
Abstract
Changes in cellular ion levels can modulate distinct signaling networks aimed at correcting major disruptions in ion balances that might otherwise threaten cell growth and development. Salt-inducible kinase 1 (SIK1) and salt overly sensitive 2 (SOS2) are key protein kinases within such networks in mammalian and plant cells, respectively. In animals, SIK1 expression and activity are regulated in response to the salt content of the diet, and in plants SOS2 activity is controlled by the salinity of the soil. The specific ionic stress (elevated intracellular sodium) is followed by changes in intracellular calcium; the calcium signals are sensed by calcium-binding proteins and lead to activation of SIK1 or SOS2. These kinases target major plasma membrane transporters such as the Na+,K+-ATPase in mammalian cells, and Na+/H+ exchangers in the plasma membrane and membranes of intracellular vacuoles of plant cells. Activation of these networks prevents abnormal increases in intracellular sodium concentration.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Plant and animal cells have different sodium requirements. Animal cells have evolved to profit from the sodium concentration gradient across the plasma membrane to import/export many solutes, nutrients, and water. In plant cells, protons serve the equivalent role of sodium. While most mineral nutrients in animals are obtained from plant-based food, plants do not provide sufficient sodium. For most plants, including almost all crops, sodium concentrations in the soil are often too high and become toxic, a problem known as salt stress [53]. In fact, a significant portion of the world’s arable land and nearly half of the irrigated agricultural land is affected by high soil salinity [54]. Sodium in the soil inhibits the root uptake of potassium, an essential mineral element for all plants. High concentrations of sodium salts and other salts also hamper the plants’ water absorption by lowering soil water potential. When sodium accumulates in the cytoplasm of plant cells, it can inhibit metabolic enzymes and can cause oxidative stress [21].
In contrast to this chemical stress present in plant cells, animal cells are constantly challenged by an osmotic stress. Failure to cope with osmotic challenges will result in unpredictable changes in the cell’s molecular and architectural phenotype, leading to the loss of predetermined functions and eventually to cell death. Alterations in the molecular phenotype include changes in the patterns of gene expression and of the protein products derived from them; changes in the architectural phenotype arise within the cell cytoskeleton due to disproportionate increases in cell size. Alterations in the architectural phenotype are likely to have a significant effect on the organization of regulatory networks under which the molecular phenotype is controlled and thereby harm the cell’s physiological functions. The degree of cell adaptation to these challenges is proportional to the magnitude and duration of the stress.
Salt overly sensitive 2 network in plant cells
In the cytoplasm of plant cells, the sodium concentration depends on sodium import, export, and storage in vacuoles. Sodium enters root cells passively through non-selective cation channels [1] and the HKT family of K+/Na+ transporters [40]. Although little is known about the regulation of sodium import into plant cells, it is well known that sodium is exported from plant cells by the plasma membrane Na+/H+ antiporter SOS1 [35, 43], while a member of the NHX family of Na+/H+ antiporters mediates sodium storage in the vacuole [10]. The Na+/H+ antiporters are driven by an H+ motive force established by membrane H+-ATPases [34]. The expression levels as well as the activities of the antiporters are tightly regulated by salt stress. This regulation involves a calcium-responsive protein kinase pathway [54].
Root exposure to sodium stress elicits an increase in the cytosolic free Ca2+ concentration [54]. The direct trigger for this calcium signal may possibly be an elevation of cytoplasmic Na+ level due to passive Na+ entry. The calcium transporter mediating this increase in cytosolic Ca2+ is not known, so genetic evidence for the function of the calcium signal has depended on the downstream calcium-response pathway. An EF hand calcium-binding protein, known as salt overly sensitive 3 (SOS3), is the calcium sensor in plants under salt stress [25]. SOS3 has four EF hands, each of which can bind a Ca2+ [42]. Structural studies found that the fourth EF hand can also bind Mn2+, but the functional significance of this fact still remains unclear [42]. SOS3 is also myristoylated at its N-terminus. This lipid modification enables the protein and its interacting partner salt overly sensitive 2 (SOS2) to associate with the cell membrane, and this modification is required if SOS3 is to confer salt tolerance, as lack of such modification leads to salt sensitivity [17]. A loss-of-function mutation in SOS3 that reduces calcium-binding ability also renders the plants overly sensitive to salt stress [17]. SOS3 primarily works in the root, and an SOS3-like protein, SCaBP8/CBL10, is necessary for salt tolerance in the shoot [37].
