffas (Rychlewski et al. 2000), mgenthreader (McGuffin and Jones 2003), fugue (Shi et al. 2001), inub (Fischer 2000), and pcons5 (Wallner and Elofsson 2005)] predicted the same SCOP fold d.111.1.1, although the prediction scores were quite low. The crystal structure of Na-ASP-2, a PR-1 protein from the nematode parasite Necator americanus (PDB ID 1U53; Asojo et al. 2005), was ultimately selected as a 3D template for modeling, although the sequence homology between EtSAG1 and the template was only 15%. The C-terminal part of EtSAG1 adjacent to the GPI-anchor peptide appeared to have no suitable template at all and, therefore, was omitted from the model. The EtSAG1 template sequence alignment was performed manually, then Modeller9v1 (Sali and Blundell 1993) was used both for homology modeling and for the model optimization. Since Cys11 and Cys94 residues occurred close to each other, they were defined as bound. Ten model variants were generated; the best model was selected on the basis of visual inspection and evaluation of the model quality with PROCHECK (Laskowski et al. 1996). The figures were prepared using the program PyMOL (DeLano Scientific, Palo Alto, CA, USA).
Results
Effect of antibody 2H10E3 on invasion of cultured cells by sporozoites of E. tenella
The monoclonal antibody derived from hybridoma 2H10E3 and its Fab fragment were tested for the ability to inhibit the invasion of cultured MDBK cells with sporozoites of E. tenella. Pretreatment with the antibody significantly decreased the invasion capacity of the sporozoites; the observed inhibition effect was dose-dependent (Fig. 1a, b). Under the used experimental conditions, the inhibition plateau of 45–50% was reached at an antibody concentration of 25 nM (1.6 μg/well). The mAb 2H10E3-derived Fab fragment demonstrated similar sporozoite-neutralizing activity, although the invasion inhibition effect was somewhat weaker probably due to its monovalency (Fig. 1c). In a comparative study with both used at concentration of 10 μg/well, the mAb and the Fab fragment demonstrated the inhibition values of 23% and 16%, respectively.

Inhibition of sporozoite invasion in vitro by anti-Eimeria mAb 2H10E3 and its Fab fragment. a Representative flow cytometry charts used to determine the inhibitory activity of the mAb 2H10E3 on labeled sporozoites of E. tenella. Left panel no infection; middle panel infected cells (negative control); right panel infection with sporozoites preincubated with inhibitory mAb 2H10E3 at concentration of 100 nM. In this example, a 48% inhibition was observed. b Dose-dependent inhibition of sporozoite invasion in vitro by mAb 2H10E3. c Comparison of the inhibitory activity of mAb 2H10E3 and its Fab fragment at concentration 10 μg/well
Analysis of antigen-binding activity and specificity of antibody 2H10E3
Binding of mAb 2H10E3 to protein extracts from different developmental stages of E. tenella was assessed by ELISA and by Western blot analysis. The antibody specifically bound to the immobilized extract from sporulated oocysts of E. tenella. Saturation binding experiments which were performed using three independent mAb preparations (Fig. 2a) resulted in an apparent affinity constant (K D) in sub-nanomolar range, 0.73 ± 0.09 nM.

Analyses of antigen-binding activity of mAb 2H10E3 by ELISA using the oocyst extracts (a) and by Western blot analysis under non-reducing conditions (b). Lanes M molecular weight markers (the positions in kilodalton are indicated on the left); 1,2 extracts of merozoites (10 μg/lane); 3,4 extracts of oocysts (10 μg/lane); 5,6 extracts of sporozoites (1 μg/lane)
SDS-PAGE separation under non-reducing conditions followed by Western blot analysis using mAb 2H10E3 revealed three protein bands with approximate molecular masses of 24.5, 22.0, and 18.5 kDa in protein extracts from oocysts and sporozoites, but not in extracts of merozoites (Fig. 2b). Interestingly, the antigen could also be detected in supernatants of infected MDBK cells (see below). In contrast to non-reducing SDS-PAGE, no antibody binding was detected when gel analysis was repeated under reducing conditions (data not shown), thus indicating the importance of maintaining intact disulfide bonds in the structure of epitope recognized by the antibody.
