
Abstract
Main conclusion
PAE8 and PAE9 have pectin acetylesterase activity and together remove one-third of the cell wall acetate associated with pectin formation in Arabidopsis leaves. In pae8 and pae9 mutants, substantial amounts of acetate accumulate in cell walls. In addition, the inflorescence stem height is decreased.
Pectic polysaccharides constitute a significant part of the primary cell walls in dicotyledonous angiosperms. This diverse group of polysaccharides has been implicated in several physiological processes including cell-to-cell adhesion and pathogenesis. Several pectic polysaccharides contain acetyl-moieties directly affecting their physical properties such as gelling capacity, an important trait for the food industry. In order to gain further insight into the biological role of pectin acetylation, a reverse genetics approach was used to investigate the function of genes that are members of the Pectin AcetylEsterase gene family (PAE) in Arabidopsis. Mutations in two members of the PAE family (PAE8 and PAE9) lead to cell walls with an approximately 20 % increase in acetate content. High-molecular-weight fractions enriched in pectic rhamnogalacturonan I (RGI) extracted from the mutants had increased acetate content. In addition, the pae8 mutant displayed increased acetate content also in low-molecular-weight pectic fractions. The pae8/pae9-2 double mutant exhibited an additive effect by increasing wall acetate content by up to 37 %, suggesting that the two genes are not redundant and act on acetyl-substituents of different pectic domains. The pae8 and pae8/pae9-2 mutants exhibit reduced inflorescence growth underscoring the role of pectic acetylation in plant development. When heterologously expressed and purified, both gene products were shown to release acetate from the corresponding mutant pectic fractions in vitro. PAEs play a significant role in modulating the acetylation state of pectic polymers in the wall, highlighting the importance of apoplastic metabolism for the plant cell and plant growth.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Pectins are one of the major components of the primary cell walls of dicots, representing approximately 35 % of the dry mass in these cellular structures (Carpita and Gibeaut 1993; Mohnen 2008). This highly diverse group of polysaccharides has been implicated in cellular processes including cell adhesion, cell growth and pathogen perception (Anderson et al. 2012; McNeil et al. 1984; Ridley et al. 2001). Pectins are divided into four structurally distinct groups: homogalacturonan (HG), rhamnogalacturonan I (RGI), rhamnogalacturonan II and xylogalacturonan (Harholt et al. 2010). The common feature amongst these four groups is the presence of galacturonic acid in the backbone of these polymers. Homogalacturonan is the most abundant pectin polymer in the dicot wall. It consists of a backbone of α-1,4-linked galacturonic acid that can be methyl esterified at the C-6 position (Atmodjo et al. 2013). HG contributes to the general cohesiveness of the cell wall via pectic cross-linking by calcium bridges (Albersheim et al. 2011; McCann and Roberts 1996; Ralet et al. 2008). RGI is the second most abundant pectic polymer in the primary wall. It consists of a repeating disaccharide of α-1,4-d-GalA-α-1,2-l-Rha. The rhamnosyl-residue can be further substituted with side chains composed of arabinose and galactose, generating large branched structures (Atmodjo et al. 2013; Ralet et al. 2005). The biological role of RGI in the cell wall is still not clear; however, the available data suggest that it plays a structural role in wall architecture (Harholt et al. 2010; Jones et al. 2003; Ulvskov et al. 2005).
All pectic polysaccharides are to a certain extent O-acetylated (Harholt et al. 2010; Ralet et al. 2005). The acetyl-moieties are usually attached to the O-2 and O-3 positions of the galacturonic acid moiety of both HG and RGI (Ishii 1997; Keenan et al. 1985). Acetylation of pectins impacts the gelation properties of this class of polymers, which is relevant for the food industry (Ralet et al. 2003). The acetate groups could negatively interfere with the calcium cross-linking between HG polymers via steric hindrance (Ralet et al. 2003).
The degree of pectin acetylation can be modulated by two processes: addition of acetyl-substituents in the Golgi apparatus, the site of pectin synthesis and/or removal of acetyl-substituents by acetylesterases after deposition in the apoplast.
Biochemically, it has been demonstrated that acetyl-CoA provides the substrate for O-acetylation in the Golgi (Pauly and Scheller 2000). Recently, several proteins involved in the transfer of acetate to wall polymers have been identified. One of these proteins is reduced wall acetylation (RWA), a putative acetyl-substrate transporter. When knocked out, the acetylation of multiple wall polymers is affected including pectic polysaccharides (Manabe et al. 2011, 2013). Another class of proteins that impact polymer O-acetylation are the members of the TBL gene family, which represent putative polymer specific acetyltransferases as demonstrated by TBL27/AXY4 or TBL29 involved in xyloglucan O-acetylation (Gille et al. 2011b) or xylan O-acetylation, respectively (Xiong et al. 2013). A pectin acetyltransferase has not been identified to date, but it is likely that such a gene is also a member of the TBL family (Gille and Pauly 2012).
Pectin acetylesterases have first been characterized by in vitro activities of purified proteins (Bordenave et al. 1995; Breton et al. 1996; Christensen et al. 1996; Williamson 1991). The identified plant pectin acetylesterases are members of the carbohydrate esterase family 13 [CE13; (Henrissat et al. 2001)]. Members of CE13 are able to hydrolyze acetylester bonds from pectins, but the specific substrate recognized by these enzymes has yet to be demonstrated (Bordenave et al. 1995; Christensen et al. 1996; Gou et al. 2012; Orfila et al. 2012; Williamson 1991). When a poplar pectin acetylesterase was overexpressed in tobacco, the transgenic plants displayed aberrant floral structures and severely collapsed pollen grain walls demonstrating an impact of pectin acetylation on plant tissue morphology and plasticity (Gou et al. 2012). The expression of a mung bean pectin acetylesterase in potato plants resulted in tubers with stiffer and stronger wall matrixes (Orfila et al. 2012). The overexpression of fungal pectin acetylesterases in Arabidopsis resulted in the up-regulation of genes involved in wall polymer O-acetylation (RWAs) and resistance to specific fungal pathogens (Pogorelko et al. 2013). As these few examples demonstrate, the function of O-acetyl groups in the life cycle of plants is beginning to emerge. In this study, we aim at gaining more insights by studying putative pectin acetylesterases in Arabidopsis.
Materials and methods
Plant growth
Arabidopsis thaliana ecotype Col-0 seeds were stratified at 4 °C for 24 h in a 0.15 % agar solution and pipetted onto soil. Pots were placed in chambers maintained at long-day conditions (16 h light/8 h dark) with a 170–190 μmol m−2 s−1 light intensity at 22 °C. Plant tissue was harvested at 35 days. At that time also stem height measurements were made from the base of the rosettes to the highest flower in the main inflorescence. Nicotiana benthamiana plants used for transient gene expression were grown for 2 weeks, then transplanted into destination pots and grown for an additional 4 weeks until use. These plants were grown under long-day conditions (16 h light/8 h dark) with 170–190 μmol m−2 s−1 light intensity at 26 °C. All plants were fertilized once with Miracle Grow All-purpose Plant Food (Scotts) according to manufacturer’s recommendation and watered as needed.
