
Abstract
Phosphoglycerate mutases (PGM) interconvert 2- and 3-phosphoglycerate in the glycolytic and gluconeogenic pathways. A putative cofactor-independent phosphoglycerate mutase gene (iPGM) was identified in the genome sequence of the Wolbachia endosymbiont from the filarial nematode, Brugia malayi (wBm). Since iPGM has no sequence or structural similarity to the cofactor-dependent phosphoglycerate mutase (dPGM) found in mammals, it may represent an attractive Wolbachia drug target. In the present study, wBm–iPGM cloned and expressed in Escherichia coli was mostly insoluble and inactive. However, the protein was successfully produced in the yeast Kluyveromyces lactis and the purified recombinant wBm–iPGM showed typical PGM activity. Our results provide a foundation for further development of wBm–iPGM as a promising new drug target for novel anti-filarial therapies that selectively target the endosymbiont.
Similar content being viewed by others

Introduction
In recent years, obligate α-proteobacterial endosymbionts belonging to the genus Wolbachia that are present in most filarial nematode species have become the focus of intense study as a new approach to chemotherapy. Studies performed in vitro or in animal models, as well as clinical trials in humans have shown the susceptibility of Wolbachia to the tetracycline family of antibiotics. Tetracycline-mediated clearance of Wolbachia from nematode tissues correlates with a block in embryogenesis and worm development and, in certain situations, leads to a significant reduction in adult worm burdens and a reduction in associated pathology (Taylor et al. 2005; Hoerauf 2006). These studies have clearly demonstrated the feasibility of treating filarial infections through anti-Wolbachia therapies, but the protracted treatment regimens required for efficacy, coupled with contra-indications for certain individuals, render tetracycline therapy in its present form unsuitable for mass treatment in endemic areas. Therefore, there is a need to discover alternative anti-Wolbachia treatments which may be facilitated by identification of new drug targets in this endosymbiont.
The completed genome sequence of the Wolbachia endosymbiont from the filarial nematode Brugia malayi (wBm; Foster et al. 2005) has generated an unrivalled resource for identification of enzymes and processes that are either lacking in mammals or differ substantially from their mammalian counterparts, warranting their further evaluation as candidate drug targets. We have identified a putative cofactor-independent phosphoglycerate mutase (iPGM) in the catalog of proteins predicted by the wBm genome sequence. Phosphoglycerate mutase (PGM) catalyzes the interconversion of 2- and 3-phosphoglycerate (2-PG and 3-PG) in the glycolytic and gluconeogenic pathways that are essential in most organisms. PGM exists in two distinct forms, iPGM and cofactor-dependent phosphoglycerate mutase, dPGM (Fothergill-Gilmore and Watson 1989; Jedrzejas 2000). The iPGM proteins are ~57 kD monomers and promote the intramolecular transfer of the phosphoryl group between the monophosphoglycerates through a phosphoserine intermediate. Conversely, dPGM is ~27 kD and is usually active as a dimer or tetramer. It catalyzes the intermolecular transfer of the phosphoryl group between the monophosphoglycerates and the cofactor (2,3-diphosphoglycerate) via a phosphohistidine intermediate. Furthermore, there is no similarity in either the primary sequences or three-dimensional structures of iPGM and dPGM enzymes (Fothergill-Gilmore and Watson 1989; Jedrzejas 2000). Vertebrates possess only dPGM (Carreras et al. 1982), which has raised the suggestion that iPGM may represent a potential drug target in pathogenic organisms which contain only that form (Fraser et al. 1999; Galperin and Jedrzejas 2001; Zhang et al. 2004). Our genomic analysis of wBm, the Wolbachia endosymbiont from B. malayi, indicated that this bacterium encodes only iPGM which we have cloned and expressed in active form.