IFAT using mAb 2H10E3 identified the location of the corresponding antigen on the surface both of free (extracellular) sporozoites and of sporozoites after invasion into the cell (Fig. 3). Interestingly, intracellular staining has revealed a fluorescent trail behind the sporozoite, which disappeared after more than 8 h post-infection (Fig. 3b–d). This observation indicates antigen shedding outside and inside the infected cells.

Immunolocalization of the antigen recognized by mAb 2H10E3 as determined by indirect fluorescent antibody test (IFAT). Fluorescence microscopy images are shown either of extracellular sporozoites (a) or intracellular sporozoites in infected HepG2 cells 2 h (b), 6 h (c), or 8 h (d) after infection. Antigen staining was performed with mAb 2H10E3 followed by Alexa488-conjugated anti-mouse antibody. Arrows indicate trace signals seen shortly after invasion behind the invading sporozoite (≤6 h). The nuclei of HepG2 cells are colored violet
Identification of a protein target for neutralizing antibody 2H10E3
The protein extracts from sporulated oocysts were separated on 2D SDS-PAA gels under non-reducing conditions and used either for Western blot analysis or for fluorescent staining with RuBP. The first gel revealed a single spot recognized by the antibody (Fig. 4a). The corresponding stained spot was cut out from the second gel (Fig. 4b) and used for digestion with trypsin. The tryptic peptide mass fingerprint data were collected by MALDI-ToF MS analysis and the database search for the match of the six found peptides resulted in the identification of a 19-kDa sporozoite antigen of E. tenella (Rosenberg et al. 2005), GeneBank GI 56967488 (Fig. 4c). Further analysis of the same sample by ESI-Q-ToF MS/MS could determine de novo sequences of two peptides. One peptide was derived from the already found 19-kDa sporozoite antigen, but the second one appeared to be derived from the sporozoite surface protein T4 (EtSAG1; Brothers et al. 1988; Tabares et al. 2004), GeneBank GI 158877 (Fig. 5).

Identification of E. tenella antigens recognized by mAb 2H10E3 using MALDI-ToF MS analysis of protein spot 01. a Western blot analysis of 2D gel. The positively stained spot is indicated with an arrow. b RuBP-stained second 2D gel used for isolation of the positive spot (indicated by an arrow). c MS profile with indicated masses of peptides derived from the 19-kDa sporozoite antigen of E. tenella. The inset shows the amino acid sequence of the corresponding protein where found peptides are marked in bold

Identification of E. tenella antigens recognized by mAb 2H10E3 using ESI-Q-TOF MS analysis of protein spot 01. Data processing and database searching led to identification of the 19-kDa sporozoite antigen of E. tenella (a) and EtSAG1 antigen (b). The insets show the amino acid sequences of the corresponding proteins where the peptide sequences, which were used for identification in the database search, are indicated in bold. The EtSAG1 sequence is shown without a signal peptide. The protease cleavage site at EtSAG1 (RRL) is underlined
To confirm the antigen specificity of the antibody 2H10E3, the genes encoding both EtSAG1 and 19-kDa antigens of E. tenella were cloned and expressed in bacteria. Since the EtSAG1 antigen as isolated from the parasite is composed of a 17-kDa polypeptide (large subunit) and a 8-kDa polypeptide (small subunit) linked by a disulfide bridge (Brothers et al. 1988), recombinant EtSAG1 protein was produced in bacteria as a 25-kDa precursor without a signal peptide and a GPI anchor. Both recombinant antigens were isolated and purified to homogeneity (data not shown). Western blot analysis and ELISA demonstrated that the mAb 2H10E3 specifically recognized only EtSAG1; no binding to 19-kDa antigen was observed (Fig. 6). Accordingly, the antibody seemed to recognize only a large 17-kDa subunit of EtSAG1 in the oocyst extract of E. tenella (Fig. 6b). However, the number of protein bands detected by the antibody differed between the preparations of the oocyst extract (compare Figs. 2b and 6b).

