
Abstract
We reported recently that peroxisome proliferator-activated receptor β (PPARβ) activation promotes a calcineurin-dependent exercise-like remodelling characterised by increased numbers of oxidative fibres and capillaries. As physical exercise also induces myonuclear accretion, we investigated whether PPARβ activation alters myonuclear density. Transgenic muscle-specific PPARβ over-expression induced 14% increase of myonuclear density. Pharmacological PPARβ activation promoted rapid and massive myonuclear accretion (20% increase after 48 h), which is dependent upon calcineurin/nuclear factor of activated T cells signalling pathway. In vivo bromodeoxyuridine labelling and proliferating cell nuclear antigen immunodetection revealed that PPARβ activation did not promote cell proliferation, suggesting that the PPARβ-promoted myonuclear accretion involves fusion of pre-existing muscle precursor cells to myofibres rather than cell division. Finally, we showed that in skeletal muscle, ageing led to a down-regulation of PPARβ accompanied by decrease of both oxidative fibre number and myonuclear density. PPARβ pharmacological activation counteracts, at least in part, the ageing-driven muscle remodelling.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Adult skeletal muscle displays a remarkable capacity for regeneration and adaptation. Skeletal myofibres represent the largest cells in the body. They are syncitial, post-mitotic and terminally differentiated [6, 13, 37]. Muscle fibres are unable to proliferate but contain a pool of muscle precursor cells (MPCs), commonly called “satellite cells”, which reside between the basal lamina and the sarcolemma [19]. The term of muscle precursor cells is used in this report due to the heterogeneity within the satellite cell population [40].
MPCs play crucial roles in muscle physiology, particularly in muscle repair and regeneration. In response to stress-induced myofibre damages, reparation results from the fusion of MPCs with damaged but viable myofibres [25, 27, 28] and regeneration from the fusion of several MPCs within the basal lamina of the destroyed fibres for de novo myofibre formation [25, 31, 33, 44]. MPCs proliferation is a preliminary and obligatory step of these processes [26].
MPCs are also involved in adaptive response of skeletal muscle to acute and chronic changes in intensity or type of physical activity, such as muscle functional overload, treadmill running, chronic motor nerve stimulation or voluntary running [2, 16, 30, 32]. In these particular conditions, MPCs proliferation is required for modifications of myonuclear density and myofibre hypertrophy but does not seem to be necessary for fibre twitch from faster to slower phenotype [16, 29]. However, myonuclear accretion is an early event of the adaptive response to aerobic training which is characterised by a transition from glycolytic to oxidative fibres. Furthermore, Tseng et al. showed that slow fibres always had small volume/myonucleus regarding to fibre diameter and that fast/oxidative fibres with high succinate dehydrogenase (SDH) activity appeared to have lower volume/myonucleus than fast/glycolytic fibres [39]. These differences in nuclear domain size between fibre types were proposed to be related to the greater amount of proteins required for oxidative metabolism, including those implicated in mitochondrial structure and functions.
Finally, the role of MPCs in age-related muscle atrophy is poorly documented, and conflicting observations have been reported. It seems that ageing does not affect the ability of MPCs to respond to various challenges even if there might exist a delay in the proliferative responses of MPCs from aged muscle [12]. Brack et al. reported that in large fast muscle fibres, the decrease of myonuclei per muscle unit preceded muscle atrophy. Authors proposed that the age-related decrease in myofibre size may actually be a compensatory response to an inadequate capacity for myonuclear replacement [4]. Bruusgaard et al. reported that aged mice display a reduction of muscle size as well as a reduction in myonuclear number [5]. In aged mouse muscles, authors have visualised many fragmented nuclei suggesting increased apoptosis and have proposed that reduction of myonuclei in old muscle could result in a reduction of regenerative capacity of MPCs [5]. This hypothesis is in contrast with previous observations made in muscle inactivity experiments, in which fibre size reduction precedes the decrease in myonuclei number [45].
We previously described the effect of peroxisome proliferator-activated receptor (PPARβ) over-expression or pharmacological activation in mouse tibialis anterior (TLA), a fast/oxidative-glycolytic muscle [10, 17]. In both models, we reported an impressive muscle remodelling characterised by 37% hyperplasia and an increase in oxidative fibre and capillary number [10, 18]. In PPARβ agonist-treated animals, this remodelling takes place within only 2 days [10]. Because of the observed oxidative and angiogenic remodelling as well as the improvement of metabolic profiles of PPARβ agonist-treated animals and humans [15, 21, 36, 41], it was proposed that activation of the PPARβ pathway mimics, at least partially, the adaptive response of muscle to aerobic training. Muscle hyperplasia is indicative of MPC activation [14]. Furthermore, we have actually reported an increase of Myf5 and MyoD1 proteins immediately after pharmacological PPARβ activation using GW0742, also suggesting the possible activation of MPCs [10].
As physical exercise and ageing are two physiological situations affecting myonuclear densities, we asked how PPARβ over-expression or pharmacological activation affects myonuclear number in the TLA muscle from young and aged mice. Furthermore, we investigated whether or not MPC proliferation was required. We report here a new striking phenotype for both PPARβ over-expressing and PPARβ agonist-treated mice. In both cases, the number of nuclei per unit of fibre length is significantly increased. This augmentation of myonuclear density follows the same kinetic as the previously described muscle oxidative and angiogenic remodelling induced by PPARβ pharmacological activation, starting at day 1 and being completed at day 2 of treatment [10]. Interestingly, myonuclear accretion does not require cell proliferation. As previously described for other PPARβ-dependent remodelling [10], the increase in myonuclear density is prevented by inhibition of the calcineurin pathway. Finally, we show that PPARβ is down-regulated in aged mouse muscles and that pharmacological PPARβ activation, using GW0742, restores in large part the age-related loss of myonuclei.
Materials and methods
Animals
Animals were maintained in a 12/12 h lighting cycle and received food (UAR, France) and water ad libitum. All experimental procedures were conducted according to French legislation. Ten-week-old male C57BL6J (Janvier, France) and 10 weeks and 19 month-old male B6D2 (maintained in our animal facility) were used in the various experiments. We always used four animals per condition to obtain statistical values. Animals were killed after the indicated times by cervical dislocation, and tibialis anterior muscles were harvested immediately after killing.
