
Abstract
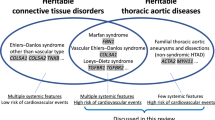
Heritable connective tissue diseases comprise a heterogeneous group of multisystemic disorders that are characterized by significant morbidity and mortality. These disorders do not merely result from defects in the amount or structure of one of the components of the extracellular matrix, as the extracellular matrix also serves other functions, including sequestration of cytokines, such as transforming growth factor beta (TGFβ). Indeed, disturbed TGFβ signaling was demonstrated in several heritable connective tissue diseases, including syndromic forms such as Marfan or Loeys-Dietz syndrome and non-syndromic presentations of thoracic aortic aneurysm/dissection. Because of these findings, new therapeutic targets have been unveiled, leading to the initiation of large clinical trials with angiotensin II type 1 receptor antagonists that also have an inhibiting effect on TGFβ signaling. Here, we present an overview of the clinical characteristics, the molecular findings, and the therapeutic strategies for the currently known syndromic and non-syndromic forms of thoracic aortic aneurysm/dissection.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
The extracellular matrix (ECM) is a highly organized multimolecular structure that is indispensable for the normal functioning of organ systems. Heritable connective tissue diseases (HCTD) comprise a heterogeneous group of multisystemic disorders that result from genetic defects affecting normal ECM assembly and maintenance. HCTD are characterized by significant morbidity and mortality [43]. Up to now, most genes shown to be implicated in HCTD encode structural connective tissue proteins, such as collagens, fibrillin and fibronectin, or enzymes involved in the biosynthesis or processing of those proteins. It has been generally assumed that defects in the amount or structure of one of these ECM components affect normal organization and structural integrity of the supporting connective tissues and result in the typical weakness of bones, skin, vascular or other tissues, which characterizes the individual disease phenotypes. During the past decade, however, this viewpoint was found to be an oversimplification. This became apparent when other functions were discovered for the ECM, exemplified by the sequestration function for TGFβ [36], and the identification of various non-structural genes for HCTD [5, 22, 26, 29]. This implies that the pathogenesis of HCTD and the organization of the ECM are far more complex than originally thought. Thus, the current perception of the ECM is that of a complex network that, besides its mechanical role in providing strength and support to the tissues, also acts as an important reservoir for cytokines and growth factors implicated in cellular proliferation, differentiation, migration, and survival and thus has an important regulatory function in the development and homeostasis of body organs and tissues [43]. Interestingly, this fresh view on ECM function has provided new means of therapeutic intervention. Originally, when ECM was assigned a merely structural role, the only putative therapy was replacement of the deficient ECM component by gene therapy, something that seemed unachievable. Nowadays, however, with the recognition of a regulatory function for ECM components, intervening in the relevant regulatory pathways using (existing) pharmaceuticals may provide a plausible therapeutic approach in case of deficiency of an ECM component. As such, some HCTD, including Marfan (MFS) and Loeys-Dietz (LDS) syndromes, and related disorders have evolved from “incurable” genetic disorders (apart from surgical interventions) towards diseases in which hopes for new therapies have risen. Therefore, we will focus this review on these HCTD and leave other HCTDs including the Ehlers-Danlos syndromes (EDS, with the exception of vascular EDS), skeletal dysplasias, osteogenesis imperfecta, pseudoxanthoma elasticum and others largely untouched. For these HCTD, we would like to refer to other recent reviews [9, 13, 33, 49].
Marfan syndrome
The Marfan syndrome (MFS; MIM#154700) is an autosomal dominant HCTD that occurs in approximately 1/5,000 individuals. It is a multisystemic disorder, mainly affecting the skeleton (long bone overgrowth, pectus deformity, and arachnodactyly), the eyes (lens dislocation), and the cardiovascular system. Cardiovascular pathology includes aortic root dilatation primarily at the level of the sinuses of Valsalva, dissection, and rupture, and is the leading cause of mortality [46]. Other cardiovascular manifestations include mitral valve prolapse, mitral regurgitation, (supra)ventricular arrhythmias, and systolic and diastolic left ventricular functional impairment. Each of these cardiovascular complications may present as early as infancy or may not be diagnosed until adulthood [54]. Other extra-cardiovascular manifestations include dural ectasia, striae distensae, recurrent inguinal hernias, pneumothorax, and lung emphysema.
In 1991, FBN1 (encoding fibrillin-1, a structural component of the microfibrils) was identified as the gene responsible for MFS [11]. Up to now, several hundred FBN1 mutations have been identified throughout the entire gene, but no major correlation between the nature of the mutation and the clinical phenotype has emerged [27]. Moreover, despite the presence of identical FBN1 mutations, a remarkable intrafamilial variability is observed, in addition to a marked interfamilial variability, which has led to the hypothesis that modifier genes must be involved.
Until recently, the clinical diagnosis was based on the Ghent nosology [8], but since its revision in 2010 [30], clinical geneticists have adopted the new version [12, 40, 44]. The revised Ghent nosology puts more weight on cardiovascular manifestations, ectopia lentis, and molecular FBN1 testing and summarizes the diagnostic decision making in a few simple rules depending on the absence or the presence of a family history [30].