Analyses of binding of mAb 2H10E3 to recombinant E. tenella antigens. a Binding to immobilized EtSAG1 and 19-kDa antigens (0.5 and 2 μg/well), as determined by ELISA. As a positive control, the protein extract from oocysts of E. tenella (0.4 μg/well) was used. Means and SDs of duplicates are shown. b SDS-PAGE and Western blot analysis. Lanes M molecular mass markers (values in kilodalton are shown on the left); 1 recombinant EtSAG1; 2 recombinant 19-kDa antigen; 3 oocyst extract of E. tenella
Based on the preliminary observation that the antigen was shed into the culture medium of the sporozoite-infected cells, comparative Western blot analysis was performed using supernatants of infected MDBK cells and the protein extracts of sporulated oocysts. The results demonstrated that while in oocyst extract mainly a large subunit of matured and processed EtSAG1 was recognized by mAb 2H10E3, different forms of the antigen precursor could be found in the supernatants of infected cells (Fig. 7).

Comparison of antigen species recognized by the antibody 2H10E3 in supernatants of sporozoite-infected MDBK cells and in sporulated oocysts of E. tenella. a Western blot analysis. Lanes M molecular mass markers (the values in kilodalton are shown on the left); 1 supernatant of infected MDBK cells; 2 supernatant of non-infected MDBK cells (negative control); 3 protein extract from oocysts of E. tenella. b Schematic outline of EtSAG1 precursor and of putative protein species found in supernatants of infected cells. SP signal peptide, RRL Arg-Arg-Leu cleavage site between the 17- and 8-kDa subunits of mature EtSAG1, GPI-AP GPI anchor peptide. The molecular masses in kilodalton were calculated for each protein species on the basis of its amino acid sequence using Protein Calculator v3.3 software (http://www.scripps.edu/cgi-bin/cdputnam/protcalc3) and are indicated
Analyses of species cross-reactivity of antibody 2H10E3
For analysis of cross-reactivity of the antibody 2H10E3 with other Eimeria species, complex soluble antigens were prepared from sporulated oocysts of four avian (E. tenella, E. acervulina, E. brunetti and E. maxima) and two rodent Eimeria species (E. papillata and E. nieschulzi) and tested in ELISA and Western blot analyses. The ELISA data demonstrated specific interaction of the antibody only with the extract from oocysts of E. tenella (Fig. 8). These results were confirmed by Western blot analysis (data not shown). The antibody 2H10E3 did not show any reactivity with the protein preparations from other species, thus indicating that it recognizes an epitope of the EtSAG1 antigen which is unique for E. tenella.

Species specificity of the antibody 2H10E3 as determined by ELISA using oocyst extracts from four avian (E. tenella, E. maxima, E. acervulina, E. brunetti) and two rodent (E. papillata and E. nieschulzi) Eimeria species. In parallel, binding to recombinant EtSAG1 and 19-kDa antigens was tested. As a negative control, secondary HRP-conjugated goat anti-mouse IgG antibody alone was used
Computer modeling of EtSAG1 and implications for antibody–antigen interactions
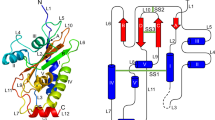
To understand the mechanism of blocking the sporozoite infectivity by the antibody 2H10E3, we have performed an in silico analysis of the EtSAG1 structure. The model was generated only in part by homology modeling since in Eimeria, the GPI-anchored surface antigens have not been yet structurally characterized and no proper pdb template was available (see “Materials and methods”). Nevertheless, the fold recognition methods allowed us to find out a template for the N-terminal part of EtSAG1. As revealed in Figs. 9 and 10, the overall fold of EtSAG1 is composed of one three-stranded beta-sheet surrounded by four alpha helices and loops which are cross-linked by three disulfide bonds. Two disulfide bridges (Cys11–Cys94 and Cys60–Cys151) provide intramolecular stabilization of the large subunit, while the bond Cys146–Cys171 links the large and small EtSAG1 chains together. Interestingly, the central beta-sheet is composed of one strand from the small subunit placed in between two strands from the large subunit (Fig. 9).