PAE vector constructs
For cloning purposes DNA amplification fragments were generated using Phusion DNA Polymerase (Finnzymes). The putative native promoter and genomic sequence for At4g19420 were cloned into pCR™8/GW/TOPO (Invitrogen) and later introduced via a SmaI/SacI restriction/ligation into pPZP221 (Hajdukiewicz et al. 1994) for complementation. For the overexpression of PAE9 in the pae9-1 background, Gateway® Cloning was used. The PAE9 (At5g23870.1) coding sequence (cds) was amplified from leaf cDNA with attB sites containing primers (Table S1) and recombined in a BP reaction with the pDONOR221 vector (Invitrogen). This vector was then recombined in an LR reaction with the binary vector pORE E4 (Coutu et al. 2007) containing the enTCUP2 promoter for overexpression (Schultink et al. 2013). The PAE9 cds was amplified from a vector (pDONOR221 containing AT5G23870.1) and tagged with 6X His at its C-terminus. This fragment was cloned into the pCR™8/GW/TOPO vector backbone. Conventional cloning, using restriction digests (XhoI/BamHI) and ligation, was used to clone the PAE9 cds tagged with 6X His into pART7 (Gleave 1992) followed by cloning into the pART27 binary vector (Gleave 1992). The PAE8 cds was amplified from Arabidopsis cDNA and Gibson cloning (Gibson et al. 2009) was used to generate PAE8cds:6XHIS in pART7 followed by conventional cloning into pART27 for protein expression in tobacco. The New England Biolabs Gibson Assembly cloning kit was used according to manufacturer’s recommendation as well as the on-line tool NEBuilder™. Binary vectors were transformed into the Agrobacterium tumefaciens strain GV3101 which was then used to dip transform (Clough and Bent 1998) A. thaliana (pae8, SALK_132026; pae9-1, SALK_046973C) or infiltrate N. benthamiana.
Protein expression, extraction, purification and western blots
For transient protein expression in tobacco, the agrobacterium infiltration buffer used contained 10 mM MES, pH 5.6, 10 mM MgCl2 and 0.15 mM acetoseryngone. The cells were suspended in OD600 1.4–2.0 and incubated for 3–4 h with acetoseryngone. Plants were then immersed in infiltration buffer containing cells and vacuum infiltrated 3 times for 3 min each. A construct overexpressing the P19 gene silencing suppressor (Voinnet et al. 2003) was always infiltrated in conjunction with the construct of interest (PAE8:6XHIS or PAE9:6XHIS in pART27) or the empty vector (pART27; final OD600 ratio of 1:0.7, respectively).
To identify and verify enzymatic activity of proteins transiently expressed in N. benthamiana proteins were extracted from pre-ground tobacco leaves (mortar and pestle in liquid nitrogen). The powder of pre-ground leaves (equivalent to ~1 mL in 2 mL Eppendorf tubes) was ground again in a Retsch ball mill (25 Hz, 2.5 min; company) and extraction buffer added (1 M NaCl; 50 mM Na2PO4, pH 8; 10 mM imidazole; 1X Halt™ Protease Inhibitor, Thermo Scientific 1861278; 2 mM β-mercaptoethanol). The suspension was incubated under gentle agitation at 4 °C for 1 h, spun down at 20,800 g for 10 min and supernatant collected for protein purification. Protein content of supernatant was measured using the Bradford assay (Bio-Rad protein assay) to normalize protein concentration for Ni–NTA bead (Qiagen 1018240) loading (20 µL of resin/2 mL protein extract). Beads were incubated with protein for 1 h at 4 °C under gentle agitation and collected into a mini spin column (Pharmacia Biotech) after a 500 g, 1 min spin down. The beads were washed 5 times with 250 µL extraction buffer, 4 times with 250 µL washing buffer (300 mM NaCl, 50 mM Na2PO4 pH 8, 20 mM imidazole) and 6 times with 50 µL elution buffer (300 mM NaCl, 50 mM Na2PO4 pH 8, 150 mM imidazole). The eluate was buffer exchanged with 50 mM amonium formate, pH 4.5 in a 500 µL Vivaspin column (MWCO of 5,000 Da, Sartorius Stedim Biotech). A final volume of ~200 µL was recovered, which was used for activity assays and western blots.
After proteins were obtained and denatured in loading buffer (NuPAGE LDS Sample Buffer 4X, Invitrogen, NP0007) at 70 °C for 10 min, 20 µL of protein was loaded onto an SDS polyacrylamide gel (10 % Criterion™ Precast Gel). These were then wet blotted onto nitrocellulose membranes using transfer buffer [0.075 % (v/v) ethanolamine, 0.0935 % (w/v) Glycine and 20 % (v/v) ethanol] at 100 V for 80 min at 4 °C. The membrane was then blocked overnight at 4 °C in 50 mM Tris Hcl, 150 mM NaCl and 0.5 % Tween (TBS-T) containing 3 % (w/v) nonfat powdered milk. Primary antibody (mouse anti 6X HIS, Fisher 50272472) was added (1:3,000, v/v) and incubated for 3 h at room temperature. This was followed by three 10-min washes with TBS-T and incubation in TBS-T containing 3 % (w/v) nonfat powdered milk with the secondary HRP conjugated antibody (Goat Anti-mouse IgG HRP Conjugate, Invitrogen, M30102; 1:3,000, v/v) for 1 h at room temperature. After another series of TBS-T washes (3) the membrane was developed by adding chemiluminescent reagents (Genscript LumiSensor) and visualized using the Fuji LAS-4000 imager.
Protein sequence analysis
Protein sequence alignments and trees were generated using the Seaview4 and the MUSCLE software packages.
Plant cell wall preparations
Stems tissue (15–20 mg of stem lower internode) was pre-ground in large metal ball containers (Retsch MM 400; 30 Hz for 30 s). For stem preparations and the reverse genetics screen (Table 1; leaves) dry plant material (20–40 mg of leaf or 10–20 mg of stem material) was ground in liquid nitrogen using 3 small metal balls in 2 mL plastic tubes twice (Retsch MM 400; 25 Hz for 2.5 min). The ground material was washed twice with 70 % ethanol (1.5 mL) by vortexing, pelleting the wall material (20,800 g for 10 min) and discarding the supernatant. The residue was washed three times with a 1:1 (v:v) methanol:chloroform solution (1.5 mL) using the same pelleting conditions as described above. The pellet was dried in a speed vacuum centrifuge at 60 °C for 15 min (Eppendorf Vacufuge™).
Plant cell wall preparations for PAE8 and PAE9 lines used for pectin digest, substrate production and size exclusion chromatography (SEC) were conducted in larger volumes. Up to 600 mg of dried 35-day-old leaf material was ground (200 mg at a time) in large metal ball mill grinders (Retsch ball mill MM 400) two times at 30 Hz for 30 s. This procedure produced a fine powder, 500 mg of which was washed 4 times with 70 % ethanol (30 mL per wash) by vortexing and pelleting of the wall material (3,220 g for 10 min). The supernatant was discarded and the pellet was washed 4 times with a 1:1 methanol:chloroform solution (30 mL per wash) using the same pelleting conditions as described above. The pellet was dried at room temperature for 48 h followed by at least 1 h in the lyophilizer.
Enzymatic de-starching and pectinase digest
Starch was removed from the small cell wall preparations (Table 1) by enzymatic digestion. A reaction consisted of 13–19 mg wall material in 1 mL McIlvane buffer, pH 5. The suspension was vortexed and incubated at 80 °C for 20 min and then cooled on ice. A reaction buffer was added to a final concentration of 0.065 μg mL−1 sodium azide; 0.65 μg mL−1 α-amylase (Sigma A-6380); 12.8 U mL−1 of Pullulanase M2 (Megazyme). The digest was incubated at 37 °C at 230 rpm for 15 h. The digest was stopped by incubation at 99 °C for 10 min. Wall material was pelleted by centrifugation (3,220g for 10 min) and the supernatant discarded. The pellet was washed with equal volumes of water (30 mL) for 3 times. Finally, a 70 % aqueous ethanol wash of equal volume was performed before the pellet was dried at room temperature followed by vacuum treatment (Eppendorf Vacufuge™).