Animals over-expressing PPARβ specifically in skeletal muscle were generated as previously described [18]. Briefly, B6D2 mice harbouring a loxP–stop–loxP–PPARβ–hygromycine construction were crossed with B6D2 mice expressing Cre recombinase under human skeletal actin (HSA) promoter [20]. All animals were maintained hemizygous for their transgene. Presence of the transgenes was verified by polymerase chain reaction (PCR) analyses of tail DNA (REDExtract-N-Amp Tissue PCR Kit, Sigma). Animals harbouring the two transgenes were used as PPARβ-overexpressing mice, while animals harbouring the HSA-Cre transgene only were used as control.
Injections
GW0742, a PPARβ-specific activator [38], was dissolved in Dulbecco’s modified Eagle’s medium/dimethyl sulphoxide (DMSO) 6% (Gibco) and injected subcutaneously once a day (9 a.m.) at 1 mg/kg. Control animals received vehicle at 9 a.m..
Cyclosporine A (Sigma) treatments were performed twice a day (9 a.m. and 6 p.m.) by subcutaneous injections at 25 mg/kg in DMSO. In these experiments, GW0742 was injected subcutaneously once a day (2 p.m.) at 1 mg/kg. Injections of vehicle were performed at 9 a.m., 2 p.m. and 6 p.m. (Control group), 9 a.m. and 6 p.m. (GW group) and 2 p.m. (CsA group).
BrdU (Sigma) treatments were performed once a day (9 a.m.) by intra-peritoneal injections of the compound at 100 mg/kg in 0.9% NaCl. In these experiments, GW0742 was injected subcutaneously once a day (2 p.m.) at 1 mg/kg. Injections of vehicle were performed at 9 a.m. and 2 p.m. in control groups.
Immunofluorescence analysis
Tibialis anterior (TLA) muscles and duodenum were harvested and frozen in tissue embedding medium (VWR International) immediately after mice were killed. Ten-micrometre cryosections were performed from the middle part of muscle or from duodenum, placed on poly-lysine coated slides (VWR International) and processed for immunofluorescence analyses. Mouse monoclonal NFATc1 (7A6, sc-7294, Santa Cruz Biotechnology) and goat anti-mouse fluorescein conjugate antibodies were both used at 1:200 dilution. For BrdU immunofluorescence, cryosections were fixed on slide according manufacturer’s protocol and then incubated 45 min with an anti-BrdU antibody (Anti-Bromodeoxyuridine-Fluorescein, Roche). Finally, slides were mounted with Vectashield containing DAPI (H-1200, Vector laboratories, Burlingame, CA, USA). Slides were viewed under an epifluorescence microscope connected to a digital camera with the Spot software (Universal Imaging).
Histological and immunohistochemical analysis
TLA muscles and duodenum were harvested, fixed in 4% paraformaldehyde (PFA) overnight, dehydrated and then embedded in paraffin. Paraffin sections (5 μm) were used for either BrdU or PCNA antigen detection using M.O.M. Kit (PK-2200, Vector Laboratories). BrdU (11170376001, Roche), PCNA (PC10, Sc-56, Santa Cruz Biotechnology) and mouse monoclonal antibodies were used in a 1:50 and 1:100 dilution, respectively. DAB (SK-4100, Vector Laboratories) served as a substrate. Nuclei were counterstained with haematoxylin. Slides were viewed under an epifluorescence microscope connected to a digital camera with the Spot software (Universal Imaging).
Single-fibre isolation
Single fibres were isolated. Both fibre length and myonuclear number were assessed as described by Brack et al. [4]. Briefly, TLA muscles were fixed in 4% PFA during 2 days at room temperature. Muscles were then treated 2 h in 40% NaOH (w/v) and agitated vigorously for 10 min. Released fibres were washed in phosphate-buffered saline (PBS; pH 7.4) and stored at 4°C in PBS 0.05% azide.
Myonuclear density analysis
After DAPI staining, images of 40 single isolated fibres per animal (four animals per group) were captured using a Z-stacking procedure on a Zeiss Apotome (×10 objective), then Z-projected using the open source ImageJ software (Wayne Rasband, National institutes for Health). Fibre diameters were measured at six locations over at least 500 μm length of the fibre, and all nuclei were counted using Olympus DP-Soft software. Values obtained were averaged to obtain mean value per fibre. Any damaged areas of the fibres were not analysed.
Sodium dodecyl sulfate polyacrylamide gel electrophoresis and Western blot
Total TLA lysates from the mice treated with GW0742 or vehicle alone (DMSO) were prepared, electrophoresed and blotted on polyvinylidene difluoride membrane. Subcellular fractionation of TLA muscles was performed using the Qproteome™ Cell Compartment Kit (Quiagen) according to manufacturer’s instructions. The following antibodies were used at the indicated dilutions for immunodetection: Pan A calcineurin (AB1695, Chemicon International) 1:1,000, polyclonal anti P85 (PI3K subunit) from rabbit (06-496, Upstate, Lake Placid, NY, USA) 1:1,000, polyclonal anti CREB from rabbit (kind gift of Dr. M. Montminy, San Diego, USA) 1:2,000, polyclonal anti Rho GDIa from rabbit (A-20, sc-360, Santa Cruz Biotechnology) 1:500, polyclonal anti acetyl-histone 3 from rabbit (06-599, Upstate, Lake Placid, NY,USA) 1:1,000, monoclonal NFATc1 from mouse (7A6, sc-7294, Santa Cruz Biotechnology) 1:200, monoclonal NFATc3 from mouse (F-1, sc-8405, Santa Cruz Biotechnology) 1:200, peroxidase-coupled anti-rabbit and peroxidase-coupled anti-mouse secondary antibodies (Vector Laboratories) 1:5,000.
Quantitative reverse transcription PCR
Reverse transcription was performed using 0.2 μg of total RNA from muscle of 10 week-old and 19 month-old C57Bl6 mice (five animals per group), a Core kit (RT-RTCK-03, Eurogentec) according to manufacturer’s instruction and a mix of random primers (9 mers) and oligodT. qPCR was performed on SDS7900HT (Applied Biosystem) using Mesagreen qPCR kit for SYBR (Eurogentec) and the following primers:
-
PPARβ: 5′-AGATGGTGGCAGAGCTATGACC-3′; 5′-TCCTCCTGTGGCTGTTCC-3′.
-
Catalase: 5′-GGATCCTGACATGGTCTGGG-3′; 5′-TGGAGAGACTCGGGACGAAG-3′.
-
PDK4: 5′-GCATTTCTACTCGGATGCTCAATG-3′; 5′-CCAATGTGGCTTGGGTTTCC-3′.
