
Abstract
Cellular membranes, notably eukaryotic plasma membranes, are equipped with special proteins that actively translocate lipids from one leaflet to the other and thereby help generate membrane lipid asymmetry. Among these ATP-driven transporters, the P4 subfamily of P-type ATPases (P4-ATPases) comprises lipid flippases that catalyze the translocation of phospholipids from the exoplasmic to the cytosolic leaflet of cell membranes. While initially characterized as aminophospholipid translocases, recent studies of individual P4-ATPase family members from fungi, plants, and animals show that P4-ATPases differ in their substrate specificities and mediate transport of a broader range of lipid substrates, including lysophospholipids and synthetic alkylphospholipids. At the same time, the cellular processes known to be directly or indirectly affected by this class of transporters have expanded to include the regulation of membrane traffic, cytoskeletal dynamics, cell division, lipid metabolism, and lipid signaling. In this review, we will summarize the basic features of P4-ATPases and the physiological implications of their lipid transport activity in the cell.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Cellular membranes are organized as bilayers consisting of two leaflets, which are structurally formed by hundreds of different lipid species. In eukaryotic cells, the distribution of lipids is heterogeneous, and the membranes of different organelles have different lipid compositions. Furthermore, there are striking differences in the lipid distribution across the bilayer. Lipids in the late Golgi, endosome, and plasma membrane adopt an asymmetrical distribution with the aminophospholipids phosphatidylserine (PS) and phosphatidylethanolamine (PE) concentrated in the cytosolic leaflet and phosphatidylcholine (PC) and the sphingolipids (i.e., sphingomyelin and glycosphingolipids) enriched in the exoplasmic leaflet [38, 96]. This lipid asymmetry has been implicated in numerous cellular processes. For example, asymmetric distribution of specific lipids may induce membrane curvature, a prerequisite for vesicle formation in the secretory pathway, and controlled disruption of lipid asymmetry is critical in physiological processes such as blood coagulation, apoptosis, cytokinesis, cell fusion, and host–virus interactions.
Growing evidence indicates that the asymmetric transbilayer distribution of lipids is largely determined by a diverse group of lipid translocators that use the energy of ATP hydrolysis to move specific lipids across the bilayer. These translocators include ATP-dependent flippases and floppases, which catalyze the inward movement of phospholipids from the extracellular/luminal leaflet to the cytoplasmic leaflet and the outward movement of other lipids, respectively [20, 25]. The specificity of each of these transporters defines the resulting asymmetry that is generated. Current biochemical evidence indicates that these proteins are primarily members of the P-type and ATP-binding cassette (ABC) family of transporters. In addition to these energy-dependent translocators, certain eukaryotic cells contain phospholipid scramblases; putative membrane proteins that upon activation facilitate a rapid bidirectional movement of phospholipids across the two plasma membrane leaflets, disrupting the lipid asymmetry created by the ATP-dependent translocators. The molecular identity of the scramblase activity has not been unequivocally established yet, but several candidates have been identified [79, 87, 88, 103, 112]. This review focuses on lipid flippases belonging to the P4 subfamily of P-type ATPases (P4-ATPases). Several excellent reviews have recently treated different aspects of P4-ATPases [18, 77, 86, 94]. In this review, we will first provide an overview on the functional implications of their activities for the cell and then highlight the current focus of some of the research efforts in understanding the transport mechanism of P4-ATPases.
P-type ATPase superfamily
The P-type ATPase family comprises a large number of evolutionarily related membrane-bound pumps with the common feature that they form a phosphorylated intermediate during their catalytic cycle, hence the designation P type (Fig. 1; [57]). Based on sequence similarity, the P-type ATPase family is divided into five subfamilies (P1–P5) with different transport specificities [58]. Among the members, heavy metal-transporting P1-ATPases are in charge of detoxifying the cytoplasm and loading of heavy metals in specific cell compartments. Prominent members of P2-ATPases include the Na+/K+-ATPase that maintains electrochemical gradients for Na+ and K+ across the plasma membrane of animal cells, the human H+/K+-ATPase which is primarily responsible for the acidification of the stomach contents, and the sarcoplasmic reticulum Ca2+-ATPase which restores Ca2+ levels in the sarcoplasmic reticulum after muscle contraction. In both plants and fungi, plasma membrane-localized proton-pumping P3-ATPases extrude H+ from the cell to generate a proton-motive force and a pH gradient across the plasma membrane. While P1-, P2-, and P3-ATPases are found in all three branches of life, members of the P4-ATPases are only present in eukaryotic organisms and represent by far the largest P-type ATPase subfamily. In humans, there are 14 genes that encode P4-ATPases, while in yeast, there are five: Drs2, Dnf1, Dnf2, Dnf3, and Neo1. Mutations in one of the human members of this subfamily, ATP8B1, give rise to the rare autosomal recessive diseases progressive familial intrahepatic cholestasis (PFIC12 or Byler disease) and the related but less severe benign recurrent intrahepatic cholestasis (BRIC1) [10]. These diseases result in defects in bile salt secretion in the liver canaliculi, leading to episodes of jaundice and severe pruritus, together with non-hepatic symptoms that can include growth defects, diarrhea, and hearing loss [45]. ATP8B1 is expressed in several epithelial tissues and, notably, in the canalicular membrane of the liver [28] and the stereocilia membrane in hair cells [83]. Studies in mouse models show that P4-ATPases fulfil multiple important physiological functions; deficiencies result in a wide variety of neurological phenotypes, liver disease, immunological problems, type 2 diabetes, and diet-induced obesity (for recent reviews, see [18, 94]). How all these distinct phenotypes relate to a defective flippase activity remains to be elucidated. Accumulating evidence reveals that P4-ATPases operate as heterodimers in combination with protein subunits from the ligand-effect modulator (LEM)3 / cell division cycle (CDC50) family to flip phospholipids from the exofacial to cytosolic side of cell membranes. This active, unidirectional flip of specific phospholipid species against their concentration gradient has been implicated in generating lipid asymmetry, in scavenging exogenous lipids, and in inducing membrane curvature.

Membrane topology of P2- and P4-ATPases and their subunits. P-type ATPases consist of an actuator (A), a phosphorylation (P), a nucleotide-binding domain (N), and 10 transmembrane spanning helices. The P domain contains the canonical aspartic acid phosphorylated during the reaction cycle. The beta subunits associated with P2-ATPases are type II membrane proteins with one transmembrane segment, a short cytoplasmic tail, and a large, heavily glycosylated ectodomain with three disulfide bridges. In some cases, a gamma subunit belonging to the FXYD protein family is associated to the P2-ATPases. The CDC50 subunits of P4-ATPases consist of two membrane-spanning domains with a large extracellular loop containing four possible N-linked glycosylation sites and two disulfide bridges. Both the membrane and extracellular domains of CDC50 are required for assembly with the P4-ATPase [17, 68]
Physiological implications of P4-ATPase-catalyzed phospholipid transport
P4-ATPases and phospholipid asymmetry
Studies in yeast, parasites, and higher eukaryotes uncovered that P4-ATPases localize to the plasma membrane and are found in various intracellular compartments of the late secretory and endocytic pathways (Table 1). This implies that P4-ATPases act at multiple cellular sites to establish and maintain phospholipid asymmetry (Fig. 2(a)). Indeed, studies on the five members of this subfamily in the yeast Saccharomyces cerevisiae support this notion. For example, loss of the yeast plasma membrane-associated P4-ATPases Dnf1p and Dnf2p causes an aberrant exposure of endogenous aminophospholipids at the cell surface [15, 65]. Removal of the Golgi-associated P4-ATPases Drs2p and Dnf3p disrupts aminophospholipid transport and asymmetry in post-Golgi secretory vesicles [1, 56]. Thus, membrane aminophospholipid asymmetry appears to be established in late Golgi compartments and maintained at the plasma membrane by the help of P4-ATPases. How the PC translocase activity of several P4-ATPases relates to membrane asymmetry is still unknown, but certain cell types might restrict PC to the cytoplasmic leaflet as well. An asymmetric transbilayer lipid distribution provides the two membrane leaflets of organelles with different characteristics necessary for their respective physiological functions. Furthermore, membrane lipid asymmetry might be involved in the proper sorting of membrane proteins destined for the plasma membrane [34]. It is, therefore, not surprising that a system for sensing lipid asymmetry has been reported in yeast [41].