Mutations in two other genes, SOS2 and SOS1, also render the mutant plants overly sensitive to salt stress [50, 52]. SOS2 encodes a serine/threonine protein kinase with a catalytic region similar to the yeast sucrose non-fermenting 1 (SNF1) and the animal AMP-activated protein kinase (AMPK) [24]. SNF1 and AMPK are protein kinases that monitor cellular energy balance and regulate metabolism [14]. In plants, there are three subfamilies of protein kinases that are related phylogenetically to SNF1 and AMPK. These are referred to as SnRK1, SnRK2, and SnRK3 proteins [15]. The SnRK1s are orthologous to SNF1 and AMPK and also function in regulating metabolism. The SnRK2s are activated by hyperosmotic stress and the stress hormone abscisic acid, while the SnRK3s regulate the activities of various membrane transporters [11]. The founding member of the SnRK3 subfamily is SOS2 [15, 24]. The regulatory region on the C-terminal part of SOS2 is unrelated to the regulatory regions of SNF1 and AMPK (Fig. 1). The regulatory region of SOS2 has an autoinhibitory role in controlling the protein kinase activity [12]. This region is where the positive regulator SOS3 and the negative regulator type 2C protein phosphatase ABI2 bind [12, 30, 41] (Fig. 1). The SOS3-binding domain is a 34-amino acid sequence known as the FISL motif, and the ABI2-binding sequence is known as the protein phosphatase interaction (PPI) motif. The function of ABI2 in the sodium regulation pathway is to dephosphorylate and de-activate SOS2 or SOS1 [30].

Diagram of SOS2 domain structure and protein interactions. SOS2 has an N-terminal kinase catalytic domain similar to the catalytic domains of yeast SNF1 and animal AMPK. The regulatory domain of SOS2 contains a FISL motif that binds to the calcium-binding protein SOS3 and a PPI motif that binds to the type 2C protein phosphatase ABI2
SOS2 is active in substrate phosphorylation only when plants are exposed to salt stress. SOS2 activity depends on SOS3 and calcium [13]. One substrate of SOS2 is SOS1, the plasma membrane Na+/H+ antiporter that exports Na+ from the cytoplasm [35, 38] (Fig. 2). Other substrates of SOS2 may include vacuolar Na+/H+ antiporters and H+-ATPases because the activation of their transport activities under salt stress requires SOS2 [2, 36]. The SOS3–SOS2–SOS1 regulatory module can be expressed in yeast cells and is functional in the heterologous system, where it can confer salt tolerance by maintaining low cytoplasmic Na+ [38]. In addition to the plasma membrane Na+/H+ antiporter, vacuolar Na+/H+ antiporter, and vacuolar H+-ATPase, another transporter regulated by SOS2 is the vacuolar Ca2+/H+ antiporter CAX1 [6] (Fig. 1). In this case, however, SOS2 activates CAX1 by physical interaction, and the activation does not require SOS2 kinase activity.