-
36B4: 5′-TCCAGGCTTTGGGCATCA-3′; 5′-CTTTATCAGCTGCACATCACTCAGA-3′.
Data were all normalised using 36B4 as housekeeping gene.
Statistical analysis
The effect of the duration of treatment was performed by a one-way analysis of variance (ANOVA) test. Two-way ANOVA tests were performed for comparisons between groups (age and treatment; age and transgenicity). When significant changes were observed in ANOVA tests, Fisher’s protected least significant difference post hoc test was applied to locate the source of significant differences. When two groups were compared, we used a Student’s t test. Analyses were performed with Stat’View Abacus Concept version 5. Hierarchical ascendant classifications were performed for the determination of fibre groups according to nuclei/mm of fibre length. Results are presented as means ± SD with significance accepted when p < 0.05.
Results
PPARβ pathway activation promotes an increase in myonuclear density in TLA muscle
The effects of PPARβ over-expression on myonuclear density were first investigated by quantification of myonuclear number per millimetre of fibre length in diamidino-2-phenyl-indole (DAPI)-stained isolated fibres, prepared from TLA muscle of adult B6D2 PPARβ transgenic mice and control littermates (Tg and Ct; Fig. 1a). These experiments revealed that muscle-specific PPARβ over-expression leads to a 14% increase of nuclei number per millimetre of fibre length (92 ± 1.32, n = 160 fibres for controls versus 105 ± 3.2, n = 160 fibres for PPARβ transgenic mice, p = 0.003). Using ascendant hierarchical classification, we defined three groups of myofibres, i.e. those containing less than 85 nuclei/mm (F < 85), those containing a number of nuclei comprised between 85 and 100 nuclei/mm (85 < F < 100) and those containing more than 100 nuclei/mm (F > 100). As shown in Fig. 1b, PPARβ over-expression induced major changes in myofibre distribution in these three groups with reduction of myofibre numbers in both the F < 85 (31% to 10%) and 85 < F < 100 (41% to 35%) groups, while the number of fibres in the F > 100 group is notably increased (28% to 55%).

Muscle-specific PPARβ over-expression promotes an increase of myonuclear density. a Representative isolated DAPI-stained fibres harvested from tibialis anterior of 10-week-old control (Ct) and PPARβ transgenic (Tg) B6D2 mice. Global myonuclear density is indicated as myonuclei number per millimetre of fibre length (n = 160, four animals per group, 40 fibres per animal). b Distribution of myofibres population in TLA from control and PPARβ overexpressing mice. Myonuclei in isolated DAPI-stained fibres (n = 160, four animals per group, 40 fibres per animal) harvested from tibialis anterior of 10-week-old control (Ct) and PPARβ transgenic (Tg) B6D2F1 mice were quantified and pooled using ascendant hierarchical classification in three groups of fibres containing (1) less than 85 nuclei, (2) from 85 to 100 nuclei and (3) more than 100 nuclei/mm of fibre length. Percentage of each group, mean number of nuclei in each group (Mean) and standard deviation (s.d.) are reported for control (Ct) and PPARβ transgenic (Tg) mice
We then investigated the effects of pharmacological PPARβ activation on myonuclear density by treating 10-week-old C57Bl6 male mice with GW0742, a PPARβ-specific agonist [38]. The number of myonuclei/mm fibre length was determined at increasing times after GW0742 or vehicle injections. We first noticed a significant strain difference concerning the myonuclear density (92 ± 1.32 and 86 ± 1.40, in B6D2 and C57Bl6 mice, respectively) as well as the distribution in the three predefined groups between B6D2 and C57Bl6 mice (compare Ct in Fig. 1b to Fig. 2b). As expected, vehicle treatment did not affect myonuclear density nor distribution in the three predefined groups of fibres (not shown). On the contrary, PPARβ activation promoted a fast increase in myonuclear density with an 11% (p < 0.0001) increase after 1 day and a maximal augmentation of 20% (p < 0.0001) reached after 2 days of treatment (Fig. 2a). Classification of fibres in the three predefined groups revealed that, as previously observed in PPARβ transgenic mice, pharmacological PPARβ activation resulted in time-dependent and notable changes in fibre group distribution in favour of fibres with a high myonuclear density. This shift towards the F > 100 group is progressive from 8% to 34% after 1 day and to a maximum of 62% reached after 2 days, with a parallel decrease of percentage of fibres in both F < 85 group (47% to 28% after 1 day and to 15% after 2 days) and 85 < F < 100 group (45% to 38% after 1 day and to 23% after 2 days; Fig. 2b).

PPARβ pharmacological activation induces an increase of myonuclear density. a Quantification of myonuclei number in isolated DAPI-stained fibres (n = 160, four animals per group, 40 fibres per animal) harvested from tibialis anterior of 10-week-old C57Bl6 mice for different times of treatment with vehicle (Ct) or PPARβ agonist GW0742 (1 day (1d), 2 days (2d) and 4 days (4d)). *p < 0.001, different from vehicle-treated group (Ct); pound sign p < 0.01, different from 1 day treated animal group (1d). b Distribution of myofibres population counted in a using the same classification as in Fig. 1b. Percentage of each group, mean number of nuclei in each group (Mean) and standard deviation (s.d.) are reported for each period of treatment with GW0742
Pharmacological PPARβ activation does not promote cell division in TLA muscle
The fast and important increase in myonuclear density as well as hyperplasia suggests major events of MPC activation and fusion to myofibres. However, the quickness of myonuclear accretion in response to PPARβ activation argues against a process requiring cell proliferation. To investigate directly the effect of PPARβ activation on cell proliferation in TLA muscle, we explored by immunochemistry the expression of a well-known mitosis marker, the proliferating cell nuclear antigen (PCNA), in muscles from animals receiving or not GW0742 for various times. Sections of the duodenum from control animals were used as positive control and displayed, as expected, an important labelling in the crypts (Fig. 3a). In contrast, nearly no PCNA-positive myonuclei were detectable in sections from control animals (Fig. 3b). As shown in Fig. 3, PPARβ activation did not increase the number of PCNA-positive myonuclei, whatever the period of treatment with GW0742 was, i.e. 5 h (Fig. 3c), 24 h (Fig. 3d), or 48 h (Fig. 3e, f). However, after 48 h of PPARβ agonist treatment, several fibres with centrally located myonuclei can be observed as a mark of myonuclear accretion (Fig. 3e and f). These results suggest that PPARβ-promoted myonuclear accretion did not involve proliferation of MPCs. Finally, we did detect some PCNA-positive cells but almost only in blood vessels (Fig. 3d, h–j at high magnification).