Cellular functions involving P4-ATPases. P4-ATPases appear to exert their cellular functions by combining an enzymatic phospholipid translocation activity with an enzyme-independent action. These functions are not mutually exclusive. Active transport of lipids from the exoplasmic to the cytosolic membrane leaflet can maintain lipid asymmetry (a), scavenge lipids (b), and drive membrane budding by generating a lipid imbalance across the bilayer and/or a membrane environment permissive for vesicle budding (c). Enzyme-independent functions of P4-ATPases include recruitment of proteins involved in coat assembly (d), cellular signaling, and cytoskeleton regulation (e). See text for details
P4-ATPases as lipid scavengers
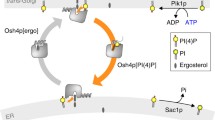
Some P4-ATPases are characterized as plasma membrane flippases with relatively broad phospholipid specificity. It is, therefore, tempting to speculate functions for these P4-ATPases beyond their role in maintaining membrane lipid asymmetry. In the yeast S. cerevisiae, for example, extracellular lysophospholipids can be taken up by cells and used as substrates for regeneration of phospholipids [73]. Lysophosphatidylethanolamine (lyso-PE) and lysophosphatidylcholine (lyso-PC) are transported into the cell and subsequently acylated to PE and PC, respectively, by the ALE1-encoded lysophospholipid acyltransferase, which also utilizes lysophosphatidic acid as a substrate [43, 90]. Loss of both plasma membrane P4-ATPases Dnf1p and Dnf2p or of their subunit Lem3p blocks the uptake of radiolabeled lyso-PC or lyso-PE and inhibits lyso-PC- or lyso-PE-dependent growth, respectively, supporting a nutrient scavenger role for these pumps [72, 73]. Notably, parasitic protozoa and helminths have developed unique metabolic pathways that allow them to survive and multiply by scavenging nutrients from the host but are unable to synthesize the majority of their own lipids de novo (reviewed in [6]). There is evidence suggesting that several parasites can take up host phospholipids in vivo and from the growth medium in vitro. Whether P4-ATPases are used by parasites for lipid acquisition remains to be demonstrated. Studies on mammalian cells uncovered a role of the human P4-ATPase beta subunit CDC50A (TMEM30a), in the uptake of the inflammatory lipid platelet-activating factor (PAF, a short-chain PC) and other exogenous short-chain choline phospholipids, implying that P4-ATPases in complex with their beta subunit can participate in inward translocation of lipid signaling molecules from outside of the cell (Fig. 2(b)) [14].
P4-ATPases as motors in vesicle formation
Genetic studies in yeast, worms, parasites, plants, and mammals have uncovered an unexpected role of P4-ATPases in vesicular trafficking. In yeast, removal of the plasma membrane-associated P4-ATPases Dnf1p and Dnf2p causes a cold-sensitive defect in the formation of endocytic vesicles [65]. Inactivation of the Golgi-resident P4-ATPase Drs2p rapidly blocks the formation of a clathrin-dependent class of post-Golgi secretory vesicles carrying exocytic cargo [13, 32, 39], while temperature-sensitive variants of the endosome-associated P4-ATPase Neo1p cause defects in receptor-mediated endocytosis, vacuolar biogenesis, and vacuolar protein sorting [40, 102]. Likewise, deletion of the P4-ATPase MgATP2 in the rice plant pathogenic fungus Magnaporthe grisea decreases the secretion of extracellular enzymes and results in abnormal Golgi-like cisternae [33].
Trafficking defects associated with aberrant P4-ATPase function are also observed in higher eukaryotes. The Caenorhabditis elegans P4-ATPase transbilayer amphipath transporter 1 (TAT-1) is required for yolk uptake in oocytes and for an early step of fluid-phase endocytosis in the intestine [22, 74]. TAT-1 forms a complex with the Cdc50 family protein CHAT-1, and both proteins are important in maintaining normal endocytic sorting/recycling by promoting membrane tubulation of the early endosome [12]. Further, TAT-5 has been linked to the regulation of ectosome shedding [100]. Loss of ALA3, a Golgi-resident P4-ATPase in Arabidopsis thaliana, causes a defect in the production of slime vesicles containing polysaccharides and enzymes for secretion [66]. In mouse mastocytoma cells, knockdown of ATP8B5 profoundly perturbs the structural organization of the Golgi complex and causes loss of constitutive secretion at lower temperature [107]. Recently, ATP8A1 in complex with CDC50A was linked to the generation of membrane ruffles in cell motility [44], and studies in neuronal PC12 cells and rat hippocampal neurons indicate a role for ATP8A2 in promoting neurite outgrowth [108].
All these studies indicate that P4-ATPases are required to support vesicle formation in multiple parts of the intracellular trafficking pathways. At least two models, not mutually exclusive, have been proposed to explain how P4-ATPases contribute to vesicle formation (Fig. 2(c)). One model is that P4-ATPases directly catalyze an inward-directed phospholipid translocation across the lipid bilayer, which creates an imbalance in phospholipid numbers between the two leaflets. In turn, this causes an inward bending of the membrane leading to budding and vesicle formation, which is stabilized by recruitment of coat proteins (e.g., clathrin or COPII proteins). Consistent with this hypothesis, insertion of exogenous phospholipids in the exoplasmic leaflet of the plasma membrane and their subsequent translocation to the cytosolic leaflet by a lipid flippase causes dramatic shape changes of red blood cells [21, 78]. Similarly, lipid flipping can provoke the formation of endocytic-like vesicles [53] and accelerates endocytosis [30]. Direct participation of ATP-driven lipid transport in vesicle budding is further supported by the observation that giant proteoliposomes formed from erythrocyte membrane fragments undergo budding in the presence of ATP [29].
A second model is that ATP-driven lipid translocation by P4-ATPases is needed to create a membrane environment permissive for vesicle budding. A high local concentration of aminophospholipids in the cytosolic leaflet may favor the recruitment or activity of peripherally associated proteins with a critical function in vesicle budding, such as small ADP ribosylation factor (Arf) GTP-binding proteins, clathrin, coat protein complex II, amphiphysin, and endophilins. Consistent with such a functional role is the observation that the yeast plasma membrane P4-ATPases Dnf1p and Dnf2p in complex with their Lem3p subunit regulate the fast membrane dissociation of cell division cycle protein 42, a small GTPase with a central role in establishing cell polarity in eukaryotic cells [23, 76]. As discussed in more detail below, various P4-ATPase family members in yeast differ in their substrate specificity. The biological implications of this are unknown, but it could be important for the recruitment or function of cytosolic proteins.
P4-ATPases as membrane scaffolds
In addition to their function as lipid flippases, P4-ATPases appear to serve also transport-independent functions (Fig. 2(d, e)). The yeast P4-ATPases Drs2p and Neo1p were found to interact with cytosolic proteins such as guanine nucleotide exchange factors and small GTPases that are crucial for the recruitment of coat proteins during membrane budding [7, 11, 15, 31, 92, 102]. Furthermore, a systematic search for Drs2p-binding partners yielded proteins involved in phosphoinositide metabolism [70]. Phosphoinositides are important signaling molecules in membrane traffic that help establish organelle identity through recruitment of effector proteins. Further, blocking ATP8B1 expression in polarized Caco-2 cells was shown to result in a profound disorganization of the apical actin cytoskeleton and a substantial loss in microvilli without affecting aminophospholipid transport and asymmetry across the apical bilayer [97]. These results suggest that ATP8B1 may provide a molecular scaffold in the apical membrane to recruit structural components or modulators of the actin cytoskeleton involved in microvilli formation. Consistent with a putative scaffold function, ATP8B1 expression on the apical membrane of Caco-2 cells increases dramatically during differentiation, concomitantly with cell polarization.
Notably, also the catalytic alpha subunits of the Na+/K+-ATPase and the closely related H+/K+-ATPase interact with ankyrin, a cytoskeletal protein implicated in epithelial junction formation [106, 111]. In a similar manner in yeast, functionality of both Golgi-localized Drs2p and plasma membrane Dnf1p is dependent on their interaction with Sla1p [48], a protein that is part of an endocytic coat/adaptor complex with clathrin. Sla1p binds to proteins involved both in endocytosis and in regulation of actin dynamics, allowing for activation of proteins involved in actin polymerization [99].
Linking the cytoskeleton to specialized areas of the plasma membrane may thus be a feature shared among several members of the P-type ATPase superfamily. Thus, intracellular P4-ATPases that seem to act without a beta subunit, e.g., Neo1p in yeast or ATP9B in mammals [89], could serve a lipid transport-independent function as scaffold switches to recruit structural components or effectors.
P4-ATPases display different substrate specificities
Initial biochemical evidence indicated that P4-ATPases flip aminophospholipids only. For example, the first P4-ATPase to be characterized was identified as the result of purification of an aminophospholipid translocase activity [91]. This protein, also known as ATPase II or, currently, ATP8A1, was isolated from bovine chromaffin granules and displays a striking similarity to the yeast protein Drs2p. The observation that removal of Drs2p caused a specific defect in the inward translocation of fluorescently labelled PS across the yeast plasma membrane provided further support for a role of P4-ATPases in translocation of di-acyl aminophospholipids. Although the function of Drs2p as an aminophospholipid translocase was subsequently questioned [52, 80], lipid transport assays with purified Golgi membranes containing a temperature-sensitive drs2ts allele indicated that Drs2p is directly coupled to flipping of PS acyl labelled with the fluorescent probe nitrobenzoxadiazole (NBD) [56]. Indeed, recent reconstitution of NBD-PS translocase activity with purified Drs2p demonstrates that this enzyme catalyzes PS translocation [113].