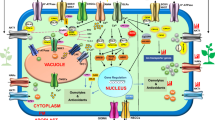
Salt sensing in plant cells. A build-up of Na+ in the cytoplasm triggers a cytosolic Ca2+ signal, which is sensed by SOS3. SOS2 is then activated by Ca2+–SOS3 to convert inactive SOS1 to an active form by phosphorylation. SOS2 also activates vacuolar membrane transporters to promote Na+ storage in the vacuole
Salt-inducible kinase 1 network in mammalian cells
Evolution has endowed mammalian cells with well-organized plasma membrane transporters that, in addition to serving diverse functions in specific organs, are capable of creating and maintaining a distinct concentration of ions inside and out of the cell. In this way, they also regulate the cell’s water content, thereby minimizing the osmotic stress imposed by the extracellular environment and permitting cell survival. Among the plasma membrane transporters, the Na+,K+-ATPase is the first barrier in this set of organized structures that support mammalian cell survival [18]. In the absence of Na+,K+-ATPase molecules at the plasma membrane, cells normally burst, or if its availability/activity is not properly controlled, the subsequent perturbation in ionic gradients will ultimately lead to cell death due to the fact that a myriad of cellular functions will fall in disarray [33]. To prevent such malfunction, mammalian cells possess organized signaling structures at the molecular and structural level. These organized signaling structures control the activity of membrane transporters that ensure cell survival and maintain specific organ functions. The regulatory networks remain largely unknown, and the elucidation of their molecular identity and organization constitutes an important and present challenge.
The Na+,K+-ATPase function, while serving a critical role in the evolution of mammalian cells by enabling their volume control, also has an impact on secondary cell functions, such as membrane potential, vectorial transport, secretion, and contractility. Because those secondary functions depend on Na+,K+-ATPase-mediated ion gradients, regulatory signals must discriminate between the house-keeping and the regulated (secondary) functions of Na+,K+-ATPase. Indeed, each tissue responds to regulatory signals in an organ-specific manner [3, 4, 27, 39, 46].
How are the changes in intracellular sodium translated into activation of Na+,K+-ATPase? In vitro studies have demonstrated that the Na+,K+-ATPase operates at one-third of its maximal capacity in intact cells [45], leading to the assumption that increases in intracellular sodium would exploit the Na+,K+-ATPase reserve capacity by increasing the catalytic activity of units present at the plasma membrane. Although attractive, this model leaves fundamental questions unanswered. As sodium rises above normal levels within cells, which Na+,K+-ATPase molecules within the cell plasma membrane will increase their activities? How many Na+,K+-ATPase molecules will respond by increasing their catalytic activities? How quickly would the Na+,K+-ATPase molecules respond? How long would this activation last? The existence of a cell network that decodes sodium signals could represent an answer to all of these questions.
In general, regulatory signaling networks are easily switched on and off, and they do provide specificity by associating with their targets [16]. In response to activation of G-protein-coupled receptors, the intracellular signals controlling cell Na+,K+-ATPase activity assemble throughout multiple interacting (binding) domains within Na+,K+-ATPase isoforms [3, 7, 28, 51]. Because of the latter, we hypothesize that unknown proteins might, as part of a regulatory network, associate with the Na+,K+-ATPase and regulate its function in response to changes (increases) in intracellular sodium.
Mass spectrometry revealed that the Na+,K+-ATPase associates with SIK1 [44]. SIK is a sucrose non-fermenting-like kinase [19] that belongs to the AMPK family [14] and whose expression is specifically regulated by salt intake [49].Three SIK isoforms have been identified: SIK1 is located in chromosome 21 whereas SIK2 (QIK) and SIK3 (QSK) are located in chromosome 11. SIK1 is ubiquitously distributed, and SIK2 and SIK3 have tissue preferences [9, 31]. Their amino acid sequences revealed a rather conserved kinase domain with significant differences within their regulatory region except for a ubiquitin domain (Fig. 3). Whereas it is still not known whether the association of SIK1 with the Na+,K+-ATPase is direct or is mediated by a linker protein, it is clear that SIK1 adds functionality to the network that controls the increases in Na+,K+-ATPase activity in response to acute/short-term changes in sodium permeability. The association of SIK1 with the Na+,K+-ATPase is not increased when the intracellular concentration of sodium rises [44], suggesting that a limited pool of Na+,K+-ATPase molecules interact with this kinase. The increases in Na+,K+-ATPase activity are true increases in the catalytic properties of the enzyme because the number of molecules within the plasma membrane remains the same regardless of the variations in intracellular sodium [44]. SIK1 activity increases after 5 min of incubation with 5 μM monensin [44], and this corresponds to an increase of ~5–6 mM in intracellular sodium.