Originally, it was assumed that the pathophysiology of MFS was due to structural deficiency of fibrillin-1, leading to weakened microfibrils. This hypothesis provided a plausible explanation for the vascular pathology and the lens dislocation, but other features such as skeletal overgrowth and thickening of the cardiac valves remained unexplained. Subsequently, the study of MFS mouse models suggested an important role for the cytokine transforming growth factor beta (TGFβ) in the pathogenesis of MFS; Fbn1 deficient mice showed increased TGFβ activation and signaling and TGFβ antagonism rescued various MFS phenotypes [19, 36, 37]. The current pathogenetic model for MFS therefore suggests that the fibrillin-1 deficient state leads to dysregulation of the TGFβ signaling cascade. Although historically, studies have focused on canonical TGFβ signaling, growing evidence now shows that non-canonical signaling pathways such as those involving the MAPKs (mitogen-activated protein kinases), including the extracellular signal-regulated kinase (ERK1/2) and the Jun N-terminal kinase (JNK) also have an important role in aneurysm development [20].
Loeys-Dietz syndrome
In 2005, a novel autosomal dominant HCTD with widespread systemic involvement was described by Loeys and Dietz. The Loeys-Dietz syndrome (LDS; MIM#609192) is typically characterized by the triad of hypertelorism (widely spaced eyes), bifid uvula/cleft palate, and arterial tortuosity with aortic aneurysm and dissection [29]. Additional manifestations include craniosynostosis, Chiari malformation, club feet, patent ductus arteriosus, and aneurysms/dissection throughout the arterial tree. LDS type 1 patients present with typical craniofacial features (craniosynostosis, cleft palate, or hypertelorism), while LDS type 2 patients mostly lack the craniofacial features but present with cutaneous findings (velvety and translucent skin, easy bruising, and atrophic scars). Despite some clinical overlap with MFS, including aortic aneurysm, arachnodactyly, pectus deformity, dural ectasia, and scoliosis, LDS patients do not present with significant long bone overgrowth or lens dislocation. Aneurysm development in LDS is more aggressive compared to that observed in MFS, i.e., on average, dissection and rupture occur at younger ages and at smaller aortic diameters than in MFS patients leading to a mean age at death of 26.0 years [32]. This implies that earlier surgical intervention (at smaller aortic diameters than in MFS patients) is indicated. Clinicians should also be aware of the increased risk of pregnancy-related complications. So far, no set of minimal diagnostic criteria have been established for LDS and the diagnosis should thus be confirmed through molecular genetic testing.
LDS is caused by mutations in genes encoding transforming growth factor-beta receptor 1 or 2 (TGFBR1 or TGFBR2) [29]. No clinical distinction can be made between TGFBR1 or TGFBR2 mutation carriers. Comparable to MFS, a remarkable inter-individual and intrafamilial clinical variability is observed. The mutations mainly affect the serine-threonine kinase domain of both receptors and lead to loss-of-function. Interestingly and paradoxically, TGFβ signaling was enhanced in aortic walls and fibroblast cultures of LDS patients [29]. After the finding of increased TGFβ signaling in a MFS mouse model [36], the discovery that mutations in TGFBR1/2 were the cause of another aneurysmal syndrome and the fact that these loss-of-function mutations led to a paradoxical activation of TGFβ signaling provided further proof for an important role of TGFβ signaling in aneurysma development [29].
Other disorders in the MFS/LDS spectrum
The aneurysm-osteoarthritis syndrome or Loeys-Dietz syndrome, type 1C
Van de Laar et al. [51] recently described another HCTD, designated aneurysms-osteoarthritis syndrome (AOS) or Loeys-Dietz syndrome, type 1C (MIM#613795). AOS is characterized by aneurysms, dissections, and tortuosity throughout the arterial tree in addition to craniofacial (including hypertelorism and abnormal palate/uvula), skeletal (including arachnodactyly and scoliosis), and cutaneous (including striae and velvety skin) features and thus perfectly fits in the phenotypic spectrum of LDS. A distinguishing feature, however, is the presence of early-onset osteoarthritis. AOS is caused by mutations in the gene encoding SMAD3, a component of the canonical TGFβ signaling pathway [51–53]. Again, dysregulation of TGFβ signaling was demonstrated when studying aortic walls from AOS patients. All investigated markers of TGFβ signaling, including pSMAD2, SMAD3, and CTGF (connective tissue growth factor) were significantly increased in these tissues [51].
Vascular Ehlers-Danlos syndrome
EDS comprises a clinically and genetically diverse group of HCTD, characterized by congenital fragility of the connective tissues. The Villefranche nosology recognizes six subtypes based on clinical characteristics, inheritance pattern, biochemical, and molecular findings [1]. The most common types of EDS are the classic, the hypermobile and the vascular type, while the kyphoscoliotic, the arthrochalasis, and the dermatosparaxis type are rather rare. Here, we restrict ourselves to the vascular type of EDS (MIM#130050) since the latter is associated with a high risk for life threatening complications and as a consequence with a decreased life expectancy. We refer to a recent review for details on the other EDS types [9].
Typical clinical manifestations of vascular EDS include thin, translucent skin, characteristic facial appearance, vascular fragility demonstrated by extensive bruising and easy bleeding and spontaneous arterial/intestinal/uterine ruptures [43]. Vascular EDS is caused by mutations in COL3A1 (type III collagen α-chain 1) [47]. These mutations consist mostly of missense mutations that lead to the substitution of essential glycine residues within the triple helical domain of the type III collagen chain. Interestingly, in a cohort of 40 patients displaying a vascular EDS-like phenotype but normal collagen III biochemistry, 30 % carried TGFBR1/2 mutations [32], suggesting on the one hand that vascular EDS closely resembles LDS but on the other hand that TGFBR mutations may cause a broad spectrum of diseases associated with aortic aneurysms. To the best of our knowledge, no demonstration of dysregulated TGFβ signaling in vascular EDS has been published so far.