Further studies on individual P4-ATPase family members from fungi, plants, and animals have revealed that P4-ATPases differ in their substrate specificities and transport lipid substrates other than di-acyl aminophospholipids (Table 1). In the yeast S. cerevisiae, for example, removal of the P4-ATPases Dnf1p and Dnf2p abolishes inward translocation of fluorescently labelled PS, PE, and even PC across the plasma membrane [65], but see also [84]. Moreover, Dnf1p and Dnf2p are also capable of translocating the natural lipids lyso-PE and lyso-PC across the plasma membrane [72, 73]. In addition, the double Δdnf1Δdnf2 mutant cells are defective for the uptake of alkylphosphocholine derivatives and are, therefore, resistant to the toxic effects of these drugs [65]. In fact, recent data suggest that Dnf1p prefers lysolipids rather than di-acyl phospholipids as its substrate [5]. Similarly, Leishmania parasites deficient for Dnf1p orthologs are defective in inward translocation of PE, PC, and alkylphosphocholine derivatives at the plasma membrane [63, 101].
Studies in the plant A. thaliana further substantiate the notion that members of the P4-ATPase family do not serve exclusively as aminophospholipid-specific translocators. The Arabidopsis genome encodes 12 P4-ATPases, designated ALA1 to ALA12 (for aminophospholipid ATPase) [2]. So far, only three ALA proteins have been partially characterized: ALA1, ALA2, and ALA3. ALA1 localizes to the plant plasma membrane, while ALA2 and ALA3 are located to the pre-vacuolar compartment and the Golgi apparatus, respectively. Complementation studies in yeast mutants lacking the P4-ATPases Dnf1p, Dnf2p, and Drs2p revealed that ALA2 specifically transports PS, while ALA3 has broader specificity and facilitates transport of PS, PE, and PC, but not of the lyso-PC derivative miltefosine [50, 66].
In mammals, at least 14 P4-ATPases [36], designated ATP8A1 through ATP11C, have been identified, but their substrate specificities are still poorly defined. Among the P4-ATPases expressed in mammalian cells, ATP8A1, ATP8A2, ATP8B1, ATP8B3, and ATP11C have, so far, been connected to PS translocation. ATP8A1 is dependent on PS and PE for ATPase activity [27, 59] and is able to translocate fluorescent PS upon expression in yeast [82]. Likewise, ATP8A2, a P4-ATPase highly expressed in the brain, testes, and retina, exhibits PS-dependent ATPase activity and the ability to translocate fluorescent PS, and to some extend PE, in proteoliposomes [16, 17]. Deficiency of ATP8B1, a P4-ATPase located in the canalicular membrane of liver cells, is accompanied by enhanced recovery of PS, but not PC or PE in bile [61], and heterologous expression of ATP8B1 restores the non-endocytic uptake of NBD-PS in PS transport-defective CHO mutant cells [60, 93]; see also [97]. In the spermatozoa of mice, ATP8B3 is necessary for PS asymmetry and fertilization [98]. ATP11C, a P4-ATPase essential for B cell development, was found to play a crucial role in PS translocation [109]. Notably, recent data show that also mammalian P4-ATPases do not only include aminophospholipid-specific translocators; ATP8B5 from mouse testes was found to transport PC and PE upon heterologous expression in yeast [107]. Recently, a role of ATP8B1 as a cardiolipin flippase [71] has been suggested but requires further investigations [62].
Based on sequence similarity, P4-ATPases have been further subdivided into six classes (designated classes 1–6) carrying conserved class-specific amino acid sequences [35, 37]. However, it seems unlikely that these sequence similarities are linked to functional similarities as the subdivision into classes does not correlate directly with the different substrate specificities so far reported for various P4-ATPase members (Table 1). For example, class 1 P4-ATPases comprise flippases that, in some organisms, are specific for PS (e.g., C. elegans TAT-1) and, in others, also transport PE (e.g. S. cerevisiae Drs2p) and, in others, PC as well (but barely PS, as Leishmania donovani LdMT). All eukaryotic organisms analyzed contain genes encoding class 1 and class 2 P4-ATPases (Table 1). C. elegans and Arabidopsis express class 5 P4-ATPases as well, while mammals and Drosophila express both class 5 (ATP10) and class 6 (ATP11) P4-ATPases. In contrast, class 3 and class 4 P4-ATPases are solely expressed in yeast (Table 1).
The requirement for a beta subunit for P4-ATPases
The catalytic subunit of P4-ATPases associates with a beta subunit from the Cdc50 family, which is required for functional maturation of the enzyme. Structurally, P4-ATPase beta subunits contain two transmembrane domains and a large exoplasmic loop that is heavily glycosylated (Fig. 1). This resembles a fusion of the two subunits for the Na+/K+-ATPase, the beta subunit with one transmembrane domain and a large glycosylated ectodomain and the gamma subunit with one transmembrane domain and very short cytosolic and luminal extensions. P4-ATPase beta subunits have, therefore, been suggested to be analogous to the Na+/K+-ATPase beta and gamma subunits [67, 69]. The role of the P4-ATPase beta subunit also seems to be similar to that of the Na+/K+-ATPase accessory subunits. They act as chaperones required for the P4-ATPase to leave the endoplasmic reticulum (ER), and they seem to affect the catalytic properties of the complex, being required for lipid translocation.
The function as chaperone was demonstrated by the fact that the association of Cdc50 proteins with P4-ATPases is a prerequisite for stability and ER export of the transporter complex [8, 31, 50, 51, 60, 64, 66, 75]. Notably, yeast Neo1p as well as the related mammalian P4-ATPases ATPA9 and ATP9B are able to exit the ER independent of CDC50 proteins. In co-immunoprecipitation studies, none of these proteins formed a stable complex with CDC50 proteins, and experimental evidence that they catalyze phospholipid transport is lacking. This suggests that Neo1p, ATP9A, and ATP9B possess a biochemical activity that is different from the other P4-ATPases, and for which, they might not require a Cdc50-binding partner.
Based on genetic studies in yeast, it was suggested that the beta subunit is required not only for ER exit but also for proper localization of the P4-ATPase to the membrane. Thus, yeast Drs2p and its beta subunit, Cdc50p, are localized to the trans-Golgi network, while Dnf1p and Dnf2p, sharing a common beta subunit, Lem3p, localize both to the plasma membrane. However, this does not seem to be the case in multicellular organisms. Plant ALA1, ALA2, and ALA3 can use three different beta subunits for ER exit, and each P4-ATPase will reach a different final subcellular localization in the presence of each of these subunits [50, 51]. Likewise, several human P4-ATPases can interact with all three identified human Cdc50p homologues to exit the ER [8]. It is a possibility that CDC50 proteins from multicellular organisms have evolved to interact with P4-ATPases in a different way than their yeast counterparts. In a yeast cell, all P4-ATPases and beta subunits are expressed at the same time within the same cell, and it might be necessary for proper function that each beta subunit will only recognize one interacting partner. In multicellular organisms, however, expression of each protein can be controlled both temporally and spatially to avoid undesired interactions. Thus, it might be more effective for the cell to maintain a number of interchangeable beta subunits that will be ready at each given moment to assist ER exit of a number of P4-ATPases expressed under different conditions and fulfilling distinct physiological roles.
The P4-ATPase beta subunit appears to also serve a role in lipid translocation. It has been suggested that transmembrane flipping might occur at the interface between a P4-ATPase and its Cdc50-binding partner, an arrangement in which Cdc50 proteins would contribute directly to the transport specificity of the complex [16, 69, 113]. This idea is consistent with the observation that the yeast trans-Golgi P4-ATPases Drs2p and Dnf3p, which exhibit different translocation profiles [1], interact with different Cdc50 homologues. Drs2p interacts with Cdc50p [75] and Dnf3p with Crf1p [31], whereas the plasma membrane P4-ATPases Dnf1p and Dnf2p, which have the same substrate specificity [65], both interact with Lem3p [31, 75]. However, co-expression studies of different plant Cdc50 proteins and P4-ATPases show no influence of the beta subunit on lipid specificity, implying that the determinants for substrate specificity primarily [50]. Recent studies uncovered a series of residues involved in defining the substrate specificity of P4-ATPases within the catalytical subunit [4], consistent with this notion. Studies on yeast Drs2p suggest that Cdc50 proteins directly participate in the P4-ATPase reaction cycle [46] and perhaps help create a high-affinity phospholipid-binding site in the membrane domain of P4-ATPases [86]. Similar findings have been reported for the closely related Na+/K+-ATPase where the subunit affects the K+ affinity of the pump. Whether all P4-ATPases require a CDC50-binding partner to accomplish lipid transport is unknown.