For larger cell wall preparations the de-starching digest included 150–170 mg wall material in McIlvane buffer, pH 5 (~13 mg mL−1). The suspension was vortexed and incubated at 80 °C for 20 min and then cooled on ice. A reaction buffer was added with a final concentration of 0.065 μg mL−1 sodium azide; 0.65 μg mL−1 α-amylase (Sigma A-6380); and 12.8 U mL−1 of Pullulanase M2 (Megazyme). The digest was incubated at 37 °C at 230 rpm for 15 h. The digest was stopped by incubation at 80 °C for 20 min. Wall material was pelleted by centrifugation (3,220g for 10 min) and the supernatant discarded. The pellet was washed with equal volumes of water (30 mL) for 3 times. Finally, a 70 % aqueous ethanol wash of equal volume was performed before the pellet was dried at room temperature followed by vacuum treatment (Eppendorf Vacufuge™).
De-starched wall material was treated with pectinases to generate the pectic extract. The enzymatic reaction (per mL) was conducted with 6 mg of material in 50 mM ammonium formate, pH 4.5, containing 0.2 μg sodium azide, 2 mU of endopolygalacturononase M2 (EC3.2.1.15; Megazyme) and 0.04 mU of pectin methyl esterase (EC3.1.1.11; Novozymes, Christgau et al. 1996). The digest was incubated for 17.5 h at 37 °C with 230 rpm agitation and stopped by incubation at 80 °C for 20 min. The pellet was spun down (3,220 g for 10 min) and the supernatant, representing the pectic extract, was filtered through a 0.45-μm syringe filter (mini sart high flow 16533, Satorius Stedin). The pellet was washed 3 times with 15 mL water before drying.
Pectin fractionation using SEC
The pectic extract was freeze-dried and resuspended in 50 mM amonium formate, pH 4.5 (1 mL). The concentrated pectic extract was subjected to size exclusion chromatography using a superpose 12 10/300 GL column (Amersham Biosciences). The column was connected to an Akta Purifier FPLC (General Electric) or a PL-GPC 50 (Varian Inc.) chromatographer. The column was equilibrated in 50 mM amonium formate, pH 4.5, using a 0.4-mL min−1 flow. Samples were injected manually using a 100-μL loop. Fractions were collected in 30-s intervals in a 96-well microtiter plate (Greiner bio-one) and lyophilized. The resulting pectic fractions were resuspended in water (100 μL) and used for subsequent analysis. Dextran standards of known sizes were used to calibrate the column.
Acetic acid measurements of de-starched walls, pectic extract and pectin fractions
Wall preparations (10 mg/mL), pectic extract or pectic fractions were saponified by adding an equal volume of 1 M NaOH and incubation for 1 h at 26 °C, under gentle agitation of 600 rpm. The de-esterified samples were neutralized with an equal volume of 1 M HCl. The reaction was pelleted for 10 min at 20,800 g and the acetic acid content of the supernatant (10–50 μL) determined using the Acetic Acid Kit (K-Acet, Megazyme) as described (Gille et al. 2011a).
Activity assays
Recombinant protein derived from tobacco leaves (PAE9 and PAE8) was tested for activity against pectin fractions. The added protein was equalized based on protein content and was incubated for 18 h with 40 μL of each pectic fraction in a total volume of 60 μL (50 mM ammonium formate, pH 4.5, for PAE9 and pH 5.0 for PAE8). After incubation the acetate released was measured using the Acetic Acid Kit (K-ACET, Megazyme).
Transcriptional analysis of A. thaliana (RT-PCR and Q-RT-PCR)
For RT-PCR analysis total RNA was extracted from plant tissue using the Plant RNeasy kit from Qiagen according to manufacturer’s protocol. Total RNA (3.5 µg) was treated with DNAse (Roche 04716728001) in a 20-µL reaction using 1 µL DNAse and 2 µL of 10 X buffer. Treatment was carried out at 37 °C for 15 min and followed by a 70 °C denaturation step for 15 min. The Superscript III First Strand Synthesis kit from Invitrogen was used to synthesize cDNA and non-RT controls from 250 ng of total RNA. The cDNA (2 µL) was then used in JumpStart Red Taq ReadyMix (Sigma) PCR reactions. The polypyrimidine tract-binding protein 1 [PTB, (Gille et al. 2009)] gene was used as an expression control.
For Q-RT-PCR analysis total RNA was extracted from plant tissue using the Plant RNeasy kit from Qiagen according to manufacturer’s protocol. DNase treatment for Q-RT-PCR was performed using the Ambion TURBO DNA-free™ kit. Treated total RNA (200 ng) was then used for cDNA synthesis using the M-MLV Reverse Transcriptase kit (Invitrogen 28025-013) following manufacturer’s instructions. The templates (1 µL) were used for Q-RT-PCR reactions with the Thermo Scientific Maxima SYBR Green/ROX Q-PCR Master Mix. Reactions were set up according to instructions from the manual and Q-RT-PCR reactions were run on a StepOnePlus Real-Time PCR System from applied Biosciences. Cycling conditions commenced with 10 min at 95 °C followed by 40 cycles of 95 °C for 15 s, 60 °C for 30 s and 72 °C for 30 s. Relative abundance of transcripts was calculated by the Applied Biosciences software using the PTB gene as the internal control.
Monosaccharide analysis using high-performance anion-exchange chromatography with pulse amperometric detection (HPAEC-PAD)
Wall material, pectic extract and remaining residue and pectic fractions were hydrolyzed in 250 μL of 2 M tri-fluoroacetic acid (TFA) at 121 °C for 90 min. The reaction was dried under a flow of nitrogen. The sample was washed two times with 300 µL isopropanol and dried under nitrogen stream each time. The hydrolyzed samples were resuspended in water and subjected to a CarboPac PA20 (neutral sugars) or CarboPac A200 (uronic acids) columns using 3 separate gradient programs to resolve all sugars of interest. For neutral sugar separation a flow rate of 0.4 mL min−1 with the following gradient was used: 2 mM NaOH for 20 min; 100 mM NaOH flush for 5 min; and 2 mM NaOH for 5 min for re-equilibration. In parallel a second method was used for neutral sugar separation: 18 mM NaOH for 15 min; 100 mM NaOH flush for 5 min; and 18 mM NaOH for 7 min for re-equilibration. For the separation of uronic acids the following gradient was used: 0.1 M NaOH with a gradient of 50–200 mM sodium acetate from 0 to 10 min; 200 mM NaOH flush for 2 min; 50 mM NaOH for 2.9 min. For quantification purposes standard curves for all measured sugars (fucose, galactose, glucose, xylose, mannose, rhamnose, arabinose, galacturonic acid and glucuronic acid) were used with the following concentrations (μg mL−1): 2, 1, 0.5, 0.25, 0.125, 0.0625, 0.03125. To account for possible sugar degradation during the TFA hydrolysis process standard mixtures of known content (6, 3, 1.5, 0.75 and 0.375 μg/individual sugar) of each analyzed sugar were hydrolyzed under the same conditions in parallel with tested samples. Measurements obtained for tested samples were corrected based on measured degradation of standards.
Statistical analysis
Statistical analysis was performed using one-way ANOVA and Tukey’s test (level of significance: P < 0.05; SigmaPlot for Windows Version 11.0) or Student’s t test (P value as indicated; Microsoft Excel tool).