Arterial tortuosity syndrome
The arterial tortuosity syndrome (ATS; MIM#208050) is an autosomal recessive HCTD that is characterized by tortuosity, elongation, stenosis, and aneurysm formation in the major arteries. Patients often die at a young age. Features in common with LDS include arachnodactyly, hypertelorism, cleft palate and/or bifid uvula, joint laxity or contractions, and micro/retrognathia. ATS is caused by loss-of-function mutations in SLC2A10, encoding GLUT10, which belongs to the glucose transporter family. ATS has been associated with enhanced TGFβ signaling, as demonstrated by increased pSMAD2 and CTGF immunostaining in patients' arterial walls [5].
Cutis laxa
Hereditary cutis laxa (CL) delineates a heterogeneous group of rare HCTD, characterized by the presence of loose, sagging, inelastic skin in addition to systemic manifestations of variable severity [43]. Both autosomal dominant (ADCL) and autosomal recessive forms (ARCL) exist. ADCL is relatively benign compared to ARCL. In ADCL, the typical loose, sagging skin can be accompanied by gastrointestinal diverticulae, hernias, and genital prolapse. Also, pulmonary artery stenosis, aortic aneurysm, bronchiectasis, and emphysema may occur. The major causal gene for ADCL is the elastin gene (ELN; MIM #123700), while one tandem duplication in the fibulin-5 gene (FBLN5; MIM#614434) has been described as well. Gain-of-function mutations in ELN lead to ADCL, whereas loss-of-function point mutations or contiguous gene deletions (involving ELN) give rise to isolated supravalvular aortic stenosis and Williams-Beuren syndrome, respectively.
ARCL type 1 (ARCL1) is a life-threatening disorder characterized by vascular anomalies, lung emphysema, and diverticulae of the urinary and gastrointestinal tract aside from the cutaneous manifestations. The prognosis of ARCL1 is poor as cardiopulmonary failure severely limits the lifespan of these patients. Mutations in two fibulin genes, FBLN4 (MIM#614437; also designated EFEMP2) or FBLN5 (MIM#219100), are responsible for ARCL1 [21, 28]. Vascular involvement (arterial aneurysms, arterial tortuosity), however, is most probably restricted to FBLN4 mutations [6, 42], while the cutaneous manifestations in FBLN4 patients are limited and are more pronounced in FBLN5 patients. As such, ARCL1 caused by FBLN4 mutations can be categorized within the LDS spectrum.
Another autosomal recessive syndrome, closely resembling ARCL1 and designated Urban-Rifkin-Davies syndrome (URDS; MIM#613177), was recently shown to be caused by mutations in LTBP4 [50]. LTBP4 patients present with severe gastro-intestinal and urinary tract involvement in addition to loose, sagging skin. Aside from peripheral pulmonary artery stenosis, LTBP4 patients have no vascular involvement. The gene product, LTBP4, belongs to the latent transforming growth factor beta binding proteins (LTBPs), which associate with TGFβ when secreted into the ECM and keep TGFβ in its latent form. The latent complexes play an important role in the regulation of TGFβ-mediated signaling.
Interestingly, both in ADCL caused by ELN mutations, ARCL1 caused by FBLN4 mutations and in URDS, dysregulation of TGF-beta signaling was observed [4, 42, 50].
Other types of cutis laxa and related diseases exist (ARCL2, ARCL3, wrinkly skin syndrome, gerodermia osteodysplastica, etc.). However, as so far no involvement of TGFβ signaling has been suggested for these conditions, we prefer not to go into detail and to refer to a recent review by Berk et al. [2] for a more thorough description of these HCTD.
Non-syndromic thoracic aortic aneurysm and dissection
Non-syndromic types of thoracic aortic aneurysms and dissections (TAAD) or types in which only minor additional features are present exist as well. Occasionally, mutations in FBN1 [34] and in TGFBR1/2 [48] causing TAAD have been described, perhaps representing the mildest end of the MFS/LDS phenotypic spectrum.
In addition, ACTA2 mutations have been identified in 14 % of TAAD patients [16], while MYH11 mutations have been found in TAAD patients with persistent ductus arteriosus [56]. Additional symptoms that can be found in ACTA2 mutation positive patients include persistent ductus arteriosus, bicuspid aortic valve, iris flocculi, and cerebrovascular accidents. In fact, ACTA2 mutations can also cause stroke, Moya-Moya disease, and coronary artery disease [17]. ACTA2 and MYH11 encode the smooth muscle cell specific α-actin and β-myosin heavy chain, respectively. Both proteins are indispensable components of the smooth muscle cells contractile apparatus and mutations in the encoding genes may prevent proper contraction of the smooth muscle cells. Interestingly, enhanced TGFβ signaling was demonstrated in aortic tissue derived from both ACTA2 and MYH11 patients [41]. Additionally, Gomez et al. [15] found a dysregulation of TGFβ signaling in ascending aortic walls of non-syndromic thoracic aneurysm patients.
Recent new findings
From the above, a rule seems to have emerged: TGFβ signaling is significantly associated with both syndromic and non-syndromic types of thoracic aortic aneurysms. More importantly, as TGFβ antagonism can attenuate or prevent the various phenotypes, a causal relationship seems highly plausible. However, the URDS, one of the disorders mentioned above, is an exception to this rule, i.e., enhanced TGFβ signaling was found, but URDS patients do not present with thoracic aortic aneurysms. Here, a few additional examples are elaborated to illustrate that increased TGFβ signaling does not always lead to thoracic aortic aneurysm development and that the overall picture is thus far more complex.