Regulation of P4-ATPase activity
In light of their physiological relevance, P4-ATPases are expected to be highly regulated. However, not much is known about this aspect. Studies in yeast have provided initial evidence that P4-ATPases are targets of kinase-dependent phosphorylation, a common mode of regulation of P-type ATPases. Yeast Dnf1p and Dnf2p can be phosphorylated in vitro by flippase kinase 1 (Fpk1p), and in vivo experiments have shown that Fpk1p and its homologue Fpk2p are required for normal levels of inward-directed phospholipid transport across the plasma membrane of yeast [54]. The specific amino acid position phosphorylated by Fpk1p and the direct impact of this phosphorylation on Dnf1/2p flippase activity remain to be established. Interestingly, Drs2p and Dnf3p also get phosphorylated by Fpk1p in vitro, but to a lesser extent. Neo1p, which has not yet been shown to transport phospholipids, is not phosphorylated by Fpk1p. The physiological significance of these results remains to be determined, and currently, it is unknown whether other kinases interact with these P4-ATPases.
The activity of Drs2p in the trans-Golgi was recently found to be regulated through interaction with cytosolic proteins and specific lipids that play a critical role in membrane budding (Table 2). For example, the guanine nucleotide exchange factor Gea2p and phosphatidylinositol 4-phosphate bind to regions in the C-terminal cytosolic tail of Drs2p, and this binding is required for Drs2p activity [11, 42, 55]. Furthermore, binding of the small GTPase Arl1p (ADP ribosylation factor-like protein 1) to the N-terminal tail of Drs2p stimulates its flippase activity [92]. Collectively, these findings imply that the C-terminal tail of Drs2p contains an autoinhibitory domain and that Drs2p flippase activity is tightly coupled to the vesicle budding machinery at the trans side of the Golgi. The challenge is now to dissect the order and regulation of the dynamic and multiple interactions between Drs2p and its partners that eventually result in membrane budding. Additionally, further studies will be necessary to unravel how the activity of other flippases such as Dnf1/2p is regulated through interaction with lipids and cytosolic proteins.
The mechanism of P4-ATPase-catalyzed phospholipid transport
Even though available data support a direct role of P4-ATPases in lipid translocation, it is unclear how these enzymes acquired the ability to translocate lipids instead of much smaller ions. Early studies on aminophospholipid translocases pointed at the glycerol backbone and the lipid head group as the key recognition elements for the flippase. More recent data indicate that several P4-ATPases are capable of transporting a broad range of lipid substrates, including synthetic alkylphospholipids lacking a glycerol backbone, and define the phosphoryl head group as the key element for substrate selection by these transporters (Table 1). Thus, P4-ATPases must at least provide a sizeable hydrophilic pathway for the polar head group to pass through the membrane.
Recent mutational studies suggest two pathways by which P4-ATPases could transport their substrate: (1) the classical pathway with the lipid transported through the interior of P4-ATPases in analogy with the cation transport mechanism of well-characterized P2-ATPases and (2) a nonclassical pathway at the protein–membrane interface (Fig. 3). In the thoroughly studied Ca2+- and Na+/K+-transporting P2-ATPases, for which a wealth of crystal structures are available [9], the cation-binding sites are present in small central cavities primarily formed by charged or polar residues in the center parts of transmembrane segments TM4, TM5, and TM6 (Fig. 3). Contributing to cavity formation, all “classical” cation-translocating P-type ATPases possess one or more highly conserved proline residues in TM4 that function as helix breakers that allow backbone carbonyl oxygen atoms to be exposed and participate in cation coordination [57]. Notably, all P4-ATPases also contain a conserved proline in the middle of TM4 (Pro-507 in Drs2p) suggesting the presence of a central cavity in these pumps with carbonyl oxygen contributing to coordination of a hydrophilic group. Besides this feature, P4-ATPases have only a few conserved residues in their transmembrane segments that share properties with those in the membrane domain of P2-ATPases. Most notably is the presence of two conserved asparagine residues in TM5 and TM6 (Asn-1019 and Asn-1050 in Drs2p), which in P2-ATPases provide oxygen groups for ion coordination. In P2-ATPases, conserved acidic group(s) positioned in the middle of TM4 and TM6 secure charge neutralization of the transported cation. In contrast, P4-ATPases have not preserved acidic residues in their predicted transmembrane segments. However, a basic lysine residue (corresponding to Lys-1018 in Drs2p and Lys-873 in the mammalian photoreceptor P4-ATPase ATP8A2) is conserved in all P4-ATPases and situated in the middle of TM5 at the same position as cation-coordinating residues in P2-ATPases. As mutating this residue has a dramatic impact on the activity of ATP8A2, it has been proposed that P4-ATPases have evolved a canonical substrate-binding site centrally located in the membrane domain of the pump, which, with the lipid head group, is deeply embedded within the protein [19]. It has been hypothesized that the role of lysine is to neutralize an acidic group in the transported phospholipid molecule, which could be its phosphate group [19]. Given the large size of a phospholipid as compared to ions, this model implies a considerable flexibility of P4-ATPases to accommodate their substrate and, especially, the lipid tail when it traverses the membrane. Interestingly, long-chain thapsigargin analogs do adjust their fatty acid chain deeply between the transmembrane sections of the Ca2+ pump sarcoendoplasmic reticulum calcium ATPase (SERCA), consistent with high structural flexibility [24, 81, 105]. Furthermore, the E1–sarcolipin structure of the P2-SERCA displays a deep, funnel-shaped, and negatively charged path that leads to the Ca2+ sites located halfway through the membrane bilayer, and phospholipid head groups have been proposed to assist Ca2+ entry to these sites [104]. A similar funnel might have evolved into a phospholipid head group-loading pathway in P4-ATPases.

Schematic overview of two proposed phospholipid transport pathways in P4-ATPases. In the classical model, the lipid is transported through a space in the transporter analogous to the cation transport mechanism of well-characterized P2-ATPases. Here, an occluded state is expected with the transported lipid deeply buried in a central cavity (red) within the P4-ATPase with the entrance and exit pathways closed. By contrast, in the external surface model, the lipid is transported at a cleft on the membrane-facing surface, and only the lipid head group is protected from the lipid environment. The presence of two substrate-selecting gates (green) acting sequentially on opposite sides of the membrane has been reported [4]. In both cases, the relative positioning of the transmembrane segments critical for phospholipid binding/transport is highlighted on a homology model of Dnf1p based on the crystallized Na+/K+-ATPase in the E2P conformation [3]. The rest of the structure is shown in surface representation
In the nonclassical transport pathway proposed for P4-ATPases (Fig. 3), interaction between the pump and the phospholipid involves only the hydrophilic head group and amino acid residues all along the membrane-facing surface of the protein [3]. In this “sliding card” model, only the head group of the phospholipid makes a direct contact with the P4-ATPase protein during transport, while the acyl chains remain within the surrounding lipid environment of the membrane at all stages [4]. In line with this model mutational studies of yeast Dnf1p identified amino acid residues involved in substrate selection at the lumenal side of TM1 and the cytosolic sides of TM1, TM2, TM3, and TM4 [3]. Mutation of the Drs2p residue corresponding to Lys-873 in ATP8A2 reduced Drs2p activity but did not change the specificity of the pump. Dnf1p residues shown to be involved in phospholipid recognition at the cytosolic side of the membrane can be mapped to a region of SERCA that, in the E2 conformation, forms a binding pocket for the head group of a PE molecule [4]. This pocket is not accessible in the E1 conformation, implying that in the SERCA Ca2+-ATPase, the PE molecule has to dynamically enter and exit as it proceeds through the catalytic cycle. This original phospholipid head group-binding pocket might have evolved in P4-ATPases to generate a cleft for lipid translocation.
While the two models appear to oppose each other, it is useful to consider a mechanistic model that combines the two. According to such a model, P4-ATPases function mechanistically according to the same principles as P2-ATPases, but with some modifications. In both classes of pumps, the transported ligand enters from one side of the membrane through a half-channel leading to a ligand-binding cavity in the middle of the plane of the membrane. After occlusion of the ligand, which involves closure of the entrance pathway, a major conformational change results in the opening of an exit half channel through which the ligand is able to leave the pump protein. What distinguishes P4-ATPases from P2-ATPases in this context is that the transported ligand is so large that the pump protein cannot accommodate all of its mass within its structure. Therefore, part of the ligand, in this case the lipid tail, has to protrude out from the flippase during passage through the membrane and, during this process, never makes contact with the pump protein. To enable this, both the entrance and exit half channels are open towards the lipid bilayer and take form as clefts. In any case, conclusive evidence for the nature of the phospholipid head group-binding site(s) and the transport pathway of P4-ATPases potentially requires successful crystallization and structural elucidation of a P4-ATPase, which has not been achieved to date.