Two ribbon views of the EtSAG1 model. The large and small polypeptide subunits are in green and in red, respectively. The C terminus of the small subunit was extended with a dotted line to show the location of the GPI anchor on the sporozoite membrane. The strands of the anti-parallel beta-sheet (a–c) and alpha helices (A–D) are indicated. The putative disulfide bridges between cysteines at positions 11-94, 60-151, and 146-171 are shown in yellow, magenta, and cyan, respectively. N- and C-terminal amino acids of the large (positions 1 and 158) as well as of the small subunits (positions 161 and 188) are indicated

Amino acid sequence (a) and a planar secondary structure diagram (b) of EtSAG1 heterodimer. The disulfide bond-forming cysteines are connected with dashed lines. In a, the protease cleavage site (RRL) is shown in black and underlined. In b, N and C termini of the large (green) and small (red) subunits of EtSAG1 are indicated. The alpha helices and beta strands are shown as cylinders (or bars in a) and arrows, respectively
The essential part of the outward facing surface of EtSAG1 was found to be highly positively charged, whereas the part adjacent to the GPI anchor is charged negatively (Fig. 11). Limited trypsin digests experiments under non-denaturing conditions demonstrated that the epitope recognized by the antibody 2H10E3 was destroyed upon cleavage of the recombinant EtSAG1 somewhere between Arg68 and Lys77 (data not shown). This finding indicates that the antibody most probably recognizes the loop between residues 60 and 80 (Fig. 10a) which is involved in the formation of the basic surface of EtSAG1.

Side view and bottom view of the EtSAG1 electrostatic potential surface. The relative charge from negative to neutral to positive is colored from red to white to blue, respectively. The projections of some charged amino acids are indicated
Discussion
The sporozoites of E. tenella, one of the most pathogenic coccidian parasites of poultry, invade intestinal cells of chickens and cause diarrhea during their development. Despite their significance for animal production and human health, the process of sporozoite invasion into host cells by Eimeria spp. and other apicomlexan parasites is not yet completely understood. For elucidation of the process of invasion, the antigen specificity of an inhibitory monoclonal antibody (2H10E3) has been analyzed. Invasion inhibition assays in vitro clearly demonstrated reduced sporozoite invasion in the presence of this monoclonal antibody. We aimed, therefore, at the identification of the recognized sporozoite antigen to clarify the process and mechanism of parasite invasion as well as to develop potentially alternative immunization strategies. After separation of complex antigen extracts from the Eimeria oocysts in 2D SDS-PAA gels, two antigen candidates have been identified based on mass spectrometric data. Characterization of the corresponding recombinant counterparts confirmed that EtSAG1 was the only target antigen, whereas the 19-kDa sporozoite antigen of E. tenella was attributed to be as a co-eluting protein of similar molecular weight and isoelectric point. EtSAG1 has already been found previously to be a molecular target for a number of sporozoite-neutralizing antibodies using similar 2D electrophoresis approach (Brothers et al. 1988; de Venevelles et al. 2004). However, identification of the corresponding antigen was performed in our study using much smaller amounts of the parasite material, i.e., only 35 μg of oocyst extract vs. 0.1–1.0 mg in earlier reports (de Venevelles et al. 2004; Sutton et al. 1989). To confirm the antigen specificity, we expressed EtSAG1 as a soluble recombinant protein correctly folded in bacterial periplasm, which could be purified under non-denaturing conditions in reasonable yields. Previously described attempts to generate recombinant EtSAG1 resulted in the accumulation of the protein product in bacterial inclusion bodies, thus requiring additional refolding in vitro for recovery of the functional antigen (Brothers et al. 1988).
EtSAG1 is a GPI-anchored protein which can be released from the sporozoite membrane by treatment with bacterial phosphatidylinositol-specific phospholipase C (Tabares et al. 2004). However, antigen shedding was observed by us and others in fluorescent studies of extracellular sporozoites when the sporozoites were fixed on a glass slide after moving time of 10 min. The antigen was detected both on the surface of sporozoites and on the slide itself. Indirect evidence of EtSAG1 shedding was also provided by the fact that this antigen was obviously present in the microneme fraction used for the immunization of mice, leading to hybridoma 2H10E3. A similar observation was previously made for another GPI-anchored sporozoite surface antigen of E. tenella, EtSAG13, which initially had been characterized as a potential microneme protein, EtMIC6, as it co-purifies with the microneme organelles on sucrose density gradients (Tabares et al. 2004; Tomley and Soldati 2001). In the present study, we demonstrated for the first time shedding of EtSAG1 from the surface of intracellular sporozoites by both Western blot analysis and IFAT. The IFAT experiments clearly revealed a fluorescent trail transiently appearing behind the invading sporozoite. In addition, EtSAG1 could also be seen on the surface of intracellular sporozoites. However, the function of detached and surface-bound EtSAG1 inside the invaded cell remains still unclear. A role in signal transduction across the plasma membranes was assigned earlier to a number of GPI-anchored proteins (Sharom and Radeva 2004). It is also known that GPI anchor can be cleaved by endogenous and exogenous PI-specific phospholipases, thus leading to the release of the extracellular part of the receptor (Sharom and Lehto 2002). The phenomenon of trail formation due to the GPI-anchored surface antigens was also observed by others when analyzing specificity of antibodies against Eimeria merozoites (Bauer et al. 1995).