In the stiff skin syndrome (SSS; MIM#184900), a rare, autosomal dominant condition of congenital scleroderma (thickened skin) associated with short stature, domain-specific FBN1 mutations have been identified. All mutations were positioned in two FBN1 exons that encode the fourth TGFβ binding protein-like domain (TB4). This domain contains the RGD (arginine–glycine–aspartic acid) motif, which mediates integrin binding. SSS patients do not show the typical MFS symptoms, including long bone overgrowth, thoracic aortic aneurysms, ectopia lentis and joint laxity; nor do MFS patients ever present with skin fibrosis. Still, increased TGFβ concentration and signaling was observed in the dermis of SSS patients [31]. Given the restricted nature of the mutations and limited affected organs observed in SSS (mainly skin), it was hypothesized that SSS mutations may lead to a gain-of-microfibrillar-function, while most MFS mutations clearly result in loss of function [31].
Recently, mutations restricted to two exons encoding the TB5 domain of FBN1 were shown to lead to acromicric (AD; MIM#102370) and autosomal dominant geleophysic dysplasia (GD; MIM#614185) [24]. Both syndromes are characterized by severe short stature, short extremities, and stiff joints. Thoracic aortic aneurysms are not part of the phenotypic spectrum, although cardiac valves stenosis and insufficiency have been associated with GD. As such, the AD and GD spectrum again opposes the MFS spectrum. Still, enhanced TGFβ signaling was demonstrated irrefutably in fibroblasts of these patients. As AD had been shown previously to be caused by mutations in ADAMTSL2 [25] and a direct interaction between FBN1 and ADAMTSL2 was found, the authors suggested that a dysregulation of the FBN1/ADAMTSL2/TGFβ interrelationship lay at the basis of the AD/GD phenotypes.
These few examples further illustrate the complexity of the TGFβ signaling pathways and their regulatory levels. Most likely, several factors, including the nature and the location of the mutation, the nature of the affected gene product, its spatiotemporal expression levels and the influence of other paracrine or autocrine factors influence the final outcome. Additional research will thus be necessary to understand all contributing factors in order to be able to predict aneurysm formation.
Treatment strategies
Traditionally, management of MFS patients consisted of a regular evaluation of their aortic diameters and therapeutic treatment with beta-blockers [45]. For MFS patients, surgical intervention is indicated when the aortic diameter exceeds 5 cm or when the aneurysmal growth exceeds 1 cm/year. For LDS, surgical intervention at smaller aortic diameters is recommended. In addition, as the aneurysms in LDS patients can be found throughout the arterial tree, more extensive imaging is mandatory.
Interestingly, the identification of enhanced TGFβ signaling in MFS mouse models has led to the identification of losartan as an exciting new possible therapy for MFS [18, 19], as losartan also has an inhibiting effect on TGFβ signaling besides being an antagonist of the angiotensin II type 1 receptor. Indeed, losartan outperformed the beta-blocker atenolol when applied to a MFS mouse model [19]. This exiting finding led to a preliminary observational study in seventeen pediatric MFS patients in whom other therapy had failed to prevent progressive aortic root dilatation [3]. These patients were treated with losartan during 12 to 47 months, leading to a reduction in rate of aortic root diameter growth from 3.54 ± 2.87 mm/year to 0.46 ± 0.62 mm/year. As this preliminary study had provided proof that losartan can be efficient in human patients as well, several large, randomized clinical trials on MFS patients have been initiated since. A first trial is comparing atenolol with losartan in approximately 600 MFS patients during a 3-year period. Unless the trial is stopped early for treatment benefit, results can be expected by 2014 [23]. Additional trials with various designs and inclusion criteria are currently ongoing in Belgium, France, Italy, The Netherlands, Taiwan, and the United Kingdom [10, 14, 35, 39]. The next few years will thus reveal whether losartan will become the preferred therapeutic in MFS patients.
Because of the less frequent nature of related disorders with aneurysm formation and TGFβ signaling upregulation (LDS, AOS, ATS, ADCL, ARCL1, TAAD), it is unlikely that large, randomized trials can be organized for these conditions. For these rarer conditions, evidence may be gathered through the study of relevant animal models, after which cautious treatment of patients may be considered. Prudence is indeed called for, because in contrast to the findings in thoracic aortic aneurysms, it was shown in a validated mouse model of abdominal aortic aneurysm formation (Ang II-induced; [7]), that TGFβ activity actually protects against aortic aneurysm progression and that TGFβ neutralization promotes aortic aneurysm formation [55].
Although no causal treatment exists for EDS, prophylactic measures are essential, in particular for the vascular type of EDS because of its serious nature. Individuals with vascular EDS should avoid contact sports and isometric exertion (like weightlifting). Usage of anti-coagulentia and other agents that interfere with platelet function should also be avoided. Likewise, invasive procedures, including angiography, catherizations and surgical interventions are contra-indicated. Recently, it was demonstrated that the beta-blocker celiprolol could prevent major complications in patients with vascular EDS [38], giving new hope for treatment of this devastating condition.
Much of the information that we have reviewed here, is also summarized in Table 1 (clinical synopsis) and Fig. 1 (defects within components of the ECM and the TGFβ signaling pathway).