Materials and methods
Cloning of wBm–iPGM
The wBM–iPGM open reading frame was amplified from a bacterial artificial chromosome, BMBAC39G04 (Foster et al. 2004), known to contain the gene, using Phusion High-Fidelity DNA Polymerase (New England Biolabs) and the following primers: Forward 5′-GATCTACTCGAGATGAACTTTAAGTCAGTTGTTTTATG-3′ (XhoI site underlined) and Reverse 5′-ATAAGAATGCGGCCGCTTACACAATCAGTGAACTACCTGT-3′ (NotI site underlined). The PCR product was cloned between the corresponding sites of the vectors, pKLMF-EK and pKLMF-FX (GenBank accession nos. FJ010196 and FJ010197, respectively; New England Biolabs) for intracellular expression of wBm–iPGM bearing an in-frame N-terminal maltose binding protein (MBP) fusion partner, separated by either an enterokinase (EK) or Factor Xa (FX) protease cleavage site, in the yeast Kluyveromyces lactis. The integrity of the cloned sequences was confirmed by DNA sequencing.
Expression of wBm–iPGM in K. lactis
The construct, pKLMF-EK-wBm–iPGM, was linearized with SacII and used to transform K. lactis GG799 competent cells following the K. lactis Protein Expression Kit recommendations (New England Biolabs). Transformants containing multiply-integrated copies of the expression cassette, which may produce more recombinant protein, were identified by PCR of patched colonies using Integration Primer 4: 5′-GTTTACCTTCTTCAGTTTTCAT-3′ paired with the kit’s Integration Primer 3 and selected for production of the MBP–wBm–iPGM fusion protein. A single colony was used to inoculate 3 ml YPGlucose (10 g yeast extract, 20 g Bacto Peptone/L, 2% glucose) and cells were grown with shaking at 240 rpm at 30oC overnight. Two 1-L YPGalactose (2%) cultures were inoculated with 1 ml each of the overnight culture, and cells were grown in baffled flasks at 30°C with shaking at 200 rpm for 3 days. Cells were pelleted at 6,000 × g for 15 min at 4°C and pooled to give about 24 g wet cells. The cells were washed once in 25 ml of ice-cold Column buffer (20 mM Tris–HCl pH 7.5, 200 mM NaCl, 1 mM DTT, 5% Glycerol) and pelleted once more. The pellet was resuspended in four volumes (~100 ml) of cold Column buffer supplemented with Complete EDTA-free protease inhibitor cocktail (Roche Applied Sciences). The cell suspension was passed through a TS Series Benchtop Cell Disruptor (Constant Systems) at 40 Kpsi three times. The sample was centrifuged at 14,000 × g for 30 min at 4°C and the supernatant harvested. The cell extract was loaded on to a 10 ml amylose column according to the pMAL Protein Fusion and Purification System recommendations (New England Biolabs). Recombinant MBP–wBm–iPGM was eluted with column buffer containing 10 mM maltose and 1.5-ml fractions were collected for separation by SDS-PAGE and for PGM enzyme assays.
PGM enzyme assays
The activity of the wBm–iPGM fusion protein was measured in the forward (glycolytic) direction using an established enzyme-coupled assay (White and Fothergill-Gilmore 1992; Fraser et al. 1999; Raverdy et al. 2007). Briefly, MBP–wBm–iPGM was added to 1 ml assay buffer (30 mM Tris–HCl pH 7.0, 5 mM MgSO4, 20 mM KCl) supplemented with 0.15 mM NADH, 1 mM ADP, 1.5 mM 3-PG (Sigma P8877), and 2.5 units each of enolase (Sigma E6126), pyruvate kinase (Sigma P7768) and l-lactic dehydrogenase (Sigma L2518). PGM activity was determined indirectly by monitoring the consumption of NADH at 340 nm. PGM reactions were performed at 30°C for 5 min with data collected at 10 s intervals using a Beckman DU 640 spectrophotometer set at 340 nm. Controls included (1) a baseline reaction that lacked iPGM, (2) a reaction that also lacked iPGM but had a comparable volume of column buffer and, (3) a reaction using a comparable amount of MBP–wBm–iPGM that had been boiled for 5 min.