Concluding remarks
As is clear from this overview, phospholipid translocation and asymmetry in eukaryotic cells require P4-ATPases, and reconstitution experiments with purified enzymes demonstrate that these enzymes participate directly in lipid translocation. Yet, many fundamental questions still remain to be investigated. First, the mechanism of phospholipid transport by P4-ATPases is still an enigma. A major unresolved question is whether lipids are transported through the interior of P4-ATPases in analogy with the cation transport mechanism of well-characterized P-type ATPases such as the Na+/K+- and Ca2+-ATPases or other mechanisms should be considered. Further biochemical studies are required to unravel the enzyme kinetic parameters and the role of the CDC50 beta subunit in the catalytic cycle of these pumps. Notably, an improved, higher resolution assay for studying phospholipid transport has recently been developed [85] that can help to address these issues in more detail. Secondly, the substrate specificity of the individual P4-ATPases is still largely unknown. Its further elucidation will require transport measurements with natural lipids by P4-ATPases reconstituted into proteoliposomes, a challenging task given the difficulties of purifying membrane proteins, the water insolubility of the substrates, and the requirement for subunits and/or accessory proteins. The recent purification and functional reconstitution of two P4-ATPases [16, 113] offer hope for similar studies on other P4-ATPases. Third, we still lack a detailed understanding on how P4-ATPases participate in vesicle budding. Recent progress in membrane protein reconstitution into giant vesicles [26, 110] can help to test whether P4-ATPase-catalyzed phospholipid transport has a primary role in driving vesicle budding in the late secretory and endocytic pathway. Finally, while a number of interacting partners have been found for the yeast P4-ATPases, little or nothing is known about the interaction partners for P4-ATPases in higher eukaryotes. Several phenotypes have been associated to defects in P4-ATPase expression and activity in mammalians and plants, but the mechanisms underlying these phenotypes are largely unknown. At the same time, many P4-ATPases from higher eukaryotes do not have an assigned physiological role, most probably due to functional redundancy. Further characterization of mutant lines and screening for protein–protein interactions are required to shed some light onto these points and get a better understanding of P4-ATPase function and regulation.
References
Alder-Baerens N, Lisman Q, Luong L, Pomorski T, Holthuis JC (2006) Loss of P4 ATPases Drs2p and Dnf3p disrupts aminophospholipid transport and asymmetry in yeast post-Golgi secretory vesicles. Mol Biol Cell 17(4):1632–1642. doi:10.1091/mbc.E05-10-0912
Axelsen KB, Palmgren MG (1998) Evolution of substrate specificities in the P-type ATPase superfamily. J Mol Evol 46(1):84–101
Baldridge RD, Graham TR (2012) Identification of residues defining phospholipid flippase substrate specificity of type IV P-type ATPases. Proc Natl Acad Sci U S A 109(6):E290–E298. doi:10.1073/pnas.1115725109
Baldridge RD, Graham TR (2013) Two-gate mechanism for phospholipid selection and transport by type IV P-type ATPases. Proc Natl Acad Sci U S A 110(5):E358–E367. doi:10.1073/pnas.1216948110
Baldridge RD, Xu P, Graham TR (2013) Type IV P-type ATPases distinguish mono- versus diacyl phosphatidylserine using a cytofacial exit gate in the membrane domain. J Biol Chem 288(27):19516–19527. doi:10.1074/jbc.M113.476911
Bansal D, Bhatti HS, Sehgal R (2005) Role of cholesterol in parasitic infections. Lipids Health Dis 4:10. doi:10.1186/1476-511X-4-10
Barbosa S, Pratte D, Schwarz H, Pipkorn R, Singer-Kruger B (2010) Oligomeric Dop1p is part of the endosomal Neo1p-Ysl2p-Arl1p membrane remodeling complex. Traffic 11(8):1092–1106. doi:10.1111/j.1600-0854.2010.01079.x
Bryde S, Hennrich H, Verhulst PM, Devaux PF, Lenoir G, Holthuis JC (2010) CDC50 proteins are critical components of the human class-1 P4-ATPase transport machinery. J Biol Chem 285(52):40562–40572. doi:10.1074/jbc.M110.139543
Bublitz M, Morth JP, Nissen P (2011) P-type ATPases at a glance. J Cell Sci 124(Pt 15):2515–2519. doi:10.1242/jcs.088716
Bull LN, van Eijk MJ, Pawlikowska L, DeYoung JA, Juijn JA, Liao M, Klomp LW, Lomri N, Berger R, Scharschmidt BF, Knisely AS, Houwen RH, Freimer NB (1998) A gene encoding a P-type ATPase mutated in two forms of hereditary cholestasis. Nat Genet 18(3):219–224. doi:10.1038/ng0398-219
Chantalat S, Park SK, Hua Z, Liu K, Gobin R, Peyroche A, Rambourg A, Graham TR, Jackson CL (2004) The Arf activator Gea2p and the P-type ATPase Drs2p interact at the Golgi in Saccharomyces cerevisiae. J Cell Sci 117(Pt 5):711–722. doi:10.1242/jcs.00896
Chen B, Jiang Y, Zeng S, Yan J, Li X, Zhang Y, Zou W, Wang X (2010) Endocytic sorting and recycling require membrane phosphatidylserine asymmetry maintained by TAT-1/CHAT-1. PLoS Genet 6(12). doi: 10.1371/journal.pgen.1001235
Chen CY, Ingram MF, Rosal PH, Graham TR (1999) Role for Drs2p, a P-type ATPase and potential aminophospholipid translocase, in yeast late Golgi function. J Cell Biol 147(6):1223–1236
Chen R, Brady E, McIntyre TM (2011) Human TMEM30a promotes uptake of antitumor and bioactive choline phospholipids into mammalian cells. J Immunol 186(5):3215–3225. doi:10.4049/jimmunol.1002710
Chen S, Wang J, Muthusamy BP, Liu K, Zare S, Andersen RJ, Graham TR (2006) Roles for the Drs2p-Cdc50p complex in protein transport and phosphatidylserine asymmetry of the yeast plasma membrane. Traffic 7(11):1503–1517. doi:10.1111/j.1600-0854.2006.00485.x
Coleman JA, Kwok MC, Molday RS (2009) Localization, purification, and functional reconstitution of the P4-ATPase Atp8a2, a phosphatidylserine flippase in photoreceptor disc membranes. J Biol Chem 284(47):32670–32679. doi:10.1074/jbc.M109.047415
Coleman JA, Molday RS (2011) Critical role of the beta-subunit CDC50A in the stable expression, assembly, subcellular localization, and lipid transport activity of the P4-ATPase ATP8A2. J Biol Chem 286(19):17205–17216. doi:10.1074/jbc.M111.229419
Coleman JA, Quazi F, Molday RS (2013) Mammalian P4-ATPases and ABC transporters and their role in phospholipid transport. Biochim Biophys Acta 1831(3):555–574. doi:10.1016/j.bbalip.2012.10.006
Coleman JA, Vestergaard AL, Molday RS, Vilsen B, Peter Andersen J (2012) Critical role of a transmembrane lysine in aminophospholipid transport by mammalian photoreceptor P4-ATPase ATP8A2. Proc Natl Acad Sci U S A 109(5):1449–1454. doi:10.1073/pnas.1108862109
Daleke DL (2007) Phospholipid flippases. J Biol Chem 282(2):821–825. doi:10.1074/jbc.R600035200
Daleke DL, Huestis WH (1985) Incorporation and translocation of aminophospholipids in human erythrocytes. Biochemistry 24(20):5406–5416
Darland-Ransom M, Wang X, Sun C-L, Mapes J, Gengyo-Ando K, Mitani S, Xue D (2008) Role of C. elegans TAT-1 protein in maintaining plasma membrane phosphatidylserine asymmetry. Science 320(5875):528–531. doi:10.1126/science.1155847
Das A, Slaughter BD, Unruh JR, Bradford WD, Alexander R, Rubinstein B, Li R (2012) Flippase-mediated phospholipid asymmetry promotes fast Cdc42 recycling in dynamic maintenance of cell polarity. Nat Cell Biol 14(3):304–310. doi:10.1038/ncb2444