To clarify the mechanism of invasion inhibition caused by the EtSAG1-specific antibody, we generated a 3D structural model of EtSAG1 as a member of EtSAG protein family. Recent analysis of available EST sequences of E. tenella allowed identification of full-length genes encoding 23 different GPI-anchored EtSAGs that clearly fall into two distinct groups, A (EtSAGs 1–12) and B (EtSAGs 13–23; Tabares et al. 2004). Screening of all available sequence databases with the 23 EtSAG sequences, using a variety of search algorithms, did not identify any homologous proteins from other organisms. Accordingly, no pdb template could be found for homology modeling of the whole protein. It appears that “EtSAG fold” does not bear an evolutionary relationship to any known protein structure. The model generated in the present study sheds the light on structure–functional organization of EtSAGs which are characterized by the presence of six conserved cysteines.
The site and host specificity that avian Eimeria sporozoites and other apicomplexan parasites exhibit for invasion in vivo suggests that specific interactions between the sporozoites and the target host cells may mediate the invasion process. Therefore, host cell receptors facilitating the parasite entry into the target cell have been postulated, although not yet identified (reviewed in Augustine 2001a). On the other hand, there is proven ability of Eimeria sporozoites to invade into a broad range of avian and mammalian cells in vitro (Augustine 2001b). Although sporozoite motility, its structural and secreted antigens appear to provide the mechanisms for propelling the sporozoite into the host cell, the host cell seems to possess characteristics by which the sporozoites recognize and interact with the cell as a prelude to invasion. There is a growing body of evidence that EtSAG1 is closely involved into initiation of the infection process. For example, it was demonstrated in a recent study that out of six sporozoite-neutralizing antibodies selected from the phage-displayed antibody library, four specifically recognized EtSAG1 (Zimmermann et al., manuscript in preparation). In addition, we demonstrated specific binding of the recombinant EtSAG1 to MDBK cells by Western blot analysis of the cell lysates after incubation with the recombinant protein followed by thorough washing (data not shown). The identity of the EtSAG1 ligand (and other SAG proteins of Eimeria) has been a subject of extensive speculations (Augustine 2001a). Our structure of EtSAG1 provides significant insight into this issue and serves as a template for further functional investigations. The outward-facing surface of the antigen was found to comprise an expanded positively charged patch. This structural feature provides a hint that negatively charged sulfated proteoglycans may be ligands for Eimeria SAGs, as it was recently shown for structurally different SAG1 and SAG3 of the much better studied Toxoplasma gondii (He et al. 2002). Attachment of Toxoplasma to target cells in vitro is mediated at least in part by recognition of negatively charged cellular heparan sulfate proteoglycans (Jacquet et al. 2001; Ortega-Barria and Boothroyd 1999). Our hypothesis that sulfated proteoglycans on the surface of the host cell are responsible for attachment of Eimeria sporozoites has been confirmed by early observations that parasite invasion could be abrogated by pretreatment of the host cell with polycations, such as poly-l-histidine and poly-l-lysine (Augustine 1980) or cationized ferritin (Augustine and Danforth 1984).
As it was already mentioned, EtSAG1 proved to be an immunodominant sporozoite antigen and has been used in a number of studies either for active immunization approaches or as a target for passive immunization. The conventional immunization route is the vaccination using either attenuated strains or protective antigens (Allen and Fetterer 2002; Dalloul and Lillehoj 2005; Shirley et al. 2005, 2007). As an alternative strategy, coccidiosis could be prevented or mitigated by oral, intraperitoneal, or intravenous delivery of antibodies that inhibit parasite invasion. However, EtSAG1 has no close homologs in other Eimeria species, and therefore, EtSAG1-specific antibodies have limited practical applicability. We have demonstrated in our study that antibody 2H10E3 specifically interacted only with sporozoites of E. tenella; no binding to other Eimeria species was observed. Although EtSAG1 is a sporozoite antigen and is not expressed by merozoites, its close similarity with other group A members of EtSAG family, most of which are merozoite-specific (Tabares et al. 2004), indicates that the results of this study may contribute to better understanding of infection processes in coccidiosis and to developing generalized prevention strategies.