GW0742-promoted myonuclear remodelling is independent of cell proliferation in tibialis anterior. Immunohistochemical localization of PCNA, a well-known mitotic marker, in a section of duodenum as positive control, b, g section of TLA muscles from vehicle-treated animals, c–j sections of TLA muscles from animals receiving GW0742 for various times: c, h 5 h, d, i 24 h, e, f, j 48 h. Note the important labelling in the crypts and the quasi absence of positive myonuclei in both control and GW0742-treated animals. PCNA signals were often detected in blood vessels (arrows in d, h–j) but rarely in myonuclei (arrows in c). Finally, note the elevated number of centrally located nuclei (arrowhead in e, f). Scale bars, 50 μm
To confirm these observations, bromodeoxyuridine (BrdU) incorporation into DNA was used. In vivo BrdU labelling technique allows identification of MPCs that have proliferated, migrated and either incorporated into existing myofibres or having been implicated in the formation of new fibres [16, 35]. BrdU incorporation into DNA was determined by two different methods, indirect immunofluorescence on cryosections and immunochemistry on paraffin-embedded sections. As shown in Fig. 4a, b, sections from the duodenum of control animals that received for 1 (Fig. 4b) or 2 days (Fig. 4a) daily injections of BrdU validated the method to follow cell proliferation in vivo, as BrdU-positive cells are detected both in crypts, where the cells are proliferating, and in the lower parts of the villi that contain epithelial cells, which have proliferated in the crypts (Fig. 4b) and then migrated during their differentiation towards the villous apex (Fig. 4a). Data presented in Fig. 4 confirmed that PPARβ-promoted muscle remodelling does not implicate cell proliferation as the number of BrdU-positive nuclei remained very low in muscle from animals treated for 2 days with GW0742. Less than 1% of nuclei were BrdU-positive, and no significant difference was found compared to muscles from untreated animals. Furthermore, as previously observed, several myofibres containing central nuclei can be evidenced in muscles from animals treated for 48 h with GW0742 (Fig. 4c, e, f). Interestingly, these nuclei remained BrdU-negative. As central nuclei are marks of the fusion of MPCs to fibres and/or newly formed myofibres, these observations also strongly support the conclusion that myonuclear accretion and fibre hyperplasia promoted by PPARβ activation took place without MPC proliferation.

GW0742-promoted myonuclear remodelling does not require cell division in tibialis anterior. Mice were injected with BrdU and or not with GW0742. Duodenum (a, b) or TLA muscle (c–f) harvested from 24 h (b) or 48 h (a, c–f) post GW0742 treatment, were either frozen in tissue-embedding medium or fixed, dehydrated and embedded in paraffin. Frozen sections on slides were stained with anti-BrdU antibody coupled to fluorescein (a, c, e) and mounted using Vectashield containing DAPI as described in “Materials and methods”. Positive cells are detected in blood vessels, and very few myonuclei are labelled (c, e). Arrowheads indicate myofibres with central nucleus (c, e). Paraffin sections were stained with anti-BrdU antibody (b, d, f) as described in “Materials and methods”, and nuclei were counterstained with haematoxylin. Very few myonuclei are labelled (d, and arrow in f); on the contrary, a large number of myofibres with one or more central nuclei are visible (d, e, f). Note the increased BrdU labelling in the duodenum sections between 24 h (b) and 48 h (a) of the BrdU pulse. Scale bar, 50 μm
Positive effects of PPARβ activation on myonuclear density are dependent of the calcineurin/NFAT pathway
We previously provided evidences that the active calcineurin pathway is required for the myogenic and angiogenic responses to PPARβ activation in the adult mouse [10]. To test whether an active calcineurin pathway was required for the PPARβ-promoted myonuclear accretion, we explored the effects of co-administration of cyclosporine A (CsA), a potent inhibitor of calcineurin/nuclear factor of activated T-cells (NFAT) pathway, on TLA myonuclear density in mice treated by GW0742 for 2 days.
As shown in Fig. 5a, b, CsA administration alone neither affected the myonuclear density nor the distribution in the three defined fibre groups. On the contrary, CsA administration totally blunted the PPARβ-dependant increment of global myonuclear density (from 86 to 104 nuclei/mm in the absence and to 87 nuclei/mm in presence of CsA; Fig. 5a). Calcineurin inhibition strongly reduced the redistribution of myofibres towards the group with the highest myonuclear density (shift from 8% to 62% in the absence of CsA versus shift from 8% to 20% when CsA was co-administrated; Fig. 5b).

Positive effects of PPARβ on myonuclear density are calcineurin dependent. a Quantification of nuclei number in isolated DAPI-stained fibres harvested from tibialis anterior of 10-week-old C57Bl6 animals (n = 160, four animals per group, 40 fibres per animal), treated 2 days with vehicle (Ct), cyclosporine A (CsA), PPARβ agonist GW0742 (2d) or PPARβ agonist GW0742 and cyclosporine A (2d + CsA). *p < 0.01, different from Ct, CsA and 2d + CsA groups. b Distribution of myofibres population counted in a using the same classification as in Fig. 1b. Percentage of each group, mean number of nuclei in each group (Mean) and standard deviation (s.d.) are reported for each treatment
Our data also indicated that in TLA muscle, GW0742 treatment did not promote an increase of calcineurin at the protein level (Fig. 6a) but rather provokes an induction of its phosphatase activity as attested by the rapid nuclear translocation of nuclear factor of activated T cells C1 and C3. Indeed, enhanced NFATc1 nuclearization can be evidenced as soon as 5 h after GW0742 treatment (Fig. 6b, c). It remained high at 24 h of treatment and returned to control value after 48 h (Fig. 6c). NFATc3 nuclearization can be evidenced after 24 h of PPARβ agonist treatment, and it returned to control values after 48 h of treatment.