Denmeade SR, Mhaka AM, Rosen DM, Brennen WN, Dalrymple S, Dach I, Olesen C, Gurel B, Demarzo AM, Wilding G, Carducci MA, Dionne CA, Moller JV, Nissen P, Christensen SB, Isaacs JT (2012) Engineering a prostate-specific membrane antigen-activated tumor endothelial cell prodrug for cancer therapy. Sci Transl Med 4(140), 140ra186. doi:10.1126/scitranslmed.3003886
Devaux PF, Lopez-Montero I, Bryde S (2006) Proteins involved in lipid translocation in eukaryotic cells. Chem Phys Lipids 141(1–2):119–132. doi:10.1016/j.chemphyslip.2006.02.007
Dezi M, Di Cicco A, Bassereau P, Levy D (2013) Detergent-mediated incorporation of transmembrane proteins in giant unilamellar vesicles with controlled physiological contents. Proc Natl Acad Sci U S A 110(18):7276–7281. doi:10.1073/pnas.1303857110
Ding J, Wu Z, Crider BP, Ma Y, Li X, Slaughter C, Gong L, Xie XS (2000) Identification and functional expression of four isoforms of ATPase II, the putative aminophospholipid translocase. Effect of isoform variation on the ATPase activity and phospholipid specificity. J Biol Chem 275(30):23378–23386. doi:10.1074/jbc.M910319199
Eppens EF, van Mil SW, de Vree JM, Mok KS, Juijn JA, Oude Elferink RP, Berger R, Houwen RH, Klomp LW (2001) FIC1, the protein affected in two forms of hereditary cholestasis, is localized in the cholangiocyte and the canalicular membrane of the hepatocyte. J Hepatol 35(4):436–443
Ezanno P, Cribier S, Devaux PF (2010) Asymmetrical stress generated by the erythrocyte lipid flippase triggers multiple bud formation on the surface of spherical giant liposomes. Eur Biophys J 39(8):1277–1280. doi:10.1007/s00249-009-0557-3
Farge E, Ojcius DM, Subtil A, Dautry-Varsat A (1999) Enhancement of endocytosis due to aminophospholipid transport across the plasma membrane of living cells. Am J Physiol 276(3 Pt 1):C725–C733
Furuta N, Fujimura-Kamada K, Saito K, Yamamoto T, Tanaka K (2007) Endocytic recycling in yeast is regulated by putative phospholipid translocases and the Ypt31p/32p-Rcy1p pathway. Mol Biol Cell 18(1):295–312. doi:10.1091/mbc.E06-05-0461
Gall WE, Geething NC, Hua Z, Ingram MF, Liu K, Chen SI, Graham TR (2002) Drs2p-dependent formation of exocytic clathrin-coated vesicles in vivo. Curr Biol 12(18):1623–1627
Gilbert MJ, Thornton CR, Wakley GE, Talbot NJ (2006) A P-type ATPase required for rice blast disease and induction of host resistance. Nature 440(7083):535–539. doi:10.1038/nature04567
Hachiro T, Yamamoto T, Nakano K, Tanaka K (2013) Phospholipid flippases Lem3p-Dnf1p and Lem3p-Dnf2p are involved in the sorting of the tryptophan permease Tat2p in yeast. J Biol Chem 288(5):3594–3608. doi:10.1074/jbc.M112.416263
Halleck MS, Lawler JJ, Blackshaw S, Gao L, Nagarajan P, Hacker C, Pyle S, Newman JT, Nakanishi Y, Ando H, Weinstock D, Williamson P, Schlegel RA (1999) Differential expression of putative transbilayer amphipath transporters. Physiol Genomics 1(3):139–150
Halleck MS, Pradhan D, Blackman C, Berkes C, Williamson P, Schlegel RA (1998) Multiple members of a third subfamily of P-type ATPases identified by genomic sequences and ESTs. Genome Res 8(4):354–361
Halleck MS, Schlegel RA, Williamson PL (2002) Reanalysis of ATP11B, a type IV P-type ATPase. J Biol Chem 277(12):9736–9740. doi:10.1074/jbc.M200240200
Holthuis JC, Levine TP (2005) Lipid traffic: floppy drives and a superhighway. Nat Rev Mol Cell Biol 6(3):209–220. doi:10.1038/nrm1591
Hua Z, Fatheddin P, Graham TR (2002) An essential subfamily of Drs2p-related P-type ATPases is required for protein trafficking between Golgi complex and endosomal/vacuolar system. Mol Biol Cell 13(9):3162–3177. doi:10.1091/mbc.E02-03-0172
Hua Z, Graham TR (2003) Requirement for neo1p in retrograde transport from the Golgi complex to the endoplasmic reticulum. Mol Biol Cell 14(12):4971–4983. doi:10.1091/mbc.E03-07-0463
Ikeda M, Kihara A, Denpoh A, Igarashi Y (2008) The Rim101 pathway is involved in Rsb1 expression induced by altered lipid asymmetry. Mol Biol Cell 19(5):1922–1931. doi:10.1091/mbc.E07-08-0806
Jacquot A, Montigny C, Hennrich H, Barry R, le Maire M, Jaxel C, Holthuis J, Champeil P, Lenoir G (2012) Phosphatidylserine stimulation of Drs2p.Cdc50p lipid translocase dephosphorylation is controlled by phosphatidylinositol-4-phosphate. J Biol Chem 287(16):13249–13261. doi:10.1074/jbc.M111.313916
Jain S, Stanford N, Bhagwat N, Seiler B, Costanzo M, Boone C, Oelkers P (2007) Identification of a novel lysophospholipid acyltransferase in Saccharomyces cerevisiae. J Biol Chem 282(42):30562–30569. doi:10.1074/jbc.M706326200
Kato U, Inadome H, Yamamoto M, Emoto K, Kobayashi T, Umeda M (2013) Role for phospholipid flippase complex of ATP8A1 and CDC50A proteins in cell migration. J Biol Chem 288(7):4922–4934. doi:10.1074/jbc.M112.402701
Klomp LW, Vargas JC, van Mil SW, Pawlikowska L, Strautnieks SS, van Eijk MJ, Juijn JA, Pabon-Pena C, Smith LB, DeYoung JA, Byrne JA, Gombert J, van der Brugge G, Berger R, Jankowska I, Pawlowska J, Villa E, Knisely AS, Thompson RJ, Freimer NB, Houwen RH, Bull LN (2004) Characterization of mutations in ATP8B1 associated with hereditary cholestasis. Hepatology 40(1):27–38. doi:10.1002/hep.20285
Lenoir G, Williamson P, Puts CF, Holthuis JC (2009) Cdc50p plays a vital role in the ATPase reaction cycle of the putative aminophospholipid transporter Drs2p. J Biol Chem 284(27):17956–17967. doi:10.1074/jbc.M109.013722
Levano K, Punia V, Raghunath M, Debata PR, Curcio GM, Mogha A, Purkayastha S, McCloskey D, Fata J, Banerjee P (2012) Atp8a1 deficiency is associated with phosphatidylserine externalization in hippocampus and delayed hippocampus-dependent learning. J Neurochem 120(2):302–313. doi:10.1111/j.1471-4159.2011.07543.x
Liu K, Hua Z, Nepute JA, Graham TR (2007) Yeast P4-ATPases Drs2p and Dnf1p are essential cargos of the NPFXD/Sla1p endocytic pathway. Mol Biol Cell 18(2):487–500. doi:10.1091/mbc.E06-07-0592
Liu K, Surendhran K, Nothwehr SF, Graham TR (2008) P4-ATPase requirement for AP-1/clathrin function in protein transport from the trans-Golgi network and early endosomes. Mol Biol Cell 19(8):3526–3535. doi:10.1091/mbc.E08-01-0025
Lopez-Marques RL, Poulsen LR, Hanisch S, Meffert K, Buch-Pedersen MJ, Jakobsen MK, Pomorski TG, Palmgren MG (2010) Intracellular targeting signals and lipid specificity determinants of the ALA/ALIS P4-ATPase complex reside in the catalytic ALA alpha-subunit. Mol Biol Cell 21(5):791–801. doi:10.1091/mbc.E09-08-0656
Lopez-Marques RL, Poulsen LR, Palmgren MG (2012) A putative plant aminophospholipid flippase, the Arabidopsis P4 ATPase ALA1, localizes to the plasma membrane following association with a beta-subunit. PLoS One 7(4):e33042. doi:10.1371/journal.pone.0033042
Marx U, Polakowski T, Pomorski T, Lang C, Nelson H, Nelson N, Herrmann A (1999) Rapid transbilayer movement of fluorescent phospholipid analogues in the plasma membrane of endocytosis-deficient yeast cells does not require the Drs2 protein. Eur J Biochem 263(1):254–263
Muller P, Pomorski T, Herrmann A (1994) Incorporation of phospholipid analogues into the plasma membrane affects ATP-induced vesiculation of human erythrocyte ghosts. Biochem Biophys Res Commun 199(2):881–887
Nakano K, Yamamoto T, Kishimoto T, Noji T, Tanaka K (2008) Protein kinases Fpk1p and Fpk2p are novel regulators of phospholipid asymmetry. Mol Biol Cell 19(4):1783–1797. doi:10.1091/mbc.E07-07-0646