References
Aden DP, Fogel A, Plotkin S, Damjanov I, Knowles BB (1979) Controlled synthesis of HBsAg in a differentiated human liver carcinoma-derived cell line. Nature 282:615–616
Allen PC, Fetterer RH (2002) Recent advances in biology and immunobiology of Eimeria species and in diagnosis and control of infection with these coccidian parasites of poultry. Clin Microbiol Rev 15:58–65
Altschul SF, Madden TL, Schaffer AA, Zhang J, Zhang Z, Miller W, Lipman DJ (1997) Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res 25:3389–3402
Asojo OA, Goud G, Dhar K, Loukas A, Zhan B, Deumic V, Liu S, Borgstahl GE, Hotez PJ (2005) X-ray structure of Na-ASP-2, a pathogenesis-related-1 protein from the nematode parasite, Necator americanus, and a vaccine antigen for human hookworm infection. J Mol Biol 346:801–814
Augustine PC (1980) Effect of polyions, Ca++, and enzymes on penetration of cultured cells by Eimeria meleagrimitis sporozoites. J Parasitol 66:498–505
Augustine PC (2001a) Cell: sporozoite interactions and invasion by apicomplexan parasites of the genus Eimeria. Int J Parasitol 31:1–8
Augustine PC (2001b) Invasion of different cell types by sporozoites of Eimeria species and effects of monoclonal antibody 1209-C2 on invasion of cells by sporozoites of several apicomplexan parasites. J Eukaryot Microbiol 48:177–181
Augustine PC, Danforth HD (1984) Effects of cationized ferritin and neuraminidase on invasion of cultured cells by Eimeria meleagrimitis sporozoites. J Protozool 31:140–144
Bauer C, Dubremetz JF, Entzeroth R (1995) Characterization of surface antigens of Eimeria nieschulzi (Sporozoa, Eimeriidae) merozoites. Parasitol Res 81:230–234
Bradford MM (1976) A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem 72:248–254
Brothers VM, Kuhn I, Paul LS, Gabe JD, Andrews WH, Sias SR, McCaman MT, Dragon EA, Files JG (1988) Characterization of a surface antigen of Eimeria tenella sporozoites and synthesis from a cloned cDNA in Escherichia coli. Mol Biochem Parasitol 28:235–247
Brown PJ, Billington KJ, Bumstead JM, Clark JD, Tomley FM (2000) A microneme protein from Eimeria tenella with homology to the Apple domains of coagulation factor XI and plasma pre-kallikrein. Mol Biochem Parasitol 107:91–102
Crane MS, Murray PK, Gnozzio MJ, MacDonald TT (1988) Passive protection of chickens against Eimeria tenella infection by monoclonal antibody. Infect Immun 56:972–976
Dalloul RA, Lillehoj HS (2005) Recent advances in immunomodulation and vaccination strategies against coccidiosis. Avian Dis 49:1–8
Danforth HD (1983) Use of monoclonal antibodies directed against Eimeria tenella sporozoites to determine stage specificity and in vitro effect on parasite penetration and development. Am J Vet Res 44:1722–1727
de Venevelles P, Chich JF, Faigle W, Loew D, Labbe M, Girard-Misguich F, Pery P (2004) Towards a reference map of Eimeria tenella sporozoite proteins by two-dimensional electrophoresis and mass spectrometry. Int J Parasitol 34:1321–1331
Fernando AM (1990) Eimeria: infections of the intestine. In: Long PL (ed) Coccidiosis of man and domestic animals. CRC, Boca Raton, pp 63–75
Fischer D (2000) Hybrid fold recognition: combining sequence derived properties with evolutionary information. In: Altman RB, Ak D, Hunter L, Klein TE (eds) Pacific Symp Biocomputing. World Scientific, New York, pp 119–30
He XL, Grigg ME, Boothroyd JC, Garcia KC (2002) Structure of the immunodominant surface antigen from the Toxoplasma gondii SRS superfamily. Nat Struct Biol 9:606–611
Jacquet A, Coulon L, De Neve J, Daminet V, Haumont M, Garcia L, Bollen A, Jurado M, Biemans R (2001) The surface antigen SAG3 mediates the attachment of Toxoplasma gondii to cell-surface proteoglycans. Mol Biochem Parasitol 116:35–44