Defects within components of the ECM and the TGFβ signaling pathway leading to various HCTD. The defective components are indicated with a red cross and the respective diseases are listed in the vicinity of the relevant cross
References
Beighton P, De Paepe A, Steinmann B, Tsipouras P, Wenstrup RJ (1998) Ehlers-Danlos syndromes: revised nosology, villefranche, 1997. Ehlers-Danlos National Foundation (USA) and Ehlers-Danlos Support Group (UK). Am J Med Genet 77(1):31–37
Berk DR, Bentley DD, Bayliss SJ, Lind A, Urban Z (2012) Cutis laxa: a review. J Am Acad Dermatol 66(5):842.e1–842.e17
Brooke BS, Habashi JP, Judge DP, Patel N, Loeys B, Dietz HC 3rd (2008) Angiotensin II blockade and aortic-root dilation in Marfan's syndrome. N Engl J Med 358(26):2787–2795
Callewaert B, Renard M, Hucthagowder V, Albrecht B, Hausser I, Blair E, Dias C, Albino A, Wachi H, Sato F, Mecham RP, Loeys B, Coucke PJ, De Paepe A, Urban Z (2011) New insights into the pathogenesis of autosomal-dominant cutis laxa with report of five ELN mutations. Hum Mutat 32(4):445–455
Coucke PJ, Willaert A, Wessels MW, Callewaert B, Zoppi N, De Backer J, Fox JE, Mancini GM, Kambouris M, Gardella R, Facchetti F, Willems PJ, Forsyth R, Dietz HC, Barlati S, Colombi M, Loeys B, De Paepe A (2006) Mutations in the facilitative glucose transporter GLUT10 alter angiogenesis and cause arterial tortuosity syndrome. Nat Genet 38(4):452–457
Daugherty A, Manning MW, Cassis LA (2000) Angiotensin II promotes atherosclerotic lesions and aneurysms in apolipoprotein E-deficient mice. J Clin Invest 105(11):1605–1612
Dasouki M, Markova D, Garola R, Sasaki T, Charbonneau NL, Sakai LY, Chu ML (2007) Compound heterozygous mutations in fibulin-4 causing neonatal lethal pulmonary artery occlusion, aortic aneurysm, arachnodactyly, and mild cutis laxa. Am J Med Genet A 143A(22):2635–2641
De Paepe A, Devereux RB, Dietz HC, Hennekam RC, Pyeritz RE (1996) Revised diagnostic criteria for the Marfan syndrome. Am J Med Genet 62(4):417–426
De Paepe A, Malfait F (2012) The Ehlers-Danlos Syndrome, a disorder with many faces. Clin Genet. doi:10.1111/j.1399-0004.2012.01858.x
Detaint D, Aegerter P, Tubach F, Hoffman I, Plauchu H, Dulac Y, Faivre LO, Delrue MA, Collignon P, Odent S, Tchitchinadze M, Bouffard C, Arnoult F, Gautier M, Boileau C, Jondeau G (2010) Rationale and design of a randomized clinical trial (Marfan Sartan) of angiotensin II receptor blocker therapy versus placebo in individuals with Marfan syndrome. Arch Cardiovasc Dis 103(5):317–325
Dietz HC, Cutting GR, Pyeritz RE, Maslen CL, Sakai LY, Corson GM, Puffenberger EG, Hamosh A, Nanthakumar EJ, Curristin SM et al (1991) Marfan syndrome caused by a recurrent de novo missense mutation in the fibrillin gene. Nature 352(6333):337–339
Faivre L, Collod-Beroud G, Ades L, Arbustini E, Child A, Callewaert B, Loeys B, Binquet C, Gautier E, Mayer K, Arslan-Kirchner M, Grasso M, Beroud C, Hamroun D, Bonithon-Kopp C, Plauchu H, Robinson P, De Backer J, Coucke P, Francke U, Bouchot O, Wolf J, Stheneur C, Hanna N, Detaint D, De Paepe A, Boileau C, Jondeau G (2011) The new Ghent criteria for Marfan syndrome: what do they change? Clin Genet 81(5):433–442
Forlino A, Cabral WA, Barnes AM, Marini JC (2011) New perspectives on osteogenesis imperfecta. Nat Rev Endocrinol 7(9):540–557
Gambarin FI, Favalli V, Serio A, Regazzi M, Pasotti M, Klersy C, Dore R, Mannarino S, Vigano M, Odero A, Amato S, Tavazzi L, Arbustini E (2009) Rationale and design of a trial evaluating the effects of losartan vs. nebivolol vs. the association of both on the progression of aortic root dilation in Marfan syndrome with FBN1 gene mutations. J Cardiovasc Med (Hagerstown) 10(4):354–362
Gomez D, Al Haj Zen A, Borges LF, Philippe M, Gutierrez PS, Jondeau G, Michel JB, Vranckx R (2009) Syndromic and non-syndromic aneurysms of the human ascending aorta share activation of the Smad2 pathway. J Pathol 218(1):131–142
Guo DC, Pannu H, Tran-Fadulu V, Papke CL, Yu RK, Avidan N, Bourgeois S, Estrera AL, Safi HJ, Sparks E, Amor D, Ades L, McConnell V, Willoughby CE, Abuelo D, Willing M, Lewis RA, Kim DH, Scherer S, Tung PP, Ahn C, Buja LM, Raman CS, Shete SS, Milewicz DM (2007) Mutations in smooth muscle alpha-actin (ACTA2) lead to thoracic aortic aneurysms and dissections. Nat Genet 39(12):1488–1493