Natarajan P, Liu K, Patil D, Sciorra V, Jackson C, Graham T (2009) Regulation of a Golgi flippase by phosphoinositides and an ArfGEF. Nat Cell Biol 11(12):1421–1426. doi:10.1038/ncb1989
Natarajan P, Wang J, Hua Z, Graham TR (2004) Drs2p-coupled aminophospholipid translocase activity in yeast Golgi membranes and relationship to in vivo function. Proc Natl Acad Sci U S A 101(29):10614–10619. doi:10.1073/pnas.0404146101
Palmgren M, Nissen P (2011) P-type ATPases. Annu Rev Biophys 40:243–266. doi:10.1146/annurev.biophys.093008.131331
Palmgren MG, Axelsen KB (1998) Evolution of P-type ATPases. Biochim Biophys Acta 1365(1–2):37–45
Paterson JK, Renkema K, Burden L, Halleck MS, Schlegel RA, Williamson P, Daleke DL (2006) Lipid specific activation of the murine P4-ATPase Atp8a1 (ATPase II). Biochemistry 45(16):5367–5376. doi:10.1021/bi052359b
Paulusma CC, Folmer DE, Ho-Mok KS, de Waart DR, Hilarius PM, Verhoeven AJ, Oude Elferink RP (2008) ATP8B1 requires an accessory protein for endoplasmic reticulum exit and plasma membrane lipid flippase activity. Hepatology 47(1):268–278. doi:10.1002/hep.21950
Paulusma CC, Groen A, Kunne C, Ho-Mok KS, Spijkerboer AL, Rudi de Waart D, Hoek FJ, Vreeling H, Hoeben KA, van Marle J, Pawlikowska L, Bull LN, Hofmann AF, Knisely AS, Oude Elferink RP (2006) Atp8b1 deficiency in mice reduces resistance of the canalicular membrane to hydrophobic bile salts and impairs bile salt transport. Hepatology 44(1):195–204. doi:10.1002/hep.21212
Paulusma CC, Houwen RH, Williamson PL (2011) The flip side of cardiolipin import. Nat Med 17(4):413. doi:10.1038/nm0411-413a, author reply 413–414
Perez-Victoria FJ, Gamarro F, Ouellette M, Castanys S (2003) Functional cloning of the miltefosine transporter. A novel P-type phospholipid translocase from Leishmania involved in drug resistance. J Biol Chem 278(50):49965–49971. doi:10.1074/jbc.M308352200
Perez-Victoria FJ, Sanchez-Canete MP, Castanys S, Gamarro F (2006) Phospholipid translocation and miltefosine potency require both L. donovani miltefosine transporter and the new protein LdRos3 in Leishmania parasites. J Biol Chem 281(33):23766–23775. doi:10.1074/jbc.M605214200
Pomorski T, Lombardi R, Riezman H, Devaux PF, van Meer G, Holthuis JC (2003) Drs2p-related P-type ATPases Dnf1p and Dnf2p are required for phospholipid translocation across the yeast plasma membrane and serve a role in endocytosis. Mol Biol Cell 14(3):1240–1254. doi:10.1091/mbc.E02-08-0501
Poulsen LR, Lopez-Marques RL, McDowell SC, Okkeri J, Licht D, Schulz A, Pomorski T, Harper JF, Palmgren MG (2008) The Arabidopsis P4-ATPase ALA3 localizes to the Golgi and requires a beta-subunit to function in lipid translocation and secretory vesicle formation. Plant Cell 20(3):658–676. doi:10.1105/tpc.107.054767
Poulsen LR, Lopez-Marques RL, Palmgren MG (2008) Flippases: still more questions than answers. Cell Mol Life Sci 65(20):3119–3125. doi:10.1007/s00018-008-8341-6
Puts C, Panatala R, Hennrich H, Tsareva A, Williamson P, Holthuis J (2012) Mapping functional interactions in a heterodimeric phospholipid pump. J Biol Chem 287(36):30529–30540. doi:10.1074/jbc.M112.371088
Puts CF, Holthuis JC (2009) Mechanism and significance of P4 ATPase-catalyzed lipid transport: lessons from a Na+/K+-pump. Biochim Biophys Acta 1791(7):603–611. doi:10.1016/j.bbalip.2009.02.005
Puts CF, Lenoir G, Krijgsveld J, Williamson P, Holthuis JC (2010) A P4-ATPase protein interaction network reveals a link between aminophospholipid transport and phosphoinositide metabolism. J Proteome Res 9(2):833–842. doi:10.1021/pr900743b
Ray NB, Durairaj L, Chen BB, McVerry BJ, Ryan AJ, Donahoe M, Waltenbaugh AK, O’Donnell CP, Henderson FC, Etscheidt CA, McCoy DM, Agassandian M, Hayes-Rowan EC, Coon TA, Butler PL, Gakhar L, Mathur SN, Sieren JC, Tyurina YY, Kagan VE, McLennan G, Mallampalli RK (2010) Dynamic regulation of cardiolipin by the lipid pump Atp8b1 determines the severity of lung injury in experimental pneumonia. Nat Med 16(10):1120–1127. doi:10.1038/nm.2213
Riekhof WR, Voelker DR (2006) Uptake and utilization of lyso-phosphatidylethanolamine by Saccharomyces cerevisiae. J Biol Chem 281(48):36588–36596. doi:10.1074/jbc.M608851200
Riekhof WR, Wu J, Gijon MA, Zarini S, Murphy RC, Voelker DR (2007) Lysophosphatidylcholine metabolism in Saccharomyces cerevisiae: the role of P-type ATPases in transport and a broad specificity acyltransferase in acylation. J Biol Chem 282(51):36853–36861. doi:10.1074/jbc.M706718200
Ruaud AF, Nilsson L, Richard F, Larsen MK, Bessereau JL, Tuck S (2009) The C. elegans P4-ATPase TAT-1 regulates lysosome biogenesis and endocytosis. Traffic 10(1):88–100. doi:10.1111/j.1600-0854.2008.00844.x
Saito K, Fujimura-Kamada K, Furuta N, Kato U, Umeda M, Tanaka K (2004) Cdc50p, a protein required for polarized growth, associates with the Drs2p P-type ATPase implicated in phospholipid translocation in Saccharomyces cerevisiae. Mol Biol Cell 15(7):3418–3432. doi:10.1091/mbc.E03-11-0829
Saito K, Fujimura-Kamada K, Hanamatsu H, Kato U, Umeda M, Kozminski KG, Tanaka K (2007) Transbilayer phospholipid flipping regulates Cdc42p signaling during polarized cell growth via Rga GTPase-activating proteins. Dev Cell 13(5):743–751. doi:10.1016/j.devcel.2007.09.014
Sebastian TT, Baldridge RD, Xu P, Graham TR (2012) Phospholipid flippases: building asymmetric membranes and transport vesicles. Biochim Biophys Acta 1821(8):1068–1077. doi:10.1016/j.bbalip.2011.12.007
Seigneuret M, Devaux PF (1984) ATP-dependent asymmetric distribution of spin-labeled phospholipids in the erythrocyte membrane: relation to shape changes. Proc Natl Acad Sci U S A 81(12):3751–3755
Shettihalli AK, Gummadi SN (2013) Biochemical evidence for lead and mercury induced transbilayer movement of phospholipids mediated by human phospholipid scramblase 1. Chem Res Toxicol 26(6):918–925. doi:10.1021/tx400090h
Siegmund A, Grant A, Angeletti C, Malone L, Nichols JW, Rudolph HK (1998) Loss of Drs2p does not abolish transfer of fluorescence-labeled phospholipids across the plasma membrane of Saccharomyces cerevisiae. J Biol Chem 273(51):34399–34405
Sohoel H, Jensen AM, Moller JV, Nissen P, Denmeade SR, Isaacs JT, Olsen CE, Christensen SB (2006) Natural products as starting materials for development of second-generation SERCA inhibitors targeted towards prostate cancer cells. Bioorg Med Chem 14(8):2810–2815. doi:10.1016/j.bmc.2005.12.001
Soupene E, Kemaladewi DU, Kuypers FA (2008) ATP8A1 activity and phosphatidylserine transbilayer movement. J Recept Ligand Channel Res 1:1–10
Stapelbroek JM, Peters TA, van Beurden DH, Curfs JH, Joosten A, Beynon AJ, van Leeuwen BM, van der Velden LM, Bull L, Oude Elferink RP, van Zanten BA, Klomp LW, Houwen RH (2009) ATP8B1 is essential for maintaining normal hearing. Proc Natl Acad Sci U S A 106(24):9709–9714. doi:10.1073/pnas.0807919106
Stevens HC, Malone L, Nichols JW (2008) The putative aminophospholipid translocases, DNF1 and DNF2, are not required for 7-nitrobenz-2-oxa-1,3-diazol-4-yl-phosphatidylserine flip across the plasma membrane of Saccharomyces cerevisiae. J Biol Chem 283(50):35060–35069. doi:10.1074/jbc.M802379200
Stone A, Chau C, Eaton C, Foran E, Kapur M, Prevatt E, Belkin N, Kerr D, Kohlin T, Williamson P (2012) Biochemical characterization of P4-ATPase mutations identified in patients with progressive familial intrahepatic cholestasis. J Biol Chem 287(49):41139–41151. doi:10.1074/jbc.M112.413039