Results
Mutants in the Arabidopsis pectin acetylesterase gene family have walls with increased acetate content and reduced inflorescence stem growth
The A. thaliana genome contains 12 annotated CE13 genes [Fig. 1; CAZY.org; (Henrissat et al. 2001)]. Based on the availability of T-DNA insertion lines with altered gene expression, the function of nine of those genes (At2g46930, At3g09410, At3g62060, At4g19410, At4g19420, At5g23870, At5g26670, At5g45280, At3g05910) was investigated. Gene expression in mutant lines was established by RT-PCR and at least one line per gene showed reduced expression (Table 1; Fig. 2). Since rosette leaves are rich in pectic polymers (Zablackis et al. 1995) this tissue was chosen for biochemical analyses. Wall material of 5- to 6-week-old leaves was prepared and its alkali labile acetate content determined (Table 1). Some T-DNA lines affected in gene expression exhibited a significant alteration/increase in acetate content including pae3-2 (SALK_137505C), pae7-2 (GABI_272B08) pae8 (At4g19420; SALK_132026), pae9-1 (At5g23870; SALK_046973C), pae9-2 (At5g23870; GABI_803G08) and pae11-2 (GABI_505H02). PAEs, whose second allel mutants represent a knock-down rather than a knockout allel (PAE3; PAE5; PAE7), or where one or both of the allels did not display a statistically significant increase in acetate content (PAE6, PAE10, PAE11, PAE12) were due to their genetic inconsistencies not further pursued here. Should more knock-out lines become available in the future many of those PAE would certainly be of interest for a follow-up.

Phylogenetic tree of gene family related to a pectin acetylesterase. Tree constructed using the maximum likelihood method with the Seaview4 software package, which used muscle and PhyML for alignment and tree, respectively. VLAE acetylesterase expressed in the developing corm of Amorphophallus konjac. Bar indicates relative distance between protein sequences. Black stars highlight the genes focused on in the present study

RT-PCR of pectin acetylesterase T-DNA insertion lines. RT-PCR for PAE3 transcript (pae3-1, SALK_066524C; pae3-2, SALK_137505C; 3-week-old leaves); PAE5 (pae5-1, SALK_140555; pae5-2, SALK_052303C; 3-week-old leaves); PAE6 (pae6-1, SALK_020618; pae6-2, SALK_134907; 19-day-old leaves); PAE8 (pae8, SALK_132026; 2-week-old leaves); PAE9 (pae9-1, SALK_046973C; pae9-2, GABI-803G08; 3-week-old leaves); PAE7 (pae7-1, SALK_093502C; pae7-2, GABI_272B08, 19-day-old leaves); PAE10 (pae10-1, SALK_043807; pae10-2, SAIL_802_C05; 3-week-old leaves); PAE11 (pae11-1, SALK_049340.48.65.x; pae11-2, GABI_505H02; 19-day-old leaves); PAE12 (pae12-1, GABI_018A02; pae12-2, GABI_646F06; 3-week-old leaves). PTB—housekeeping gene. WT = Col-0 plants of same age as insertion lines being tested. Primers used for each reaction are listed in Suppl. Table S2
Two knockout alleles of PAE9 were available (Figs. 2, 3a). When leaf material was tested for altered wall acetate content an approximate 20 % increase was found in both mutant alleles (Fig. 4a). Three independent PAE9 overexpression lines in the pae9-1 background revealed complementation of the wall acetate back to WT levels. The overexpression lines were shown to have at least 3 times higher transcript levels than WT plants (Fig. 3b). These results indicate that PAE9 is also responsible for modulating leaf wall acetate levels.

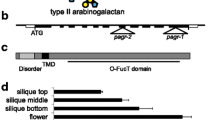
Gene models for PAE8 and PAE9 and Q-RT-PCR of complementation and overexpression lines. a Gene model for PAE8 and PAE9; boxes indicate exons (gray, translated regions); black line in between exons indicate introns. Vertical black triangles indicate position of T-DNA insertions in relation to translation start site (pae8 SALK_132026, +2,046 bp; pae9-1, SALK_046973C, +2,712 bp; pae9-2, GABI-803G08, +1,949 bp). Small black triangles indicate primer positions used for RT-PCR. b Relative quantity of the PAE8/PAE9 transcript determined by Q-RT-PCR in 35-/30-day-old leaves of T3 complementation lines. Error bars indicate minimum and maximum variation. PTB gene expression was used as an internal control for normalization. WT = Col-0 35/30-day-old leaves. n = 6

Acetate content of wall (a), pectin extract, and remaining residue (b) of PAE mutant lines. Error bars indicate standard deviation; letters indicate statistical significant differences based on one-way ANOVA (P < 0.05); n ≥ 4
Since only one pae8 knock-out allel was available (Table 1; Fig. 3a), genetic complementation of this pae8 mutant was performed by transforming the genomic WT PAE8 sequence under the control of the native PAE8 promoter into the mutant. Three independently obtained T3 complementation lines were subjected to Q-RT-PCR analysis confirming that the PAE transcript was expressed (Fig. 3b). The pae8 mutant exhibited an approximate 20 % acetate increase in its leaf walls compared to WT (Fig. 4a). The complementation lines were shown to rescue the mutant phenotype, i.e. wall acetate levels were reduced down to WT levels (Fig. 4a). These data demonstrate that PAE8 modulates the wall acetate level of leaves.
A double mutant, pae8/pae9-2, was generated and analysis indicated a 37 % increase in total wall acetate content (Fig. 4a), a near additive level compared to the two single mutants.
No morphological changes could be observed in the rosette leaves of these Arabidopsis mutants. However, publically available gene expression data indicated a high expression of both PAE8 and PAE9 in the lower internode of Arabidopsis inflorescences (Fig. S1). Indeed, upon examining the inflorescence height of 35-day-old Arabidopsis plants a reduction was observed for pae8 (10 %; Fig. 5a, b) and the pae8/pae9-2 double mutant (21 %; Fig. 5a, b). The complementation lines for pae8 restored inflorescence heights to WT levels (Fig. 5b). The double mutant exhibited twice the reduction in height of the one exhibited by the pae8 single mutant suggesting an additive effect caused by the pae9-2 mutation even though no consistent height reduction was found for the PAE9 mutants. Due to this developmental alteration acetate content was determined in the stem lower internode of the lines, but no differences in acetate content could be observed between the mutant lines and WT (Fig. 5c).

Inflorescence stem heights and stem acetate content. a General appearance of 35-day-old Arabidopsis plants; white bar 5 cm. b Inflorescence height in cm; n ≥ 26. c Acetate content in the second internode of 35-day-old stems; n ≥ 6. Error bars indicate standard deviation; letters indicate statistical significant differences based on one-way ANOVA (P < 0.05)
The pae8 and pae9 mutants have increased pectin acetate content
An attempt was made to identify the wall polymer with enhanced acetyl-substituents in the pae8 and pae9 mutants. Since VrPAE1 has been shown to have pectin acetylesterase activity (Bordenave et al. 1995; Breton et al. 1996) the initial investigation focused on the pectic polysaccharides. An enzymatic pectin digest was performed on leaf wall material to extract pectic polymers and its alkali labile acetate content was determined. Indeed, the pectic extracts derived from pae8, pae9-1, pae9-2 and pae8/pae9-2 exhibited an increased acetate content compared to WT (Fig. 4b). In the PAE8 complementation lines and in the PAE9 overexpression lines in the pae9-1 background the acetate content was equal to WT levels (Fig. 4b). The monosaccharide composition of the pectic extract of the various mutant lines revealed as expected a dominance in galacturonic acid and rhamnose (Tables 2, 3, Supplemental Fig. S2) indicative of pectins. However, a reduction in the rhamnose content of pae8 (−12 %) and pae8/pae9-2 (−17 %) in the pectin extracts was observed, but not galacturonic acid, suggesting that RGI in these mutants could be less extractable.