Karim MJ, Basak SC, Trees AJ (1996) Characterization and immunoprotective properties of a monoclonal antibody against the major oocyst wall protein of Eimeria tenella. Infect Immun 64:1227–1232
Kurowski MA, Bujnicki JM (2003) GeneSilico protein structure prediction meta-server. Nucleic Acids Res 31:3305–3307
Labbe M, de Venevelles P, Girard-Misguich F, Bourdieu C, Guillaume A, Pery P (2005) Eimeria tenella microneme protein EtMIC3: identification, localisation and role in host cell infection. Mol Biochem Parasitol 140:43–53
Lamanda A, Zahn A, Roder D, Langen H (2004) Improved ruthenium II tris (bathophenantroline disulfonate) staining and destaining protocol for a better signal-to-background ratio and improved baseline resolution. Proteomics 4:599–608
Laskowski RA, Rullmannn JA, MacArthur MW, Kaptein R, Thornton JM (1996) AQUA and PROCHECK-NMR: programs for checking the quality of protein structures solved by NMR. J Biomol NMR 8:477–486
Madin SH, Darby NB Jr (1958) Established kidney cell lines of normal adult bovine and ovine origin. Proc Soc Exp Biol Med 98:574–576
McGuffin LJ, Jones DT (2003) Improvement of the GenTHREADER method for genomic fold recognition. Bioinformatics 19:874–881
Ortega-Barria E, Boothroyd JC (1999) A toxoplasma lectin-like activity specific for sulfated polysaccharides is involved in host cell infection. J Biol Chem 274:1267–1276
Raether W, Hofmann J, Uphoff M (1995) In vitro cultivation of avian Eimeria species: Eimeria tenella. In: Eckert J, Braun R, Shirley MW, Coudert P (eds) COST 89/820, Biotechnology. Guidelines on techniques in coccidiosis research. European Commission, Belgium, pp 79–92
Refega S, Girard-Misguich F, Bourdieu C, Pery P, Labbe M (2003) Gene discovery in Eimeria tenella by immunoscreening cDNA expression libraries of sporozoites and schizonts with chicken intestinal antibodies. Vet Parasitol 113:19–33
Rosenberg B, Juckett DA, Aylsworth CF, Dimitrov NV, Ho SC, Judge JW, Kessel S, Quensen J, Wong KP, Zlatkin I, Zlatkin T (2005) Protein from intestinal Eimeria protozoan stimulates IL-12 release from dendritic cells, exhibits antitumor properties in vivo and is correlated with low intestinal tumorigenicity. Int J Cancer 114:756–765
Rychlewski L, Jaroszewski L, Li W, Godzik A (2000) Comparison of sequence profiles. Strategies for structural predictions using sequence information. Protein Sci 9:232–241
Sali A, Blundell TL (1993) Comparative protein modelling by satisfaction of spatial restraints. J Mol Biol 234:779–815
Sasai K, Lillehoj HS, Matsuda H, Wergin WP (1996) Characterization of a chicken monoclonal antibody that recognizes the apical complex of Eimeria acervulina sporozoites and partially inhibits sporozoite invasion of CD8+ T lymphocytes in vitro. J Parasitol 82:82–87
Schlesier B, Mock HP (2006) Protein isolation and second-dimension electrophoretic separation. Methods Mol Biol 323:381–391
Schubert U, Fuchs J, Zimmermann J, Jahn D, Zoufal K (2005) Extracellular calcium deficiency and ryanodine inhibit Eimeria tenella sporozoite invasion in vitro. Parasitol Res 97:59–62
Sharom FJ, Lehto MT (2002) Glycosylphosphatidylinositol-anchored proteins: structure, function, and cleavage by phosphatidylinositol-specific phospholipase C. Biochem Cell Biol 80:535–549
Sharom FJ, Radeva G (2004) GPI-anchored protein cleavage in the regulation of transmembrane signals. Subcell Biochem 37:285–315
Shi J, Blundell TL, Mizuguchi K (2001) FUGUE: sequence–structure homology recognition using environment-specific substitution tables and structure-dependent gap penalties. J Mol Biol 310:243–257