Stone A, Williamson P (2012) Outside of the box: recent news about phospholipid translocation by P4 ATPases. J Chem Biol 5:131–136. doi:10.1007/s12154-012-0078-x
Suzuki J, Fujii T, Imao T, Ishihara K, Kuba H, Nagata S (2013) Calcium-dependent phospholipid scramblase activity of TMEM16 protein family members. J Biol Chem 288(19):13305–13316. doi:10.1074/jbc.M113.457937
Suzuki J, Umeda M, Sims PJ, Nagata S (2010) Calcium-dependent phospholipid scrambling by TMEM16F. Nature 468(7325):834–838. doi:10.1038/nature09583
Takatsu H, Baba K, Shima T, Umino H, Kato U, Umeda M, Nakayama K, Shin HW (2011) ATP9B, a P4-ATPase (a putative aminophospholipid translocase), localizes to the trans-Golgi network in a CDC50 protein-independent manner. J Biol Chem 286(44):38159–38167. doi:10.1074/jbc.M111.281006
Tamaki H, Shimada A, Ito Y, Ohya M, Takase J, Miyashita M, Miyagawa H, Nozaki H, Nakayama R, Kumagai H (2007) LPT1 encodes a membrane-bound O-acyltransferase involved in the acylation of lysophospholipids in the yeast Saccharomyces cerevisiae. J Biol Chem 282(47):34288–34298. doi:10.1074/jbc.M704509200
Tang X, Halleck MS, Schlegel RA, Williamson P (1996) A subfamily of P-type ATPases with aminophospholipid transporting activity. Science 272(5267):1495–1497
Tsai PC, Hsu JW, Liu YW, Chen KY, Lee FJ (2013) Arl1p regulates spatial membrane organization at the trans-Golgi network through interaction with Arf-GEF Gea2p and flippase Drs2p. Proc Natl Acad Sci U S A 110(8):E668–E677. doi:10.1073/pnas.1221484110
Ujhazy P, Ortiz D, Misra S, Li SH, Moseley J, Jones H, Arias IM (2001) Familial intrahepatic cholestasis 1: studies of localization and function. Hepatology 34(4):768–775. doi:10.1053/jhep.2001.27663
van der Mark VA, Elferink RP, Paulusma CC (2013) P4 ATPases: flippases in health and disease. Int J Mol Sci 14(4):7897–7922. doi:10.3390/ijms14047897
van der Velden LM, Wichers CG, van Breevoort AE, Coleman JA, Molday RS, Berger R, Klomp LW, van de Graaf SF (2010) Heteromeric interactions required for abundance and subcellular localization of human CDC50 proteins and class 1 P4-ATPases. J Biol Chem 285(51):40088–40096. doi:10.1074/jbc.M110.139006
van Meer G, Voelker DR, Feigenson GW (2008) Membrane lipids: where they are and how they behave. Nat Rev Mol Cell Biol 9(2):112–124. doi:10.1038/nrm2330
Verhulst PM, van der Velden LM, Oorschot V, van Faassen EE, Klumperman J, Houwen RH, Pomorski TG, Holthuis JC, Klomp LW (2010) A flippase-independent function of ATP8B1, the protein affected in familial intrahepatic cholestasis type 1, is required for apical protein expression and microvillus formation in polarized epithelial cells. Hepatology 51(6):2049–2060. doi:10.1002/hep.23586
Wang L, Beserra C, Garbers DL (2004) A novel aminophospholipid transporter exclusively expressed in spermatozoa is required for membrane lipid asymmetry and normal fertilization. Dev Biol 267(1):203–215. doi:10.1016/j.ydbio.2003.11.004
Warren DT, Andrews PD, Gourlay CW, Ayscough KR (2002) Sla1p couples the yeast endocytic machinery to proteins regulating actin dynamics. J Cell Sci 115(8):1703–1715
Wehman A, Poggioli C, Schweinsberg P, Grant B, Nance J (2011) The P4-ATPase TAT-5 inhibits the budding of extracellular vesicles in C. elegans embryos. Curr Biol 21(23):1951–1959. doi:10.1016/j.cub.2011.10.040
Weingartner A, Drobot B, Herrmann A, Sanchez-Canete MP, Gamarro F, Castanys S, Gunther Pomorski T (2010) Disruption of the lipid-transporting LdMT-LdRos3 complex in Leishmania donovani affects membrane lipid asymmetry but not host cell invasion. PLoS One 5(8):e12443. doi:10.1371/journal.pone.0012443
Wicky S, Schwarz H, Singer-Kruger B (2004) Molecular interactions of yeast Neo1p, an essential member of the Drs2 family of aminophospholipid translocases, and its role in membrane trafficking within the endomembrane system. Mol Cell Biol 24(17):7402–7418. doi:10.1128/MCB.24.17.7402-7418.2004
Wiedmer T, Zhou Q, Kwoh DY, Sims PJ (2000) Identification of three new members of the phospholipid scramblase gene family. Biochim Biophys Acta 1467(1):244–253
Winther AM, Bublitz M, Karlsen JL, Moller JV, Hansen JB, Nissen P, Buch-Pedersen MJ (2013) The sarcolipin-bound calcium pump stabilizes calcium sites exposed to the cytoplasm. Nature 495(7440):265–269. doi:10.1038/nature11900
Winther AM, Liu H, Sonntag Y, Olesen C, le Maire M, Soehoel H, Olsen CE, Christensen SB, Nissen P, Moller JV (2010) Critical roles of hydrophobicity and orientation of side chains for inactivation of sarcoplasmic reticulum Ca2+-ATPase with thapsigargin and thapsigargin analogs. J Biol Chem 285(37):28883–28892. doi:10.1074/jbc.M110.136242
Xie Z (2003) Molecular mechanisms of Na/K-ATPase-mediated signal transduction. Ann N Y Acad Sci 986:497–503
Xu P, Okkeri J, Hanisch S, Hu RY, Xu Q, Pomorski TG, Ding XY (2009) Identification of a novel mouse P4-ATPase family member highly expressed during spermatogenesis. J Cell Sci 122(Pt 16):2866–2876. doi:10.1242/jcs.047423
Xu Q, Yang GY, Liu N, Xu P, Chen YL, Zhou Z, Luo ZG, Ding X (2012) P4-ATPase ATP8A2 acts in synergy with CDC50A to enhance neurite outgrowth. FEBS Lett 586(13):1803–1812. doi:10.1016/j.febslet.2012.05.018
Yabas M, Teh CE, Frankenreiter S, Lal D, Roots CM, Whittle B, Andrews DT, Zhang Y, Teoh NC, Sprent J, Tze LE, Kucharska EM, Kofler J, Farell GC, Broer S, Goodnow CC, Enders A (2011) ATP11C is critical for the internalization of phosphatidylserine and differentiation of B lymphocytes. Nat Immunol 12(5):441–449. doi:10.1038/ni.2011
Yanagisawa M, Iwamoto M, Kato A, Yoshikawa K, Oiki S (2011) Oriented reconstitution of a membrane protein in a giant unilamellar vesicle: experimental verification with the potassium channel KcsA. J Am Chem Soc 133(30):11774–11779. doi:10.1021/ja2040859
Zhang Z, Devarajan P, Dorfman AL, Morrow JS (1998) Structure of the ankyrin-binding domain of alpha-Na, K-ATPase. J Biol Chem 273(30):18681–18684
Zhou Q, Zhao J, Stout JG, Luhm RA, Wiedmer T, Sims PJ (1997) Molecular cloning of human plasma membrane phospholipid scramblase. A protein mediating transbilayer movement of plasma membrane phospholipids. J Biol Chem 272(29):18240–18244
Zhou X, Graham TR (2009) Reconstitution of phospholipid translocase activity with purified Drs2p, a type-IV P-type ATPase from budding yeast. Proc Natl Acad Sci U S A 106(39):16586–16591. doi:10.1073/pnas.0904293106
Acknowledgments
The authors thank the UNIK research initiative of the Danish Ministry of Science, Technology and Innovation through the “Center for Synthetic Biology” at the University of Copenhagen, the Danish National Research Foundation through the PUMPKIN Center of Excellence (DNRF85), the Danish Council for Independent Research | Natural Sciences (FNU, project number 10-083406), the Lundbeck Foundation, and the Deutsche Forschungsgemeinschaft for supporting their research.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution License which permits any use, distribution, and reproduction in any medium, provided the original author(s) and the source are credited.
About this article
Cite this article
Lopez-Marques, R.L., Theorin, L., Palmgren, M.G. et al. P4-ATPases: lipid flippases in cell membranes. Pflugers Arch - Eur J Physiol 466, 1227–1240 (2014). https://doi.org/10.1007/s00424-013-1363-4
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00424-013-1363-4