The remaining wall residue after pectin extraction was also analyzed for acetate content (Fig. 4b). All remaining residues still contained acetate with changes among the different mutants that were similar to the pectin extracts. The monosaccharide analysis of these residues revealed still the presence of the pectic monosaccharides rhamnose and galacturonic acid demonstrating that the enzymatic pectin extraction conditions used did not lead to the entire solubilisation of pectins and thus removal from the residues (Tables 2, 3; Fig. S2c).
The origin of the increase of pectic acetate in the mutants was investigated by fractionation of the pectic extract through size exclusion chromatography (Fig. 6). Distinct peaks were observed for pectic fragments of different molecular weights. The eluted pectic compounds were pooled into five fractions as indicated in Fig. 6. Monosaccharide analysis of the pectin fractions indicates a qualitative separation of pectic domains (Tables 2, 3; Fig. S3; Fig. S4). Fractions I and II contain large molecules (fraction I > 147.5 kDa; fraction II 147.5–23 kDa; Fig. 6), which are enriched for monosaccharides present in RGI structures (galacturonic acid, rhamnose, arabinose and galactose). The ratio between galacturonic acid and rhamnose in fractions I and II is larger than 1 suggesting that these fractions also contain HG. Fraction III (23.2–4.6 kDa) is enriched with galacturonic acid when compared to fractions I and II, however, still containing monosaccharides representative of RGI. In fractions IV (4.6–1.5 kDa) and V (1.5–0.6 kDa) the abundance of galacturonic acid suggests small homogalacturonan fragments. The concentration of sugars in each fraction is also consistent with the chromatogram profiles obtained (Tables 2, 3; Fig. 6; Fig. S3; Fig. S4). Based on the content of acetate per galacturonic acid the degree of acetylation of the polymers in the large-molecular-weight fractions is higher than the lower molecular weight fractions (Tables 3, 4; Fig. S3; Fig. S4). Similar chromatograms (Fig. 6) and monosaccharide content (Tables 2, 3; Fig. S3; Fig. S4) were obtained for the WT and the various mutant lines indicating that no detectable change occurred in the overall pectic polymer size of the single mutants.

Size exclusion chromatography of pectic extracts. Pectic extracts of PAE mutant lines (right titles) were applied to size exclusion chromatography. Fractions were collected according to depicted scheme (roman numerals); dashed bars indicate fraction borders. Black arrows indicate elution time of dextran standards. ox9 overexpression of the PAE9 coding sequence. comp-1 complementation with the native promoter and genomic sequence of PAE8. n ≥ 5
In pae8 all fractions with the exception of fraction III exhibit a significant increase in acetate content (Table 4), while the corresponding complementation lines restored the fraction’s acetate to WT levels (Table 4). In contrast, the pae9 mutants display an increase in acetate specifically in fraction I by approximately 30 % (Table 4). The PAE9 overexpression lines in the pae9-1 background show that acetate in this fraction is restored to WT levels (Table 4).
The pae8/pae9-2 double mutant exhibits increased acetate phenotypes in all fractions with the exception of fraction III, which is in agreement with an additive phenotype of the single mutants (Table 4). In the double mutant the rhamnose content is also reduced in fractions II (40 % reduction) and III (13 % reduction) as is the galacturonic acid content in fraction II [(30 % reduction); Tables 2, 3; Fig. S3b, c]. All results are consistent with the proposed pectin acetylesterase activities.
Recombinant PAE8 and PAE9 release acetate from mutant pectin fractions in vitro
In vitro activity assays of heterologously expressed proteins were performed to investigate the biochemical activity for PAE8 and PAE9. Both proteins were tagged (6XHis) and transiently expressed in N. benthamiana (Figs. 7b, 8b). The chimeric PAE8 and PAE9 proteins were purified using affinity chromatography, and as a control the empty vector (EV) protein extract was processed in the same manner. Both proteins expressed with slightly higher molecular weights (~55 kDa) than the predicted sizes of 45.3 and 46.8 kDa for PAE8 and PAE9, respectively (Figs. 7b, 8b). As expected, no His-tagged proteins were detected in empty vector transformed tobacco (Figs. 7b, 8b).

PAE8 releases acetate in vitro from pae8 pectin fractions. a Percentage acetate released from pectin fractions (I–V) and Amorphophallus konjac acetylated glucomannan by heterologously expressed PAE8. Alkali would release 100 % of acetate in those fractions. Pectic fractions were generated from three independent pae8 biological replicates. The same protein content was used for PAE8 and EV activity assays. Asterisk indicates significant differences based on t test (P < 0.015; n = 3); EV protein extracts purified from tobacco plants transformed with an empty vector; PAE8 protein extract purified from tobacco plants transformed with PAE8:6XHIS. b Western blot showing the presence of PAE8 protein (~55 kDa) derived from tobacco plants transformed with the PAE8:6XHIS construct. +control = multi-tag positive control (Life Technologies). EV native protein extracts purified from tobacco plants transformed with an empty vector. M Magic MarkerTM XP (Life Technologies)

PAE9 releases acetate in vitro from pae9 pectin fractions. a Percentage acetate released from pectin fraction (I–V) and Amorphophallus konjac acetylated glucomannan by heterologously expressed PAE9. Alkali would release 100 % of acetate in those fractions. Pectic fractions were generated from three independent pae9-1 biological replicates. The same protein content was used for PAE9 and EV activity assays. Asterisk indicates significant differences based on t test (P < 0.015; n = 3); EV protein extracts purified from tobacco plants transformed with an empty vector; PAE9 protein extract purified from tobacco plants transformed with PAE9:6XHIS. b Western blot showing the presence of PAE9 protein (~55 kDa) derived from tobacco plants transformed with the PAE9:6XHIS construct. EV native protein extracts purified from tobacco plants transformed with an empty vector. Marker Magic MarkerTM XP (Life Technologies)
In vitro activity assays were designed to investigate pectin acetylesterase activity by utilizing the pectin fractions derived from the mutants as substrates (Fig. 6). When PAE8 and PAE9 were incubated with the corresponding mutant pectic fractions, acetate was released from fractions I and II, but not from other fractions (Figs. 7a, 8a; Table S1). PAE9 was able to release more acetate from its corresponding substrate in vitro than PAE8. The observed activities are consistent with the phenotypes of increased acetate found in the pectic fractions I and II of pae8 and pae9 mutants (Table 4), but not for pae8 in fractions IV and V (Table 4).
The transcriptome analysis of the developing corms of Amorphophallus konjac revealed the presence of an acetylesterase within the 100 most abundant transcripts that have high protein sequence similarity to PAE8 [VLAE, vodoo lily acetyl esterase; Fig. 1; (Gille et al. 2011a)]. Since Amorphophallus konjac contains mainly O-acetylated glucomannan as a storage polymer (Chua et al. 2010; Li et al. 2005) this protein was designated as a putative glucomannan O-esterase [Fig. 1; (Gille et al. 2011a)]. Hence O-acetylated glucomannan was also tested as a substrate for PAE8 and PAE9. However, neither enzyme exhibited any activity against this substrate under the conditions tested (Figs. 7a, 8a).