Guo DC, Papke CL, Tran-Fadulu V, Regalado ES, Avidan N, Johnson RJ, Kim DH, Pannu H, Willing MC, Sparks E, Pyeritz RE, Singh MN, Dalman RL, Grotta JC, Marian AJ, Boerwinkle EA, Frazier LQ, LeMaire SA, Coselli JS, Estrera AL, Safi HJ, Veeraraghavan S, Muzny DM, Wheeler DA, Willerson JT, Yu RK, Shete SS, Scherer SE, Raman CS, Buja LM, Milewicz DM (2009) Mutations in smooth muscle alpha-actin (ACTA2) cause coronary artery disease, stroke, and Moyamoya disease, along with thoracic aortic disease. Am J Hum Genet 84(5):617–627
Habashi JP, Judge DP, Holm TM, Cohn RD, Loeys BL, Cooper TK, Myers L, Klein EC, Liu G, Calvi C, Podowski M, Neptune ER, Halushka MK, Bedja D, Gabrielson K, Rifkin DB, Carta L, Ramirez F, Huso DL, Dietz HC (2006) Losartan, an AT1 antagonist, prevents aortic aneurysm in a mouse model of Marfan syndrome. Science 312(5770):117–121
Habashi JP, Doyle JJ, Holm TM, Aziz H, Schoenhoff F, Bedja D, Chen Y, Modiri AN, Judge DP, Dietz HC (2011) Angiotensin II type 2 receptor signaling attenuates aortic aneurysm in mice through ERK antagonism. Science 332(6027):361–365
Holm TM, Habashi JP, Doyle JJ, Bedja D, Chen Y, van Erp C, Lindsay ME, Kim D, Schoenhoff F, Cohn RD, Loeys BL, Thomas CJ, Patnaik S, Marugan JJ, Judge DP, Dietz HC (2011) Noncanonical TGFbeta signaling contributes to aortic aneurysm progression in Marfan syndrome mice. Science 332(6027):358–361
Hucthagowder V, Sausgruber N, Kim KH, Angle B, Marmorstein LY, Urban Z (2006) Fibulin-4: a novel gene for an autosomal recessive cutis laxa syndrome. Am J Hum Genet 78(6):1075–1080
Kornak U, Reynders E, Dimopoulou A, van Reeuwijk J, Fischer B, Rajab A, Budde B, Nurnberg P, Foulquier F, Lefeber D, Urban Z, Gruenewald S, Annaert W, Brunner HG, van Bokhoven H, Wevers R, Morava E, Matthijs G, Van Maldergem L, Mundlos S (2008) Impaired glycosylation and cutis laxa caused by mutations in the vesicular H+ -ATPase subunit ATP6V0A2. Nat Genet 40(1):32–34
Lacro RV, Dietz HC, Wruck LM, Bradley TJ, Colan SD, Devereux RB, Klein GL, Li JS, Minich LL, Paridon SM, Pearson GD, Printz BF, Pyeritz RE, Radojewski E, Roman MJ, Saul JP, Stylianou MP, Mahony L (2007) Rationale and design of a randomized clinical trial of beta-blocker therapy (atenolol) versus angiotensin II receptor blocker therapy (losartan) in individuals with Marfan syndrome. Am Heart J 154(4):624–631
Le Goff C, Morice-Picard F, Dagoneau N, Wang LW, Perrot C, Crow YJ, Bauer F, Flori E, Prost-Squarcioni C, Krakow D, Ge G, Greenspan DS, Bonnet D, Le Merrer M, Munnich A, Apte SS, Cormier-Daire V (2008) ADAMTSL2 mutations in geleophysic dysplasia demonstrate a role for ADAMTS-like proteins in TGF-beta bioavailability regulation. Nat Genet 40(9):1119–1123
Le Goff C, Mahaut C, Wang LW, Allali S, Abhyankar A, Jensen S, Zylberberg L, Collod-Beroud G, Bonnet D, Alanay Y, Brady AF, Cordier MP, Devriendt K, Genevieve D, Kiper PO, Kitoh H, Krakow D, Lynch SA, Le Merrer M, Megarbane A, Mortier G, Odent S, Polak M, Rohrbach M, Sillence D, Stolte-Dijkstra I, Superti-Furga A, Rimoin DL, Topouchian V, Unger S, Zabel B, Bole-Feysot C, Nitschke P, Handford P, Casanova JL, Boileau C, Apte SS, Munnich A, Cormier-Daire V (2011) Mutations in the TGFbeta binding-protein-like domain 5 of FBN1 are responsible for acromicric and geleophysic dysplasias. Am J Hum Genet 89(1):7–14
Le Saux O, Urban Z, Tschuch C, Csiszar K, Bacchelli B, Quaglino D, Pasquali-Ronchetti I, Pope FM, Richards A, Terry S, Bercovitch L, de Paepe A, Boyd CD (2000) Mutations in a gene encoding an ABC transporter cause pseudoxanthoma elasticum. Nat Genet 25(2):223–227
Loeys B, Nuytinck L, Delvaux I, De Bie S, De Paepe A (2001) Genotype and phenotype analysis of 171 patients referred for molecular study of the fibrillin-1 gene FBN1 because of suspected Marfan syndrome. Arch Intern Med 161(20):2447–2454
Loeys B, Van Maldergem L, Mortier G, Coucke P, Gerniers S, Naeyaert JM, De Paepe A (2002) Homozygosity for a missense mutation in fibulin-5 (FBLN5) results in a severe form of cutis laxa. Hum Mol Genet 11(18):2113–2118
Loeys BL, Chen J, Neptune ER, Judge DP, Podowski M, Holm T, Meyers J, Leitch CC, Katsanis N, Sharifi N, Xu FL, Myers LA, Spevak PJ, Cameron DE, De Backer J, Hellemans J, Chen Y, Davis EC, Webb CL, Kress W, Coucke P, Rifkin DB, De Paepe AM, Dietz HC (2005) A syndrome of altered cardiovascular, craniofacial, neurocognitive and skeletal development caused by mutations in TGFBR1 or TGFBR2. Nat Genet 37(3):275–281