Results and discussion
Sequence analysis of wBm–iPGM
The sequence of the cloned wBm–iPGM (1,506 bp) was identical to the gene predicted by the complete Wolbachia genome. The deduced protein (~56 kD) contains the catalytic serine and 13 other critical residues indicated by structural analysis of the biochemically characterized iPGM from Bacillus stearothermophilus (Jedrzejas et al. 2000a,b; Fig. 1). wBm–iPGM has greatest similarity with the iPGM proteins predicted by the genomes of other α-proteobacteria, notably rickettsial organisms such as Anaplasma marginale and Ehrlichia ruminantium (~55%). It also has similarity to experimentally verified iPGMs from other bacteria such as E. coli (Fraser et al. 1999) and B. stearothermophilus (Chander et al. 1999; ~43%) as well as from protozoan parasites such as Trypanosoma brucei (Chevalier et al. 2000; Djikeng et al. 2007; 31%), and nematodes such as B. malayi (Zhang et al. 2004; Raverdy et al. 2007; ~40%). Lateral gene transfer events between Wolbachia and their invertebrate hosts, including filarial nematodes, appear widespread (Hotopp et al. 2007) and evidence for gene transfer of iPGM between or within archaea, bacteria, and protozoans has been reported (Liapounova et al. 2006; Johnsen and Schonheit 2007). However, the clustering of wBm–iPGM with other α-proteobacterial iPGM enzymes and a lower amino acid similarity to iPGM from B. malayi, which itself clusters with other nematode iPGMs (Zhang et al. 2004; Raverdy et al. 2007) argues against lateral gene transfer being responsible for the presence of iPGM in both Wolbachia and its nematode host.

Alignment of the deduced amino acid sequences of various iPGM enzymes. The sequence of iPGM from the Wolbachia endosymbiont of Brugia malayi (wBm; GenBank accession no. AAW70991) is aligned with the extensively studied and structurally characterized iPGM from Bacillus stearothermophilus (Bs; GenBank accession no. Q9X519; Jedrzejas et al. 2000a,b) and the characterized iPGM from Brugia malayi (Bm; GenBank accession no. AAQ97626; Zhang et al. 2004; Raverdy et al. 2007). Residues that are identical in at least two of the three sequences are shaded in black, while conserved amino acid changes are grey. The catalytic serine (@) and 13 other residues (*) involved in catalysis (Jedrzejas 2000) are conserved in all three enzymes. Alignment generated with ClustalW and displayed with BOXSHADE (www.ch.embnet.org/software/BOX_form.html)
Expression and purification of recombinant wBm–iPGM
In order to demonstrate that wBm–iPGM encodes an active PGM, recombinant enzyme was produced for biochemical studies. Numerous attempts to express wBm–iPGM in E. coli were unsuccessful despite using different expression systems that produced protein with either a N-terminal MBP tag, a C-terminal His6 tag, or untagged protein produced by intein-mediated cleavage of a chitin binding domain fusion partner. In these systems, wBm–iPGM was mostly insoluble and the small amounts of soluble protein obtained were relatively impure and inactive (data not shown). These results were surprising given that we and others have produced several active iPGM enzymes from diverse organisms in these bacterial expression systems (Fraser et al. 1999; Collet et al. 2001; Guerra et al. 2004; Zhang et al. 2004; Djikeng et al. 2007; Raverdy et al. 2007). Unlike other expressed iPGM enzymes, wBm–iPGM has a high number of cysteine residues and the formation of disulfide bonds is strongly predicted (http://scratch.proteomics.ics.uci.edu/). However, there was no difference in expression and activity of wBm–iPGM when produced in E. coli strains having either reducing or oxidizing cytoplasms. Similarly, purification in the presence of reducing agents (DTT or β-mercaptoethanol) did not yield active protein. Expression of a synthetic wBm–iPGM gene, optimized for E. coli codon usage, also failed to improve expression. Active His-tagged wBm–iPGM was finally recovered from E. coli by urea denaturation of insoluble protein and subsequent refolding, but the specific activity of the refolded protein was only 0.04 units/mg (data not shown).
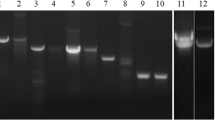
The difficulties of producing active recombinant wBm–iPGM in E. coli prompted us to develop a method for intracellular expression in yeast. We successfully produced wBm–iPGM as a MBP fusion in K. lactis. The MBP moiety serves as both a tag for purification and an aid to solubility (Kapust and Waugh 1999). MBP–wBm–iPGM was produced with a high degree of purity (Fig. 2). The apparent molecular weight (~100 kDa) is consistent with the calculated size of iPGM (~56 kDa) fused to MBP (~42 kDa). The identity of this ~100 kDa protein as a MBP fusion was confirmed by Western blot using an anti-MBP monoclonal antibody (New England Biolabs; data not shown). Similar expression and purification of wBm–iPGM was obtained using the construct pKLMF–FX–wBm–iPGM (data not shown). The method we developed for intracellular expression of MBP–wBm–iPGM in K. lactis may prove suitable for other proteins that express poorly in E. coli.