Discussion
PAE8 and PAE9 represent pectin acetylesterases based on the in vitro assays with recombinant proteins (Figs. 7a, 8a) and the elevated levels of pectin O-acetylation in the corresponding Arabidopsis mutants (Fig. 4b; Table 4). WT and mutant fractions I and II contain mainly RGI (Tables 2, 3; Fig. S3a, b), but the galacturonic acid/rhamnose ratio indicates that HG is also present, consistent with pectin structural models that HG is covalently linked to RGI (Yapo 2011). Elevated acetate levels were observed for high-molecular-weight fractions in all three single mutants (pae8, pae9-1 and pae9-2) indicating that the acetate level of RGI, but also potentially HG, is increased in those mutants. There is no increase in the galacturonic acid content in the pectic fractions of the high acetate mutants (Table 3; Fig. S3; Fig. S4), and the SEC profiles are not altered (Fig. 6) suggesting that the increased acetate content has no effect on HG digestion (Sengkhamparn et al. 2009) and fragmentation pattern. Therefore, RGI seems to be the preferred substrate for PAE8 and PAE9 in planta. Indeed, there is some evidence for reduced extractability of RGI based on the reduction of rhamnose content in the pectin extracts (pae8 and pae8/pae9-2) and fractions II and III (pae8/pae9-2; Table 2). Less RGI could represent a higher degree of acetylation in this polymer since these mutants show increased acetate levels in the pectin extract and fraction II (Table 4). Highly acetylated wall molecules are more difficult to break-down by enzymes (Busse-Wicher et al. 2014). PAE8 and PAE9 represent acetylesterases with distinct substrate specificities based on the additive effects observed in the double mutant (Fig. 4). The highly complex structure of the RGI molecule entails such diverse substrate sites for these enzymes (Nakamura et al. 2002; Ridley et al. 2001; Yapo 2011).
While the mutants (pae8, pae9-1, pae9-2 and pae8/pae9-2) harbor enzymatically extractable pectins with higher acetate contents the remaining residue still contained approximately half of the wall acetate of each line, whose levels were also altered in the mutants (Fig. 4b). The residue still contains pectins based on the monosaccharide compositional analysis (Tables 2, 3; Fig. S2c). However, the residue also contains other wall polymers such as heteroxylan and heteromannan [Fig. S2c; (Pettolino et al. 2012)], which can be O-acetylated. Therefore, there is a possibility that PAE8 and PAE9 also act on other acetylated wall polymers. Unfortunately, very few isolated, structurally defined O-acetylated wall polymers are available, because the procedures to isolate wall polymers usually entail harsh conditions such as strong alkali (Albersheim et al. 2011), resulting in the removal of acetyl substituents from isolated wall polymers. One of the substrates that is available is O-acetylated glucomannan isolated from Amorphophallus konjac (Gille et al. 2011a), which, when tested with recombinant PAE8 and PAE9, did not represent a substrate (Figs. 7a, 8a). While both enzymes do not exhibit mannan acetylesterase activity the possibility that they act on other wall polymers than RGI cannot be excluded.
According to the phylogenetic tree of the Arabidopsis pectin acetylesterase family PAE8 and PAE9 seem to be the only genes without close paralogs, whereas the other 10 genes clade in 5 pairs (Fig. 1). In Arabidopsis such recent genome duplication events have led to genetic redundancy in some traits (Vision et al. 2000). Hence, PAE8 and PAE9 seem to represent functionally non-redundant proteins. Indeed, the mutant analyses demonstrated that in corresponding mutants of only these two genes a consistent acetate phenotype could be observed as well as an additive acetate increase in the double mutant (Fig. 4a). The generation of double mutants of the other members of the PAE family which clade together and might thus be functionally redundant should prove their substrate specificity.
Some of the first mutants identified to be involved in pectin biosynthesis exhibited dwarf phenotypes, hence named QUASIMODO (Bouton et al. 2002). The qua1 mutant (putative galacturonic acid transferase) exhibits a shortened inflorescence size and general dwarfed phenotype (Bouton et al. 2002). Another mutant believed to be involved in the methylesterification of homogalacturonan, qua2, also shows defects in plant development and growth (Mouille et al. 2007). These findings parallel with our observation that mutants impaired in normal pectin acetylation result in plants with shorter inflorescences (Fig. 5a, b). The growth phenotype identified in this study is manifested in the stem (Fig. 5a, b), which is a tissue that contains only low amounts of pectins when compared to leaves (Xiong et al. 2013). However, it has been reported that pectin biosynthetic genes can be highly expressed in the vasculature (Harholt et al. 2006; Orfila et al. 2005). Pectin structures like galactan are also found in some types of tissues like tension wood which predominantly harbor secondary walls (Arend 2008). These observations suggest that pectin could have an important role in plant tissue development, even in tissues, where it is not highly abundant such as Arabidopsis inflorescence stems. Hence, the inability to detect a wall acetate increase in the mutant stems (Fig. 5c) despite a high expression of PAE8 and PAE9 in this tissue does not imply that the reduced stem height is not related to PAE activity.
Plant pectin acetylesterases act on wall polymers after their synthesis and partially remove once added O-acetyl substituents (Gou et al. 2012; Orfila et al. 2012). The pae8/pae9-2 double mutant phenotype leads to pectins with a 37 % increase in pectin O-acetylation (Fig. 4a) demonstrating the importance of PAEs to modulate the degree of this substituent on pectin. This finding parallels the action of pectin methylesterases (PME) which have been shown to de-methylesterify pectins post synthesis (Micheli 2001). The demethylesterification of HG by PME35 was shown to contribute to the mechanical strength of stem tissues in Arabidopsis (Hongo et al. 2012). It has also been shown that PME genes play a role in pathogen resistance. PME mutants in Arabidopsis promoted enhanced growth of Pseudomonas syringaea without altering total plant PME activity, associating specific pectin de-methylation patterns to pathogen resistance (Bethke et al. 2014). In strawberries a direct correlation between wall PME activity and fruit tissue softening has been established (Draye and Van Cutsem 2008). These results highlight the impact of post synthesis modifications on wall biology.
The loss of function of acetylesterase genes in the pae8/pae9-2 double mutant leads to a 37 % increase in total wall acetate levels (Fig. 4a)—a plant biomass feature that has not been achieved previously. The ability to engineer plant biomass with increased acetate content conveys advantageous attributes for biorefinery processes and human health aspects. Increased levels of acetate in hydrolysate from biomass can act as an antimicrobial agent in industrial yeast fermentation processes increasing hygiene and thus overall efficiency (Wei et al. 2013). The use of solventogenic bacteria in fermentation processes of high acetate biomass hydrolysate can help increase solvent formation (Anbarasan et al. 2012). As part of the human diet acetate in indigestible fibers (hemicelluloses and pectin) is released in the colon. Elevated acetate levels in the colon have been associated with the suppression of appetite and obesity control (Frost et al. 2014).
Author contributions
AdeS carried out experiments, analyzed the data and wrote the manuscript; PAH identified PAE8 knockout lines, SG characterized acetate content in pae8; MP conceptualized research, analyzed the data and wrote the manuscript.