Loeys BL, Schwarze U, Holm T, Callewaert BL, Thomas GH, Pannu H, De Backer JF, Oswald GL, Symoens S, Manouvrier S, Roberts AE, Faravelli F, Greco MA, Pyeritz RE, Milewicz DM, Coucke PJ, Cameron DE, Braverman AC, Byers PH, De Paepe AM, Dietz HC (2006) Aneurysm syndromes caused by mutations in the TGF-beta receptor. N Engl J Med 355(8):788–798
Loeys BL, Dietz HC, Braverman AC, Callewaert BL, De Backer J, Devereux RB, Hilhorst-Hofstee Y, Jondeau G, Faivre L, Milewicz DM, Pyeritz RE, Sponseller PD, Wordsworth P, De Paepe AM (2010) The revised Ghent nosology for the Marfan syndrome. J Med Genet 47(7):476–485
Loeys BL, Gerber EE, Riegert-Johnson D, Iqbal S, Whiteman P, McConnell V, Chillakuri CR, Macaya D, Coucke PJ, De Paepe A, Judge DP, Wigley F, Davis EC, Mardon HJ, Handford P, Keene DR, Sakai LY, Dietz HC (2010) Mutations in fibrillin-1 cause congenital scleroderma: stiff skin syndrome. Sci Transl Med 2(23):23ra20
McCarthy EF (2011) Genetic diseases of bones and joints. Semin Diagn Pathol 28(1):26–36
Milewicz DM, Michael K, Fisher N, Coselli JS, Markello T, Biddinger A (1996) Fibrillin-1 (FBN1) mutations in patients with thoracic aortic aneurysms. Circulation 94(11):2708–2711
Moberg K, De Nobele S, Devos D, Goetghebeur E, Segers P, Trachet B, Vervaet C, Renard M, Coucke P, Loeys B, De Paepe A, De Backer J (2011) The Ghent Marfan Trial—a randomized, double-blind placebo controlled trial with losartan in Marfan patients treated with beta-blockers. Int J Cardiol http://dx.doi.org/10.1016/j.ijcard.2010.12.070
Neptune ER, Frischmeyer PA, Arking DE, Myers L, Bunton TE, Gayraud B, Ramirez F, Sakai LY, Dietz HC (2003) Dysregulation of TGF-beta activation contributes to pathogenesis in Marfan syndrome. Nat Genet 33(3):407–411
Ng CM, Cheng A, Myers LA, Martinez-Murillo F, Jie C, Bedja D, Gabrielson KL, Hausladen JM, Mecham RP, Judge DP, Dietz HC (2004) TGF-beta-dependent pathogenesis of mitral valve prolapse in a mouse model of Marfan syndrome. J Clin Invest 114(11):1586–1592
Ong KT, Perdu J, De Backer J, Bozec E, Collignon P, Emmerich J, Fauret AL, Fiessinger JN, Germain DP, Georgesco G, Hulot JS, De Paepe A, Plauchu H, Jeunemaitre X, Laurent S, Boutouyrie P (2010) Effect of celiprolol on prevention of cardiovascular events in vascular Ehlers-Danlos syndrome: a prospective randomised, open, blinded-endpoints trial. Lancet 376(9751):1476–1484
Radonic T, de Witte P, Baars MJ, Zwinderman AH, Mulder BJ, Groenink M (2010) Losartan therapy in adults with Marfan syndrome: study protocol of the multi-center randomized controlled COMPARE trial. Trials 11:3
Radonic T, de Witte P, Groenink M, de Bruin-Bon R, Timmermans J, Scholte A, van den Berg M, Baars M, van Tintelen J, Kempers M, Zwinderman A, Mulder B (2011) Critical appraisal of the revised Ghent criteria for diagnosis of Marfan syndrome. Clin Genet. doi:10.1111/j.1399-0004.2011.01646.x
Renard M, Holm T, Veith R, Callewaert BL, Ades LC, Baspinar O, Pickart A, Dasouki M, Hoyer J, Rauch A, Trapane P, Earing MG, Coucke PJ, Sakai LY, Dietz HC, De Paepe AM, Loeys BL (2010) Altered TGFbeta signaling and cardiovascular manifestations in patients with autosomal recessive cutis laxa type I caused by fibulin-4 deficiency. Eur J Hum Genet 18(8):895–901
Renard M, Callewaert B, Baetens M, Campens L, Macdermot K, Fryns JP, Bonduelle M, Dietz HC, Gaspar IM, Cavaco D, Stattin EL, Schrander-Stumpel C, Coucke P, Loeys B, De Paepe A, De Backer J (2011) Novel MYH11 and ACTA2 mutations reveal a role for enhanced TGFbeta signaling in FTAAD. Int J Cardiol http://dx.doi.org/10.1016/j.ijcard.2011.08.079
Royce PM, Steinmann B (2002) Connective tissue and its heritable disorders. Molecular, genetic, and medical aspects, 2nd edn. Wiley-Liss, Inc, New York
Sheikhzadeh S, Kade C, Keyser B, Stuhrmann M, Arslan-Kirchner M, Rybczynski M, Bernhardt A, Habermann C, Hillebrand M, Mir T, Robinson P, Berger J, Detter C, Blankenberg S, Schmidtke J, von Kodolitsch Y (2011) Analysis of phenotype and genotype information for the diagnosis of Marfan syndrome. Clin Genet. doi:10.1111/j.1399-0004.2011.01771.x
Shores J, Berger KR, Murphy EA, Pyeritz RE (1994) Progression of aortic dilatation and the benefit of long-term beta-adrenergic blockade in Marfan's syndrome. N Engl J Med 330(19):1335–1341
Silverman DI, Burton KJ, Gray J, Bosner MS, Kouchoukos NT, Roman MJ, Boxer M, Devereux RB, Tsipouras P (1995) Life expectancy in the Marfan syndrome. Am J Cardiol 75(2):157–160