GW0742 induces calcineurin A-dependent NFAT nuclearization. a Calcineurin and P85 Western blot analysis after control treatment (DMSO 5H) or different times of GW0742 treatment (2, 5, 8, and 24 h). P85 is used as loading control. b Immunohistochemical localization of NFATc1 using cryosections of TLA muscles from mice treated 5 h with DMSO (DMSO 5H), GW0742 (GW 5H) or cyclosporine A and GW0742 (CsA + GW 5H). Note the increased NFATc1 nuclearization only in GW 5H-treated animals. c TLA muscles from mice treated 5 or 24 h with DMSO (DMSO 5h, DMSO 24h), or treated 5, 24, or 48 h with GW0742 (GW 5h, GW 24h, GW 48h) were used for subcellular fractionation in cytoplasmic (C), nuclear (N) and insoluble-chromatin-associated (I) proteins and subsequent Western blot analysis. Note the increased nuclear localization of NFATc1 and NFATc3 in response to PPARβ activation. CREB (nuclear), Rho GDIα (cytoplasmic) and acetylated histone H3 (H3, Chromatin associated) antibodies were used to evaluate the purity of each fraction. Scale bar, 50 μm
Ageing promotes a decrease in myonuclear density, while PPARβ agonist treatment restores a juvenile phenotype
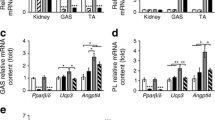
Myonuclear density is negatively regulated by ageing [4, 5]. In order to explore a possible role of PPARβ in this a process, we first investigated the effect of ageing on PPARβ expression by comparing the amounts of the corresponding mRNA in mouse TLA from adult (10-week-old) and aged (19-month-old) mice. As shown in Fig. 7, PPARβ mRNA was down-regulated by more than 30% (p = 0.03, n = 5) in aged mouse muscles when compared to young mouse muscles. To check whether the decreased expression of PPARβ impacts on gene regulation, we also quantified mRNA for two PPARβ-target genes, namely catalase, that plays a crucial role in the protection against oxidative stress [23], and pyruvate dehydrogenase kinase 4 (PDK4) that is involved in metabolic regulations [7]. These experiments revealed that in aged mice, both genes were down-regulated (40%, p = 0.01, and 75%, p = 0.001 for catalase and PDK4, respectively; Fig. 7a). However, PPARβ pathway remains active in aged mice as 4 days of GW0742 treatment promoted an up-regulation of these target genes (1.76-fold, p = 0.034, and 6.19-fold, p = 0.047 for catalase and PDK4, respectively; Fig. 7b). As expected, GW0742 treatment did not modify PPARβ expression.

Ageing reduces expression of PPARβ, catalase and PDK4 mRNAs. a Quantification of PPARβ, catalase and PDK4 using mRNAs from muscles of 10-week-old (white bar) and 19-month-old (dark bar) mice (five animals per group) and qPCR analyses as described in “Materials and methods”. *p < 0.05, different from 10-week-old mice group; pound sign p < 0.005, different from 10-week-old mice group. b Quantification of PPARβ, catalase and PDK4 using mRNAs from muscles of 20-month-old mice treated (cross-hatched bar) or not (dark bar) during 4 days with GW0742 (four animals per group) and qPCR analyses as described in “Materials and methods”. *p < 0.05, different from 20-month-old untreated mice group. Results in a and b were normalised using 36B4 as housekeeping gene
The effect of ageing on myonuclear density was next investigated in TLA muscles from young and aged B6D2 control mice (Ct and 19Ct in Fig. 8a, b). As shown in Fig. 8a and in agreement with other reports [5], global myonuclear density was slightly but significantly lower in aged mice when compared to young adult animals (−5%, p = 0.0057). The effect of ageing on myonuclear density is further evidenced when evaluating the fibre distribution profile in the previously defined groups. In aged mice, the number of fibres in the F < 85 group was increased by 1.35-fold (31% to 42%), whereas the fibre numbers in F > 100 and 85 < F < 100 groups were reduced 1.22- and 1.17-fold, respectively compared to those of young mice (Fig. 8b). Furthermore, the mean value in the F < 85 group seems to be lower in aged animals than in young adult animals, whereas other mean values remain stable (Fig. 8b).

Ageing promotes a 5% decrease of myonuclear density, while PPARβ agonist treatment restores a juvenile phenotype. a Quantification of nuclei number in isolated DAPI-stained fibres harvested from tibialis anterior of 10-week-old (Ct, Tg, GW) and 19-month-old (19Ct, 19Tg, 19GW) B6D2 mice (n = 160, four animals per group, 40 fibres per animal). Mice were either treated with vehicle only (Ct, 19Ct) or untreated PPARβ transgenic mice (Tg, 19Tg) or treated for 4 days with GW0742 (GW, 19GW). *p < 0.01, different from 10-week-old mice groups (Ct, Tg, GW); pound sign p < 0.05, different from vehicle-treated groups (Ct, 19Ct). b Distribution of myofibres population counted in a using the same classification as in Fig. 1b. Percentage of each group, mean number of nuclei in each group (Mean) and standard deviation (s.d.) are reported for each experimental condition
To explore a possible beneficial action of PPARβ pathway activation on myonuclear density, we next investigated the effects of muscle specific PPARβ over-expression on the ageing-related loss of myonuclei. In elderly PPARβ transgenic animals, we have observed a significant reduction in global myonuclear density (−7%, p < 0.0052) and a shift towards fibres that contain fewer nuclei (Tg and 19Tg in Fig. 8a, b) when compared to their younger counterparts. Despite this reduction, global myonuclear density in aged transgenic mice remained in the same order of value than that found in young untreated mice (compare CT and 19Tg in Fig. 8a). Furthermore, the percentage of fibres in the F > 100 group remained significantly higher in old transgenic animals than in control young animals (F > 100 = 28% and 46% in CT and 19Tg, respectively. Fig. 8b). Finally, we studied the effects of pharmacological PPARβ activation on myonuclear density in young and old B6D2F1 mice (GW and 19GW in Fig. 8a, b). In young control mice, 4 days treatment with GW0742 resulted in an increase (14%, p = 0.048) in the global myonuclear density (Fig. 8a) and redistribution of fibres in favour of the F > 100 group (28% to 67%, Fig. 8b). This augmentation is similar to that observed in PPARβ-over-expressing transgenic mice (compare CT, TG and GW in Fig. 8a) but is less important than the observed 20% increase in young C57Bl6-treated mice (Fig. 2a). Interestingly, in aged B6D2F1 mice (19GW), 4 days administration of PPARβ agonist led not only to a 9% (p = 0.0318) increase in the myonuclear density (compare 19Ct and 19GW in Fig. 8a) but also to a redistribution of fibres in the group which contained more than 100 nuclei/mm (F > 100; 23% to 38%; Fig. 8b). The percentage of fibres in the F < 85 group was reduced (42% to 25%), whereas the 85 < F < 100 group remained stable (Fig. 8b). Interestingly, treatment allowed aged mice to reach similar global myonuclear density as 10-week-old control mice (compare 19GW and Ct in Fig. 8a), almost the same fibre distribution profile (Fig. 8b) and slightly better mean values in the three groups, indicating that PPARβ activation could compensate the age-related loss of myonuclei.