Shirley M (1995) Cryopreservation of avian Eimeria stages. In: Eckert J, Braun R, Shirley MW, Coudert P (eds). Guidelines on techniques in coccidiosis research. The European Commission DGXII, Luxembourg, pp 95–96
Shirley MW, Smith AL, Tomley FM (2005) The biology of avian Eimeria with an emphasis on their control by vaccination. Adv Parasitol 60:285–330
Shirley MW, Smith AL, Blake DP (2007) Challenges in the successful control of the avian coccidia. Vaccine 25:5540–5547
Stotish RL, Wang CC (1975) Preparation and purification of merozoites of Eimeria tenella. J Parasitol 61:700–703
Sutton CA, Shirley MW, Wisher MH (1989) Characterization of coccidial proteins by two-dimensional sodium dodecyl sulphate–polyacrylamide gel electrophoresis. Parasitology 99(Pt 2):175–187
Tabares E, Ferguson D, Clark J, Soon PE, Wan KL, Tomley F (2004) Eimeria tenella sporozoites and merozoites differentially express glycosylphosphatidylinositol-anchored variant surface proteins. Mol Biochem Parasitol 135:123–132
Tomley F (1997) Techniques for isolation and characterization of apical organelles from Eimeria tenella sporozoites. Methods 13:171–176
Tomley FM, Soldati DS (2001) Mix and match modules: structure and function of microneme proteins in apicomplexan parasites. Trends Parasitol 17:81–88
Uchida T, Kikuchi K, Takano H, Ogimoto K, Nakai Y (1997) Monoclonal antibodies inhibiting invasion of cultured cells by Eimeria tenella sporozoites. J Vet Med Sci 59:721–723
Wallach M, Pillemer G, Yarus S, Halabi A, Pugatsch T, Mencher D (1990) Passive immunization of chickens against Eimeria maxima infection with a monoclonal antibody developed against a gametocyte antigen. Infect Immun 58:557–562
Wallner B, Elofsson A (2005) Pcons5: combining consensus, structural evaluation and fold recognition scores. Bioinformatics 21:4248–4254
Whitmire WM, Kyle JE, Speer CA, Burgess DE (1988) Inhibition of penetration of cultured cells by Eimeria bovis sporozoites by monoclonal immunoglobulin G antibodies against the parasite surface protein P20. Infect Immun 56:2538–2543
Williams RB (1999) A compartmentalised model for the estimation of the cost of coccidiosis to the world's chicken production industry. Int J Parasitol 29:1209–1229
Witzel K, Surabhi GK, Jyothsnakumari G, Sudhakar C, Matros A, Mock HP (2007) Quantitative proteome analysis of barley seeds using ruthenium(II)-tris-(bathophenanthroline-disulphonate) staining. J Proteome Res 6:1325–1333
Zgrzebski G (1994) Vergleichende Untersuchung zur Lokalisation von Antigenen der Oberfläche, der refraktilen Körper, der Rhoptrien, des apikalen Poles und der Amylopektin-Granula in verschiedenen Entwicklungsstadien (Sporozoiten, Merozoiten) von Eimeria tenella (Protozoa, Coccidia) in vitro. Dissertation. Rheinische Friedrich-Wilhelms-Universität, Bonn, p 234
Acknowledgments
We thank Prof. R. Entzeroth (TU Dresden) for providing us with the hybridoma 2H10E3 and with oocysts from the rodent Eimeria species, Prof. F. Tomley (IAH Compton) for oocysts of E. tenella (strain Houghton) and Prof. A. Daugschies (University of Leipzig) for the oocysts from avian Eimeria spp. different from E. tenella. We also thank Annegret Wolf for her excellent technical assistance.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Jahn, D., Matros, A., Bakulina, A.Y. et al. Model structure of the immunodominant surface antigen of Eimeria tenella identified as a target for sporozoite-neutralizing monoclonal antibody. Parasitol Res 105, 655–668 (2009). https://doi.org/10.1007/s00436-009-1437-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00436-009-1437-6