Superti-Furga A, Gugler E, Gitzelmann R, Steinmann B (1988) Ehlers-Danlos syndrome type IV: a multi-exon deletion in one of the two COL3A1 alleles affecting structure, stability, and processing of type III procollagen. J Biol Chem 263(13):6226–6232
Tran-Fadulu V, Pannu H, Kim DH, Vick GW 3rd, Lonsford CM, Lafont AL, Boccalandro C, Smart S, Peterson KL, Hain JZ, Willing MC, Coselli JS, LeMaire SA, Ahn C, Byers PH, Milewicz DM (2009) Analysis of multigenerational families with thoracic aortic aneurysms and dissections due to TGFBR1 or TGFBR2 mutations. J Med Genet 46(9):607–613
Uitto J, Bercovitch L, Terry SF, Terry PF (2011) Pseudoxanthoma elasticum: progress in diagnostics and research towards treatment: summary of the 2010 PXE International Research Meeting. Am J Med Genet A 155A(7):1517–1526
Urban Z, Hucthagowder V, Schurmann N, Todorovic V, Zilberberg L, Choi J, Sens C, Brown CW, Clark RD, Holland KE, Marble M, Sakai LY, Dabovic B, Rifkin DB, Davis EC (2009) Mutations in LTBP4 cause a syndrome of impaired pulmonary, gastrointestinal, genitourinary, musculoskeletal, and dermal development. Am J Hum Genet 85(5):593–605
van de Laar IM, Oldenburg RA, Pals G, Roos-Hesselink JW, de Graaf BM, Verhagen JM, Hoedemaekers YM, Willemsen R, Severijnen LA, Venselaar H, Vriend G, Pattynama PM, Collee M, Majoor-Krakauer D, Poldermans D, Frohn-Mulder IM, Micha D, Timmermans J, Hilhorst-Hofstee Y, Bierma-Zeinstra SM, Willems PJ, Kros JM, Oei EH, Oostra BA, Wessels MW, Bertoli-Avella AM (2011) Mutations in SMAD3 cause a syndromic form of aortic aneurysms and dissections with early-onset osteoarthritis. Nat Genet 43(2):121–126
van de Laar IM, van der Linde D, Oei EH, Bos PK, Bessems JH, Bierma-Zeinstra SM, van Meer BL, Pals G, Oldenburg RA, Bekkers JA, Moelker A, de Graaf BM, Matyas G, Frohn-Mulder IM, Timmermans J, Hilhorst-Hofstee Y, Cobben JM, Bruggenwirth HT, van Laer L, Loeys B, De Backer J, Coucke PJ, Dietz HC, Willems PJ, Oostra BA, De Paepe A, Roos-Hesselink JW, Bertoli-Avella AM, Wessels MW (2012) Phenotypic spectrum of the SMAD3-related aneurysms-osteoarthritis syndrome. J Med Genet 49(1):47–57
van der Linde D, van de Laar IMBM, Bertoli-Avella AM, Oldenburg RA, Bekkers JA, Mattace-Raso FUS, van den Meiracker AH, Moelker A, Tanghe HLJ, van Kooten F, Frohn IME, Timmermans J, Moltzer E, Cobben JM, Van Laer L, Loeys B, De Backer J, Coucke PJ, De Paepe A, Wessels MW, Roos-Hesselink JW (2012) Cardiovascular phenotype of the recently discovered aneurysms-osteoarthritis syndrome (AOS) caused by SMAD3 mutations. J Am Coll Cardiol (in press)
van Karnebeek CD, Naeff MS, Mulder BJ, Hennekam RC, Offringa M (2001) Natural history of cardiovascular manifestations in Marfan syndrome. Arch Dis Child 84(2):129–137
Wang Y, Ait-Oufella H, Herbin O, Bonnin P, Ramkhelawon B, Taleb S, Huang J, Offenstadt G, Combadiere C, Renia L, Johnson JL, Tharaux PL, Tedgui A, Mallat Z (2010) TGF-beta activity protects against inflammatory aortic aneurysm progression and complications in angiotensin II-infused mice. J Clin Invest 120(2):422–432
Zhu L, Vranckx R, Khau Van Kien P, Lalande A, Boisset N, Mathieu F, Wegman M, Glancy L, Gasc JM, Brunotte F, Bruneval P, Wolf JE, Michel JB, Jeunemaitre X (2006) Mutations in myosin heavy chain 11 cause a syndrome associating thoracic aortic aneurysm/aortic dissection and patent ductus arteriosus. Nat Genet 38(3):343–349
Acknowledgements
The authors' research is supported in part by funding from the Fund for Scientific Research Flanders (FWO, Belgium) [G.0221.12], and the National Marfan Foundation (USA); B.L.L. is senior clinical investigator of the Fund for Scientific Research, Flanders (FWO, Belgium).
Conflict of interest
The authors have no financial relationship with the organizations listed in the acknowledgement section.
Open Access
This article is distributed under the terms of the Creative Commons Attribution License which permits any use, distribution, and reproduction in any medium, provided the original author(s) and the source are credited.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 2.0 International License (https://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Van Laer, L., Proost, D. & Loeys, B.L. Educational paper. Eur J Pediatr 172, 997–1005 (2013). https://doi.org/10.1007/s00431-012-1773-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00431-012-1773-x