Ageing promotes a decrease in oxidative myofibre number, while PPARβ agonist treatment restores a juvenile phenotype
To characterise the impact of ageing on fibre type composition and the effects of PPARβ activation on this parameter, we used cross-sections around mid-portion of the TLA muscles from 10-week-old and 22-month-old animals treated or not for 4 days with GW0742 for in situ staining of succinate dehydrogenase activity, which is a marker of mitochondrial complex II content. In accordance with our previous observations [10], TLA from young adult C57Bl6 mice contains about 46% of SDH-positive fibres and 54% of SDH-negative fibres, and GW0742 treatment promotes an increment of the oxidative phenotype characterised by increased proportion of SDH-positive fibres (Table 1). TLA from aged animals displayed a less oxidative phenotype with decreased proportion of SDH-positive fibres (39.3 % in aged versus 46.3% in young animals). Interestingly, 4 days of GW0742 administration promoted an increase of SDH-positive fibre proportion in aged mouse TLA, resulting in a phenotype that is nearly similar to that observed in young untreated animals (Table 1). This PPARβ-promoted increment of oxidative capabilities of TLA from aged animals was then confirmed by the determination of citrate synthase (CS) activity as a marker of the oxidative metabolism and lactate dehydrogenase (LDH) activity as a marker of glycolytic metabolism. These experiments showed that, after 4 days of PPARβ agonist treatment, the CS specific activity was increased by 16.7% (p = 0.002, n = 4), while the LDH-specific activity remained unchanged (p > 0.5, n = 4). Comparable effects were previously reported in PPARβ transgenic animals [18].
Discussion
We previously reported that PPARβ activation or over-expression in mice induces striking skeletal muscle oxidative and angiogenic remodelling [10, 18]. We report here a new phenotype in both PPARβ transgenic mice and PPARβ agonist-treated mice, consisting in massive activation, recruitment and fusion of MPCs to both pre-existing and newly formed myofibres. Fusion of such a large number of myonuclei may represent a key event in the PPARβ-promoted oxidative muscle remodelling.
This newly described PPARβ-promoted phenotype is remarkable in magnitude and quickness. First, in B6D2F1 mice, muscle-specific PPARβ over-expression or pharmacological activation induced a 14% increase in myonuclear density when compared to control animals. This myonuclear density increment is even more pronounced in GW0742-treated C57Bl6 mice which display a 20% augmentation. Taking into account that in the TLA of C57Bl6, PPARβ agonist treatment also leads to a muscle hyperplasia corresponding to the generation of 600 new myofibres [10], it can be estimated that PPARβ activation resulted in addition of about one million of new myonuclei to each muscle. Furthermore, these events of MPC activation, recruitment and fusion could also happen in other muscles, resulting in addition of several millions of new myonuclei per animal. The quickness of myonuclear accretion after PPARβ administration is also very impressive. In both C57Bl6 and B6D2F1, GW0742 treatment promoted an increase in myonuclear density that was already detectable at day 1 and achieved at day 2 (Fig. 2). This process takes place in the same time frame that we have already determined for the PPARβ-promoted myogenic/angiogenic muscle remodelling [10]. Analysis of the fibre distribution depending on their myonuclear content per millimetre of fibre length (F < 85; 85 < F < 100; F > 100) allowed a better description of the effects of PPARβ pathway activation. Both PPARβ over-expression and its pharmacological activation highly increased the number of fibres that contain more than 100 nuclei/mm fibre length (Fig. 1b and 2b). Furthermore, in GW0742-treated animals, we observed a time-dependent shift from the two classes of low myonuclear density fibres towards high myonuclear density fibres. Indeed, in C57Bl6, the fibres that contain less than 100 nuclei (F < 85 and 85 < F < 100) represent 93% of the myofibres before the treatment, 66% after 1 day of treatment, and only 38% after 2 days of treatment (Fig. 2b). Since these fibres did not disappear, the most probable scenario is a rapid and progressive recruitment and fusion of MPCs to pre-existing and newly formed fibres.
Another remarkable feature of the PPARβ-promoted myonuclear accretion is the finding that proliferation of MPCs is not required for such a remodelling (Figs. 3 and 4). This is in contrast with other pathological or physiological situations leading to muscle remodelling, such as adaptation to exercise, overload or muscle regeneration, in which a critical and obligatory step of MPCs proliferation has been established [16, 26]. However, it should be noted that in these situations, muscle remodelling takes place within weeks and often follows muscle damage, whereas PPARβ-promoted muscle remodelling is achieved in 2 days without any histological sign of fibre deterioration. All our observations argue in favour of a model, in which the PPARβ pathway activates a pool of MPCs that are “committed” to differentiate and able to fuse to fibres omitting the obligate step of proliferation. Existence of such cells has already been suggested by others [1, 11, 24, 43].
The central role of the calcineurin/NFAT pathway in the maintenance of fibre type composition and in muscle adaptation to physical exercise has been well documented (for review, see [3]). The calcineurin pathway has also been associated with MPC activation [8, 22, 34]. As cyclosporine A administration abolished both myogenic and angiogenic responses to PPARβ activation, we previously proposed that active calcineurin pathway is required for PPARβ-promoted muscle remodelling [10]. Data presented here reinforce this model as demonstrating that treatment of mice with GW0742 did not affect calcineurin A expression but induced calcineurin A activation as shown by the rapid nuclearization of both NFATc1 and NFATc3 in TLA (Fig. 6). The finding that CsA administration impaired the PPARβ-dependent increase in global myonuclear density (Fig. 5a) as well as the redistribution of myofibres in the three defined fibre groups (Fig. 5b) strongly suggests that the calcineurin/NFAT pathway is involved directly in the PPARβ-promoted myonuclear accretion. Further work is required to fully characterise the signalling cascade that follows PPARβ-promoted NFAT activation.