Purification of MBP–wBm–iPGM expressed intracellularly in the yeast K. lactis. Fractions from the purification were analyzed by SDS-PAGE and the gel stained with Coomassie Blue. Lane 1 Protein Ladder (New England Biolabs), lane 2 K. lactis lysate, lane 3 supernatant of K. lactis lysate, lane 4 pellet of K. lactis lysate, lane 5 flow-through from amylose column, lane 6 column wash, lanes 7, 8, and 9, elution fractions. The arrowhead indicates the protein band corresponding to recombinant MBP–wBm–iPGM
PGM activity of recombinant MBP–wBm–iPGM
The activity of the wBm–iPGM fusion protein was measured in the forward (glycolytic) direction using an established enzyme-coupled assay (Raverdy et al. 2007) in which PGM activity is determined indirectly by monitoring the consumption of NADH at 340 nm. A typical PGM activity was observed (Fig. 3) which was significantly different from a baseline control that lacked recombinant wBm–iPGM (Fig. 3) or a control that used boiled enzyme. Duplicate assays were performed on three different preparations of MBP–wBm–iPGM. From the slope of the curves, specific activities ranging from 1.0 to 4.1 units/mg were calculated. One unit of PGM activity is defined as the amount that is required for the conversion of 1.0 μmol NADH to NAD per minute. The specific activity of wBm–iPGM produced in K. lactis is therefore up to 100-fold higher than that obtained in E. coli (0.04 units/mg) after denaturation and refolding of insoluble protein and is broadly similar to activities reported for other bacterial iPGM enzymes (Kuhn et al. 1993; Leyva-Vazquez and Setlow 1994; Chander et al. 1999) but lower than most eukaryotic iPGMs where specific activities are typically in the range of 50 to 400 units/mg (Chevalier et al. 2000; Guerra et al. 2004; Zhang et al. 2004). Removal of the MBP moiety by digestion of the fusion protein with enterokinase did not enhance the activity of wBm–iPGM (data not shown). iPGMs are metalloenzymes and all characterized bacterial enzymes appear to use manganese as the preferred ion (Jedrzejas and Setlow 2001). We did not observe any enhancement in MBP–wBm–iPGM activity when the standard magnesium-containing buffer was supplemented with either 1 mM cobalt or manganese (data not shown).

Activity of recombinant wBm–iPGM. Conversion of 3-PG to 2-PG by MBP–wBm–iPGM (filled circle) is measured indirectly by a decrease in NADH concentration, determined spectrophotometrically at 340 nm. Consumption of NADH is directly proportional to PGM activity. A baseline control lacking wBm–iPGM (open circle) is shown
The essential roles of Wolbachia endosymbionts in filarial nematode biology have resulted in these bacteria being considered as an Achilles’ heel of their worm hosts and proof-of-principle clinical trials using tetracycline antibiotics are testament to that view (Taylor et al. 2005; Hoerauf 2006). We have initiated studies to identify and characterize new candidate drug targets predicted by the wBm genome sequence. The identification of wBm–iPGM and production of active recombinant protein for further studies represents one of the first examples of utilizing the endosymbiont genome sequence to facilitate development of novel anti-Wolbachia approaches towards filarial disease control. The lack of similarity in primary sequence or tertiary structure between dPGM and iPGM makes it highly probable that an iPGM inhibitor would not affect the mammalian enzyme. The likelihood that iPGM inhibition would disrupt filarial biology either directly or through targeting the Wolbachia endosymbiont appears high. In all organisms where iPGM is present and the gene has been deleted or its transcript level reduced by RNAi, deleterious effects have been observed. Reduction of iPGM activity in C. elegans by RNAi resulted in embryonic lethality, larval lethality and abnormal morphology (Zhang et al. 2004). Similarly, iPGM has been shown to be essential in diverse bacterial species (Leyva-Vazquez and Setlow 1994; Morris et al. 1995; Glass et al. 2006; Gallagher et al. 2007). These studies in nematodes and bacteria demonstrate the feasibility of developing novel therapies that target the iPGM of B. malayi and/or its Wolbachia endosymbiont.