References
Albersheim P, Darvill A, Roberts K, Sederoff R, Staehelin A (2011) Plant cell walls: from chemistry to biology. Garland Science, New York
Anbarasan P, Baer ZC, Sreekumar S, Gross E, Binder JB, Blanch HW, Clark DS, Toste FD (2012) Integration of chemical catalysis with extractive fermentation to produce fuels. Nature 491:235–239. doi:10.1038/nature11594
Anderson CT, Wallace IS, Somerville CR (2012) Metabolic click-labeling with a fucose analog reveals pectin delivery, architecture, and dynamics in Arabidopsis cell walls. Proc Natl Acad Sci USA 109:1329–1334. doi:10.1073/pnas.1120429109
Arend M (2008) Immunolocalization of (1,4)-beta-galactan in tension wood fibers of poplar. Tree Physiol 28:1263–1267
Atmodjo MA, Hao Z, Mohnen D (2013) Evolving views of pectin biosynthesis. Annu Rev Plant Biol 64:747–779. doi:10.1146/annurev-arplant-042811-105534
Bethke G, Grundman RE, Sreekanta S, Truman W, Katagiri F, Glazebrook J (2014) Arabidopsis PECTIN METHYLESTERASEs contribute to immunity against Pseudomonas syringae. Plant Physiol 164:1093–1107. doi:10.1104/pp.113.227637
Bordenave M, Goldberg R, Huet JC, Pernollet JC (1995) A novel protein from mung bean hypocotyl cell walls with acetyl esterase activity. Phytochemistry 38:315–319
Bouton S, Leboeuf E, Mouille G, Leydecker MT, Talbotec J, Granier F, Lahaye M, Hofte H, Truong HN (2002) QUASIMODO1 encodes a putative membrane-bound glycosyltransferase required for normal pectin synthesis and cell adhesion in Arabidopsis. Plant Cell 14:2577–2590. doi:10.1105/Tpc.004259
Breton C, Bordenave M, Richard L, Pernollet JC, Huet JC, Perez S, Goldberg R (1996) PCR cloning and expression analysis of a cDNA encoding a pectinacetylesterase from Vigna radiata L. FEBS Lett 388:139–142
Busse-Wicher M, Gomes TCF, Tryfona T, Nikolovski N, Stott K, Grantham NJ, Bolam DN, Skaf MS, Dupree P (2014) The pattern of xylan acetylation suggests xylan may interact with cellulose microfibrils as a two-fold helical screw in the secondary plant cell wall of Arabidopsis thaliana. Plant J. doi:10.1111/tpj.12575
Carpita NC, Gibeaut DM (1993) Structural models of primary cell walls in flowering plants: consistency of molecular structure with the physical properties of the walls during growth. Plant J 3:1–30
Christensen TMIE, Nilsen JE, Mikkelsen JD (1996) Isolation, characterization and immuno localization of orange fruit acetyl esterase. In: Visser JV, Voragen AGJ (eds) Pectins and pectinases. Elsevier Science B.V, Amsterdan, pp 723–730
Christgau S, Kofod LV, Halkier T, Andersen LN, Hockauf M, Dorreich K, Dalboge H, Kauppinen S (1996) Pectin methyl esterase from Aspergillus aculeatus: expression cloning in yeast and characterization of the recombinant enzyme. Biochem J 319:705–712
Chua M, Baldwin TC, Hocking TJ, Chan K (2010) Traditional uses and potential health benefits of Amorphophallus konjac K. Koch ex N.E.Br. J Ethnopharmacol 128:268–278. doi:10.1016/j.jep.2010.01.021
Clough SJ, Bent AF (1998) Floral dip: a simplified method for Agrobacterium-mediated transformation of Arabidopsis thaliana. Plant J 16:735–743
Coutu C, Brandle J, Brown D, Brown K, Miki B, Simmonds J, Hegedus DD (2007) pORE: a modular binary vector series suited for both monocot and dicot plant transformation. Transgenic Res 16:771–781. doi:10.1007/s11248-007-9066-2
Draye M, Van Cutsem P (2008) Pectin methylesterases induce an abrupt increase of acidic pectin during strawberry fruit ripening. J Plant Physiol 165:1152–1160. doi:10.1016/j.jplph.2007.10.006
Frost G, Sleeth ML, Sahuri-Arisoylu M, Lizarbe B, Cerdan S, Brody L, Anastasovska J, Ghourab S, Hankir M, Zhang S, Carling D, Swann JR, Gibson G, Viardot A, Morrison D, Louise Thomas E, Bell JD (2014) The short-chain fatty acid acetate reduces appetite via a central homeostatic mechanism. Nat Commun 5:3611. doi:10.1038/ncomms4611
Gibson DG, Young L, Chuang RY, Venter JC, Hutchison CA, Smith HO (2009) Enzymatic assembly of DNA molecules up to several hundred kilobases. Nat Methods 6:343–U41. doi:10.1038/Nmeth.1318
Gille S, Pauly M (2012) O-acetylation of plant cell wall polysaccharides. Front Plant Sci 3:12. doi:10.3389/fpls.2012.00012
Gille S, Hansel U, Ziemann M, Pauly M (2009) Identification of plant cell wall mutants by means of a forward chemical genetic approach using hydrolases. Proc Natl Acad Sci USA 106:14699–14704. doi:10.1073/pnas.0905434106
Gille S, Cheng K, Skinner ME, Liepman AH, Wilkerson CG, Pauly M (2011a) Deep sequencing of voodoo lily (Amorphophallus konjac): an approach to identify relevant genes involved in the synthesis of the hemicellulose glucomannan. Planta 234:515–526. doi:10.1007/s00425-011-1422-z
Gille S, de Souza A, Xiong G, Benz M, Cheng K, Schultink A, Reca IB, Pauly M (2011b) O-acetylation of Arabidopsis hemicellulose xyloglucan requires AXY4 or AXY4L, proteins with a TBL and DUF231 domain. Plant Cell 23:4041–4053. doi:10.1105/tpc.111.091728
Gleave AP (1992) A versatile binary vector system with a T-DNA organizational-structure conducive to efficient integration of cloned DNA into the plant genome. Plant Mol Biol 20:1203–1207. doi:10.1007/Bf00028910
Gou JY, Miller LM, Hou G, Yu XH, Chen XY, Liu CJ (2012) Acetylesterase-mediated deacetylation of pectin impairs cell elongation, pollen germination, and plant reproduction. Plant Cell 24:50–65. doi:10.1105/tpc.111.092411
Hajdukiewicz P, Svab Z, Maliga P (1994) The small, versatile pPZP family of Agrobacterium binary vectors for plant transformation. Plant Mol Biol 25:989–994
Harholt J, Jensen JK, Sorensen SO, Orfila C, Pauly M, Scheller HV (2006) ARABINAN DEFICIENT 1 is a putative arabinosyltransferase involved in biosynthesis of pectic arabinan in Arabidopsis. Plant Physiol 140:49–58. doi:10.1104/pp.105.072744
Harholt J, Suttangkakul A, Scheller HV (2010) Biosynthesis of pectin. Plant Physiol 153:384–395. doi:10.1104/pp.110.156588
Henrissat B, Coutinho PM, Davies GJ (2001) A census of carbohydrate-active enzymes in the genome of Arabidopsis thaliana. Plant Mol Biol 47:55–72
Hongo S, Sato K, Yokoyama R, Nishitani K (2012) Demethylesterification of the primary wall by PECTIN METHYLESTERASE35 provides mechanical support to the Arabidopsis stem. Plant Cell 24:2624–2634. doi:10.1105/tpc.112.099325
Ishii T (1997) O-acetylated oligosaccharides from pectins of potato tuber cell walls. Plant Physiol 113:1265–1272
Jones L, Milne JL, Ashford D, McQueen-Mason SJ (2003) Cell wall arabinan is essential for guard cell function. Proc Natl Acad Sci USA 100:11783–11788. doi:10.1073/pnas.1832434100
Keenan MHJ, Belton PS, Matthew JA, Howson SJ (1985) A C-13-Nmr study of sugar-beet pectin. Carbohyd Res 138:168–170. doi:10.1016/0008-6215(85)85236-8
Li B, Xia J, Wang Y, Xie B (2005) Grain-size effect on the structure and antiobesity activity of konjac flour. J Agric Food Chem 53:7404–7407. doi:10.1021/jf050751q