A characteristic of ageing muscle is fibre atrophy and loss of myonuclei [4, 5]. In particular, the regression in myonuclear content seems to be a key event in the process of age-related muscle atrophy. Our data confirmed that ageing leads to a reduction in global myonuclear density and to a poor fibre distribution profile (Fig. 8a, b). Furthermore, ageing results in a reduction of TLA muscle oxidative capabilities (Table 1), down-regulation of PPARβ expression and transcriptional activity (Fig. 7). As we have demonstrated that PPARβ pathway activation promotes both oxidative phenotype [10, 18] and myonuclear accretion (Figs. 1 and 2), it is tempting to speculate that PPARβ down-regulation observed in muscle from aged animals is directly involved in the age-related reduction of both myonuclear density and oxidative phenotype in TLA. The findings showing that PPARβ over-expression or pharmacological activation (Fig. 8a, b and Table 1) counteract, at least in part, the effects of ageing on these parameters suggest that PPARβ activation could exert preventive or curative effects on both age-related muscle atrophy and reduction of oxidative phenotype.
The effect of such import of new genetic material on the functional and metabolic abilities of muscle cells of treated animals remains to be determined. However, it may represent the basis for the rapid muscle plasticity and adaptation to aerobic exercise, possibly allowing a reprogramming of muscle cells toward a more oxidative phenotype, through an increased coding efficiency for contractile proteins and genes involved in fatty acid oxidation and mitochondrial biogenesis [39]. This hypothesis could explain the role of PPARβ induction after various types of physical training [9, 18, 42]. Finally, our data indicate that, in addition to its beneficial effects on metabolism, PPARβ agonists may represent a therapeutic approach for the treatment of age-related muscle atrophy.
References
Adams GR, Haddad F, Baldwin KM (1999) Time course of changes in markers of myogenesis in overloaded rat skeletal muscles. J Appl Physiol 87:1705–1712
Allen DL, Monke SR, Talmadge RJ, Roy RR, Edgerton VR (1995) Plasticity of myonuclear number in hypertrophied and atrophied mammalian skeletal muscle fibers. J Appl Physiol 78:1969–1976
Bassel-Duby R, Olson EN (2006) Signaling pathways in skeletal muscle remodeling. Annu Rev Biochem 75:19–37
Brack AS, Bildsoe H, Hughes SM (2005) Evidence that satellite cell decrement contributes to preferential decline in nuclear number from large fibres during murine age-related muscle atrophy. J Cell Sci 118:4813–4821
Bruusgaard JC, Liestol K, Gundersen K (2006) Distribution of myonuclei and microtubules in live muscle fibers of young, middle-aged, and old mice. J Appl Physiol 100:2024–2030
Chambers RL, McDermott JC (1996) Molecular basis of skeletal muscle regeneration. Can J Appl Physiol 21:155–184
Degenhardt T, Saramaki A, Malinen M, Rieck M, Vaisanen S, Huotari A, Herzig KH, Muller R, Carlberg C (2007) Three members of the human pyruvate dehydrogenase kinase gene family are direct targets of the peroxisome proliferator-activated receptor beta/delta. J Mol Biol 372:341–355
Friday BB, Pavlath GK (2001) A calcineurin- and NFAT-dependent pathway regulates Myf5 gene expression in skeletal muscle reserve cells. J Cell Sci 114:303–310
Fritz T, Kramer DK, Karlsson HK, Galuska D, Engfeldt P, Zierath JR, Krook A (2006) Low-intensity exercise increases skeletal muscle protein expression of PPARdelta and UCP3 in type 2 diabetic patients. Diabetes Metab Res Rev 22:492–498
Gaudel C, Schwartz C, Giordano C, Abumrad NA, Grimaldi P (2008) Pharmacological activation of PPAR{beta} promotes rapid and calcineurin-dependent fiber remodeling and angiogenesis in mouse skeletal muscle. Am J Physiol Endocrinol Metab 295:E297–E304
Grounds MD, Garrett KL, Lai MC, Wright WE, Beilharz MW (1992) Identification of skeletal muscle precursor cells in vivo by use of MyoD1 and myogenin probes. Cell Tissue Res 267:99–104
Haddad F, Adams GR (2006) Aging-sensitive cellular and molecular mechanisms associated with skeletal muscle hypertrophy. J Appl Physiol 100:1188–1203
Hughes SM, Schiaffino S (1999) Control of muscle fibre size: a crucial factor in ageing. Acta Physiol Scand 167:307–312
Ishido M, Kami K, Masuhara M (2004) Localization of MyoD, myogenin and cell cycle regulatory factors in hypertrophying rat skeletal muscles. Acta Physiol Scand 180:281–289
Leibowitz MD, Fievet C, Hennuyer N, Peinado-Onsurbe J, Duez H, Bergera J, Cullinan CA, Sparrow CP, Baffic J, Berger GD, Santini C, Marquis RW, Tolman RL, Smith RG, Moller DE, Auwerx J (2000) Activation of PPARdelta alters lipid metabolism in db/db mice. FEBS Lett 473:333–336
Li P, Akimoto T, Zhang M, Williams RS, Yan Z (2006) Resident stem cells are not required for exercise-induced fiber-type switching and angiogenesis but are necessary for activity-dependent muscle growth. Am J Physiol Cell Physiol 290:C1461–1468
Luquet S, Gaudel C, Holst D, Lopez-Soriano J, Jehl-Pietri C, Fredenrich A, Grimaldi PA (2005) Roles of PPAR delta in lipid absorption and metabolism: a new target for the treatment of type 2 diabetes. Biochim Biophys Acta 1740:313–317
Luquet S, Lopez-Soriano J, Holst D, Fredenrich A, Melki J, Rassoulzadegan M, Grimaldi PA (2003) Peroxisome proliferator-activated receptor delta controls muscle development and oxidative capability. Faseb J 17:2299–2301
Mauro A (1961) Satellite cell of skeletal muscle fibers. J Biophys Biochem Cytol 9:493–495
Miniou P, Tiziano D, Frugier T, Roblot N, Le Meur M, Melki J (1999) Gene targeting restricted to mouse striated muscle lineage. Nucleic Acids Res 27:e27