That iPGM is present in Wolbachia, nematodes and several other pathogens while absent in mammals makes it an attractive target in diverse organisms. Therefore, identification of an iPGM inhibitor could lead to new therapeutic strategies, not only for filarial disease control, but additionally, for a range of infectious diseases. There is presently no known inhibitor of this enzyme. The availability of active recombinant iPGM enables further development of this target and its inclusion in high-throughput inhibitor screens.
References
Carreras J, Mezquita J, Bosch J, Bartrons R, Pons G (1982) Phylogeny and ontogeny of the phosphoglycerate mutases—IV. Distribution of glycerate-2,3-P2 dependent and independent phosphoglycerate mutases in algae, fungi, plants and animals. Comp Biochem Physiol B 71:591–597
Chander M, Setlow P, Lamani E, Jedrzejas MJ (1999) Structural studies on a 2,3-diphosphoglycerate independent phosphoglycerate mutase from Bacillus stearothermophilus. J Struct Biol 126:156–165
Chevalier N, Rigden DJ, Van Roy J, Opperdoes FR, Michels PA (2000) Trypanosoma brucei contains a 2,3-bisphosphoglycerate independent phosphoglycerate mutase. Eur J Biochem 267:1464–1472
Collet JF, Stroobant V, Van Schaftingen E (2001) The 2,3-bisphosphoglycerate-independent phosphoglycerate mutase from Trypanosoma brucei: metal-ion dependency and phosphoenzyme formation. FEMS Microbiol Lett 204:39–44
Djikeng A, Raverdy S, Foster J, Bartholomeu D, Zhang Y, El-Sayed NM, Carlow C (2007) Cofactor-independent phosphoglycerate mutase is an essential gene in procyclic form Trypanosoma brucei. Parasitol Res 100:887–892
Foster JM, Kumar S, Ganatra MB, Kamal IH, Ware J, Ingram J, Pope-Chappell J et al (2004) Construction of bacterial artificial chromosome libraries from the parasitic nematode Brugia malayi and physical mapping of the genome of its Wolbachia endosymbiont. Int J Parasitol 34:733–746
Foster J, Ganatra M, Kamal I, Ware J, Makarova K, Ivanova N, Bhattacharyya A et al (2005) The Wolbachia genome of Brugia malayi: endosymbiont evolution within a human pathogenic nematode. PLoS Biol 3:e121
Fothergill-Gilmore LA, Watson HC (1989) The phosphoglycerate mutases. Adv Enzymol Relat Areas Mol Biol 62:227–313
Fraser HI, Kvaratskhelia M, White MF (1999) The two analogous phosphoglycerate mutases of Escherichia coli. FEBS Lett 455:344–348
Gallagher LA, Ramage E, Jacobs MA, Kaul R, Brittnacher M, Manoil C (2007) A comprehensive transposon mutant library of Francisella novicida, a bioweapon surrogate. Proc Natl Acad Sci USA 104:1009–1014
Galperin MY, Jedrzejas MJ (2001) Conserved core structure and active site residues in alkaline phosphatase superfamily enzymes. Proteins 45:318–324
Glass JI, Assad-Garcia N, Alperovich N, Yooseph S, Lewis MR, Maruf M, Hutchison CA 3rd et al (2006) Essential genes of a minimal bacterium. Proc Natl Acad Sci USA 103:425–430
Guerra DG, Vertommen D, Fothergill-Gilmore LA, Opperdoes FR, Michels PA (2004) Characterization of the cofactor-independent phosphoglycerate mutase from Leishmania mexicana mexicana. Histidines that coordinate the two metal ions in the active site show different susceptibilities to irreversible chemical modification. Eur J Biochem 271:1798–1810
Hoerauf A (2006) New strategies to combat filariasis. Expert Review of Anti-infective Therapy 4:1–12
Hotopp JC, Clark ME, Oliveira DC, Foster JM, Fischer P, Torres MC, Giebel JD et al (2007) Widespread lateral gene transfer from intracellular bacteria to multicellular eukaryotes. Science 317:1753–1756