Manabe Y, Nafisi M, Verhertbruggen Y, Orfila C, Gille S, Rautengarten C, Cherk C, Marcus SE, Somerville S, Pauly M, Knox JP, Sakuragi Y, Scheller HV (2011) Loss-of-function mutation of REDUCED WALL ACETYLATION2 in Arabidopsis leads to reduced cell wall acetylation and increased resistance to Botrytis cinerea. Plant Physiol 155:1068–1078. doi:10.1104/pp.110.168989
Manabe Y, Verhertbruggen Y, Gille S, Harholt J, Chong SL, Pawar PM, Mellerowicz EJ, Tenkanen M, Cheng K, Pauly M, Scheller HV (2013) Reduced wall acetylation proteins play vital and distinct roles in cell wall O-acetylation in Arabidopsis. Plant Physiol 163:1107–1117. doi:10.1104/pp.113.225193
McCann MC, Roberts K (1996) Plant cell wall architecture: the role of pectins. In: Visser JV, Voragen AGJ (eds) Pectins and pectinases. Elsevier Science B.V, Amsterdam, pp 91–107
McNeil M, Darvill AG, Fry SC, Albersheim P (1984) Structure and function of the primary cell walls of plants. Annu Rev Biochem 53:625–663. doi:10.1146/annurev.bi.53.070184.003205
Micheli F (2001) Pectin methylesterases: cell wall enzymes with important roles in plant physiology. Trends Plant Sci 6:414–419
Mohnen D (2008) Pectin structure and biosynthesis. Curr Opin Plant Biol 11:266–277. doi:10.1016/j.pbi.2008.03.006
Mouille G, Ralet MC, Cavelier C, Eland C, Effroy D, Hematy K, McCartney L, Truong HN, Gaudon V, Thibault JF, Marchant A, Hofte H (2007) Homogalacturonan synthesis in Arabidopsis thaliana requires a Golgi-localized protein with a putative methyltransferase domain. Plant J 50:605–614. doi:10.1111/j.1365-313X.2007.03086.x
Nakamura A, Furuta H, Maeda H, Takao T, Nagamatsu Y (2002) Structural studies by stepwise enzymatic degradation of the main backbone of soybean soluble polysaccharides consisting of galacturonan and rhamnogalacturonan. Biosci Biotechnol Biochem 66:1301–1313
Orfila C, Sorensen SO, Harholt J, Geshi N, Crombie H, Truong HN, Reid JS, Knox JP, Scheller HV (2005) QUASIMODO1 is expressed in vascular tissue of Arabidopsis thaliana inflorescence stems, and affects homogalacturonan and xylan biosynthesis. Planta 222:613–622. doi:10.1007/s00425-005-0008-z
Orfila C, Dal Degan F, Jorgensen B, Scheller HV, Ray PM, Ulvskov P (2012) Expression of mung bean pectin acetyl esterase in potato tubers: effect on acetylation of cell wall polymers and tuber mechanical properties. Planta 236:185–196. doi:10.1007/s00425-012-1596-z
Pauly M, Scheller HV (2000) O-Acetylation of plant cell wall polysaccharides: identification and partial characterization of a rhamnogalacturonan O-acetyl-transferase from potato suspension-cultured cells. Planta 210:659–667
Pettolino FA, Walsh C, Fincher GB, Bacic A (2012) Determining the polysaccharide composition of plant cell walls. Nat Protoc 7:1590–1607. doi:10.1038/nprot.2012.081
Pogorelko G, Lionetti V, Fursova O, Sundaram RM, Qi M, Whitham SA, Bogdanove AJ, Bellincampi D, Zabotina OA (2013) Arabidopsis and Brachypodium distachyon transgenic plants expressing Aspergillus nidulans acetylesterases have decreased degree of polysaccharide acetylation and increased resistance to pathogens. Plant Physiol 162:9–23. doi:10.1104/pp.113.214460
Ralet MC, Crepeau MJ, Buchholt HC, Thibault JF (2003) Polyelectrolyte behaviour and calcium binding properties of sugar beet pectins differing in their degrees of methylation and acetylation. Biochem Eng J 16:191–201. doi:10.1016/S1369-703x(03)00037-8
Ralet MC, Cabrera JC, Bonnin E, Quemener B, Hellin P, Thibault JF (2005) Mapping sugar beet pectin acetylation pattern. Phytochemistry 66:1832–1843. doi:10.1016/j.phytochem.2005.06.003
Ralet MC, Crepeau MJ, Lefebvre J, Mouille G, Hofte H, Thibault JF (2008) Reduced number of homogalacturonan domains in pectins of an Arabidopsis mutant enhances the flexibility of the polymer. Biomacromolecules 9:1454–1460. doi:10.1021/Bm701321g
Ridley BL, O’Neill MA, Mohnen D (2001) Pectins: structure, biosynthesis, and oligogalacturonide-related signaling. Phytochemistry 57:929–967
Schultink A, Cheng K, Park YB, Cosgrove DJ, Pauly M (2013) The identification of two arabinosyltransferases from tomato reveals functional equivalency of xyloglucan side chain substituents. Plant Physiol 163:86–94. doi:10.1104/pp.113.221788
Sengkhamparn N, Bakx EJ, Verhoef R, Schols HA, Sajjaanantakul T, Voragen AG (2009) Okra pectin contains an unusual substitution of its rhamnosyl residues with acetyl and alpha-linked galactosyl groups. Carbohydr Res 344:1842–1851. doi:10.1016/j.carres.2008.11.022
Ulvskov P, Wium H, Bruce D, Jorgensen B, Qvist KB, Skjot M, Hepworth D, Borkhardt B, Sorensen SO (2005) Biophysical consequences of remodeling the neutral side chains of rhamnogalacturonan I in tubers of transgenic potatoes. Planta 220:609–620. doi:10.1007/s00425-004-1373-8
Vision TJ, Brown DG, Tanksley SD (2000) The origins of genomic duplications in Arabidopsis. Science 290:2114–2117
Voinnet O, Rivas S, Mestre P, Baulcombe D (2003) An enhanced transient expression system in plants based on suppression of gene silencing by the p19 protein of tomato bushy stunt virus. Plant J 33:949–956
Wei N, Quarterman J, Kim SR, Cate JH, Jin YS (2013) Enhanced biofuel production through coupled acetic acid and xylose consumption by engineered yeast. Nat Commun 4:2580. doi:10.1038/ncomms3580
Williamson G (1991) Purification and characterization of pectin acetylesterase from orange peel. Phytochemistry 30:445–449
Xiong G, Cheng K, Pauly M (2013) Xylan O-acetylation impacts xylem development and enzymatic recalcitrance as indicated by the Arabidopsis mutant tbl29. Mol Plant 6:1373–1375. doi:10.1093/mp/sst014
Yapo BM (2011) Pectic substances: from simple pectic polysaccharides to complex pectins-A new hypothetical model. Carbohyd Polym 86:373–385. doi:10.1016/j.carbpol.2011.05.065
Zablackis E, Huang J, Muller B, Darvill AG, Albersheim P (1995) Characterization of the cell-wall polysaccharides of Arabidopsis thaliana leaves. Plant Physiol 107:1129–1138
Acknowledgments
We thank Kirk Schnorr (Novozymes, Bagsvaerd, Denmark) for the generous gift of the pectin methylesterase. We also thank Döerte Kasten, Eddie Lam, Snigdha Poddar, Emilie Maddison and Nate Park (all at the University of California, Berkeley, CA, USA) for excellent technical support. This work was supported by the Energy Biosciences Institute.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution License which permits any use, distribution, and reproduction in any medium, provided the original author(s) and the source are credited.
About this article

Cite this article
de Souza, A., Hull, P.A., Gille, S. et al. Identification and functional characterization of the distinct plant pectin esterases PAE8 and PAE9 and their deletion mutants. Planta 240, 1123–1138 (2014). https://doi.org/10.1007/s00425-014-2139-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00425-014-2139-6