Oliver WR Jr, Shenk JL, Snaith MR, Russell CS, Plunket KD, Bodkin NL, Lewis MC, Winegar DA, Sznaidman ML, Lambert MH, Xu HE, Sternbach DD, Kliewer SA, Hansen BC, Willson TM (2001) A selective peroxisome proliferator-activated receptor delta agonist promotes reverse cholesterol transport. Proc Natl Acad Sci U S A 98:5306–5311
Perez-Ruiz A, Gnocchi VF, Zammit PS (2007) Control of Myf5 activation in adult skeletal myonuclei requires ERK signalling. Cell Signal 19:1671–1680
Pesant M, Sueur S, Dutartre P, Tallandier M, Grimaldi PA, Rochette L, Connat JL (2006) Peroxisome proliferator-activated receptor delta (PPARdelta) activation protects H9c2 cardiomyoblasts from oxidative stress-induced apoptosis. Cardiovasc Res 69:440–449
Rantanen J, Hurme T, Lukka R, Heino J, Kalimo H (1995) Satellite cell proliferation and the expression of myogenin and desmin in regenerating skeletal muscle: evidence for two different populations of satellite cells. Lab Invest 72:341–347
Robertson TA, Grounds MD, Mitchell CA, Papadimitriou JM (1990) Fusion between myogenic cells in vivo: an ultrastructural study in regenerating murine skeletal muscle. J Struct Biol 105:170–182
Robertson TA, Grounds MD, Papadimitriou JM (1992) Elucidation of aspects of murine skeletal muscle regeneration using local and whole body irradiation. J Anat 181(Pt 2):265–276
Robertson TA, Papadimitriou JM, Grounds MD (1992) Fusion between a myogenic cell in the satellite cell position and undamaged adult myofibre segments. Experientia 48:394–395
Robertson TA, Papadimitriou JM, Grounds MD (1993) Fusion of myogenic cells to the newly sealed region of damaged myofibres in skeletal muscle regeneration. Neuropathol Appl Neurobiol 19:350–358
Rosenblatt JD, Yong D, Parry DJ (1994) Satellite cell activity is required for hypertrophy of overloaded adult rat muscle. Muscle Nerve 17:608–613
Roy RR, Monke SR, Allen DL, Edgerton VR (1999) Modulation of myonuclear number in functionally overloaded and exercised rat plantaris fibers. J Appl Physiol 87:634–642
Sabourin LA, Rudnicki MA (2000) The molecular regulation of myogenesis. Clin Genet 57:16–25
Salleo A, La Spada G, Falzea G, Denaro MG, Cicciarello R (1983) Response of satellite cells and muscle fibers to long-term compensatory hypertrophy. J Submicrosc Cytol 15:929–940
Schultz E (1989) Satellite cell behavior during skeletal muscle growth and regeneration. Med Sci Sports Exerc 21:S181–186
Scicchitano BM, Spath L, Musaro A, Molinaro M, Rosenthal N, Nervi C, Adamo S (2005) Vasopressin-dependent myogenic cell differentiation is mediated by both Ca2+/calmodulin-dependent kinase and calcineurin pathways. Mol Biol Cell 16:3632–3641
Smith HK, Maxwell L, Rodgers CD, McKee NH, Plyley MJ (2001) Exercise-enhanced satellite cell proliferation and new myonuclear accretion in rat skeletal muscle. J Appl Physiol 90:1407–1414
Sprecher DL, Massien C, Pearce G, Billin AN, Perlstein I, Willson TM, Hassall DG, Ancellin N, Patterson SD, Lobe DC, Johnson TG (2007) Triglyceride:high-density lipoprotein cholesterol effects in healthy subjects administered a peroxisome proliferator activated receptor delta agonist. Arterioscler Thromb Vasc Biol 27:359–365
Stockdale FE, Holtzer H (1961) DNA synthesis and myogenesis. Exp Cell Res 24:508–520
Sznaidman ML, Haffner CD, Maloney PR, Fivush A, Chao E, Goreham D, Sierra ML, LeGrumelec C, Xu HE, Montana VG, Lambert MH, Willson TM, Oliver WR Jr, Sternbach DD (2003) Novel selective small molecule agonists for peroxisome proliferator-activated receptor delta (PPARdelta)—synthesis and biological activity. Bioorg Med Chem Lett 13:1517–1521
Tseng BS, Kasper CE, Edgerton VR (1994) Cytoplasm-to-myonucleus ratios and succinate dehydrogenase activities in adult rat slow and fast muscle fibers. Cell Tissue Res 275:39–49
Wagers AJ, Conboy IM (2005) Cellular and molecular signatures of muscle regeneration: current concepts and controversies in adult myogenesis. Cell 122:659–667
Wang YX, Lee CH, Tiep S, Yu RT, Ham J, Kang H, Evans RM (2003) Peroxisome-proliferator-activated receptor delta activates fat metabolism to prevent obesity. Cell 113:159–170
Watt MJ, Southgate RJ, Holmes AG, Febbraio MA (2004) Suppression of plasma free fatty acids upregulates peroxisome proliferator-activated receptor (PPAR) alpha and delta and PPAR coactivator 1alpha in human skeletal muscle, but not lipid regulatory genes. J Mol Endocrinol 33:533–544
Yablonka-Reuveni Z (1995) Development and postnatal regulation of adult myoblasts. Microsc Res Tech 30:366–380
Zammit PS, Heslop L, Hudon V, Rosenblatt JD, Tajbakhsh S, Buckingham ME, Beauchamp JR, Partridge TA (2002) Kinetics of myoblast proliferation show that resident satellite cells are competent to fully regenerate skeletal muscle fibers. Exp Cell Res 281:39–49
Zhong H, Roy RR, Siengthai B, Edgerton VR (2005) Effects of inactivity on fiber size and myonuclear number in rat soleus muscle. J Appl Physiol 99:1494–1499
Acknowledgments
GW0742 was a generous gift from T.M. Willson (GlaxoSmithKline). The CREB antibody was a gift of M. Montminy. We thank E. Lendoye, M. Aupetit, M. Radjkhumar, F. Millot, J. Paput, G. Manfroni and G. Visciano for technical assistance. We thank Dr. Kay-Dietrich Wagner for critical reading of the manuscript. This work was supported by a grant to PG from the Agence Nationale de la Recherche (ANR-05-PCOD-012). NW has a fellowship from the Fondation de France. The authors have no conflicting financial interest.
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License (https://creativecommons.org/licenses/by-nc/2.0), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
Giordano, C., Rousseau, A.S., Wagner, N. et al. Peroxisome proliferator-activated receptor β activation promotes myonuclear accretion in skeletal muscle of adult and aged mice. Pflugers Arch - Eur J Physiol 458, 901–913 (2009). https://doi.org/10.1007/s00424-009-0676-9
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00424-009-0676-9