Jedrzejas MJ (2000) Structure, function, and evolution of phosphoglycerate mutases: comparison with fructose-2,6-bisphosphatase, acid phosphatase, and alkaline phosphatase. Progress in Biophysics & Molecular Biology 73:263–287
Jedrzejas MJ, Setlow P (2001) Comparison of the binuclear metalloenzymes diphosphoglycerate-independent phosphoglycerate mutase and alkaline phosphatase: their mechanism of catalysis via a phosphoserine intermediate. Chem Rev 101:607–618
Jedrzejas MJ, Chander M, Setlow P, Krishnasamy G (2000a) Structure and mechanism of action of a novel phosphoglycerate mutase from Bacillus stearothermophilus. Embo J 19:1419–1431
Jedrzejas MJ, Chander M, Setlow P, Krishnasamy G (2000b) Mechanism of catalysis of the cofactor-independent phosphoglycerate mutase from Bacillus stearothermophilus. Crystal structure of the complex with 2-phosphoglycerate. J Biol Chem 275:23146–23153
Johnsen U, Schonheit P (2007) Characterization of cofactor-dependent and cofactor-independent phosphoglycerate mutases from Archaea. Extremophiles 11:647–657
Kapust RB, Waugh DS (1999) Escherichia coli maltose-binding protein is uncommonly effective at promoting the solubility of polypeptides to which it is fused. Protein Sci 8:1668–1674
Kuhn NJ, Setlow B, Setlow P (1993) Manganese (II) activation of 3-phosphoglycerate mutase of Bacillus megaterium: pH-sensitive interconversion of active and inactive forms. Arch Biochem Biophys 306:342–349
Leyva-Vazquez MA, Setlow P (1994) Cloning and nucleotide sequences of the genes encoding triose phosphate isomerase, phosphoglycerate mutase, and enolase from Bacillus subtilis. J Bacteriol 176:3903–3910
Liapounova NA, Hampl V, Gordon PM, Sensen CW, Gedamu L, Dacks JB (2006) Reconstructing the mosaic glycolytic pathway of the anaerobic eukaryote Monocercomonoides. Eukaryot Cell 5:2138–2146
Morris VL, Jackson DP, Grattan M, Ainsworth T, Cuppels DA (1995) Isolation and sequence analysis of the Pseudomonas syringae pv. tomato gene encoding a 2,3-diphosphoglycerate-independent phosphoglyceromutase. J Bacteriol 177:1727–1733
Raverdy S, Zhang Y, Foster J, Carlow CKS (2007) Molecular and biochemical characterization of nematode cofactor independent phosphoglycerate mutases. Mol Biochem Parasitol 156:210–216
Taylor MJ, Bandi C, Hoerauf A (2005) Wolbachia bacterial endosymbionts of filarial nematodes. Adv Parasitol 60:245–284
White MF, Fothergill-Gilmore LA (1992) Development of a mutagenesis, expression and purification system for yeast phosphoglycerate mutase. Investigation of the role of active-site His181. Eur J Biochem 207:709–714
Zhang Y, Foster JM, Kumar S, Fougere M, Carlow CK (2004) Cofactor-independent phosphoglycerate mutase has an essential role in Caenorhabditis elegans and is conserved in parasitic nematodes. J Biol Chem 279:37185–37190
Acknowledgments
We thank Dr. Don Comb for continued encouragement and support. We acknowledge New England Biolabs and National Institutes for Health/National Institute for Allergy and Infectious Diseases (SBIR Grant number 2R44 A1061865-02) for financial support. The experiments described in this work comply with the current laws of the USA.
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License (https://creativecommons.org/licenses/by-nc/2.0), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
Foster, J.M., Raverdy, S., Ganatra, M.B. et al. The Wolbachia endosymbiont of Brugia malayi has an active phosphoglycerate mutase: a candidate target for anti-filarial therapies. Parasitol Res 104, 1047–1052 (2009). https://doi.org/10.1007/s00436-008-1287-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00436-008-1287-7