Salt-inducible kinase isoforms. Schematic representation of all three isoforms with conserved and regulatory domains
The signaling events by which sodium rapidly triggers the activation of SIK1 appear to occur in the proximity of the Na+,K+-ATPase. They are initiated by a parallel influx of calcium (throughout the reverse Na+/Ca2+ exchanger), and are possibly limited to a discrete number of Na+,K+-ATPase units (Fig. 4). A transient rise in intracellular calcium, via the reverse Na+/Ca2+-exchanger, represents the coding signal for the activation of calmodulin kinases (in particular CaMK1) that in turn phosphorylate SIK1 at a threonine residue within its SNH domain (Thr-322). Activated SIK1 does not control Na+,K+-ATPase activity directly through protein phosphorylation (neither the α- nor the β-subunits contain a SIK1 consensus phosphorylation site) but rather by promoting the dephosphorylation of the Na+,K+-ATPase α-subunit. The latter also suggests that dephosphorylation does not result from a direct effect of sodium on Na+,K+-ATPase but rather from a more sophisticated signaling network. The Na+,K+-ATPase constitutively associates with a protein phosphatase 2A (PP2A) [32, 44], and it is logical to think that, if this phosphatase is constantly present in an active form, the Na+,K+-ATPase would never be subjected to phosphorylation, or that this event has to be tightly regulated. Activation of protein phosphatases is not only important for regulating the state of Na+,K+-ATPase subunit phosphorylation [23] but also for regulating the cellular mechanisms responsible for its traffic to and from the plasma membrane [8]. Increases in intracellular sodium result in activation of PP2A, and this effect requires an active SIK1 [44].

Salt sensing in mammalian cells.The SIK1 pathway is depicted with its presently known signaling components. Increases in Na+ permeability (from basal = ~9 to ~14 mM, [56]) promote transient increases in intracellular Ca2+ via the Na+/Ca2+ exchanger. Sequential activation of calmodulin, calmodulin kinase 1 (CaMK1), and salt-inducible kinase 1 (SIK1) leads to phosphorylation of a phosphomethylesterase 1 (PME-1), promoting its dissociation from the protein phosphatase 2A (PP2A), which is then activated. Dephosphorylation of the Na+,K+-ATPase α-subunit (NK-inactive) triggers the increase in its catalytic activity (NK-active)
Which signals link an activated SIK1 to activation of PP2A? In general, phosphorylation of protein phosphatases has been shown to inactivate the enzyme [20]. In addition to direct phosphorylation of PP2A subunits, methylation represents an alternative mechanism by which phosphatases can be regulated. Detailed in vitro studies revealed the existence of leucine carboxy methyltransferase and a protein phosphatase methylesterase-1 (PME-1) at the core of the molecular events controlling PP2A subunit methylation [26, 29]. PME-1 catalyzes the demethylation of the catalytic subunit, preventing the assembly of the regulatory subunit and thereby preventing activation of PP2A [26, 29]. PME-1 associates with the Na+,K+-ATPase/PP2A complex [44], and it is likely that its presence (within the complex) represses PP2A activity. Changes in intracellular sodium result in PME-1 phosphorylation (SIK1-dependent) and dissociation (SIK1-dependent) from the Na+,K+-ATPase/PP2A complex, leading to Na+,K+-ATPase α-subunit dephosphorylation [44]. That the basal Na+,K+-ATPase activity is not affected by the presence of dominant negative mutants of PP2A may indicate that only a limited pool of Na+,K+-ATPase can be regulated through this process and further suggests that the basal activity of the Na/K-pump is governed by its direct interaction with Na+ and K+ ions whereas its regulation is affected by a complex network of intermediate signals. Similarly, in cells transiently expressing a SIK1 mutant lacking kinase activity and in cells in which SIK1 expression has been suppressed by using small interfering RNA, the basal Na+,K+-ATPase activity remains unchanged [44]. These observations also raise the question of whether specialized pumps are present within the cell membrane.
Covalent modification (Ser/Thr phosphorylation) of Na+,K+-ATPase units may render these molecules insensitive to certain regulatory processes while making them available for other cell functions. These pumps may not necessarily be structurally different molecules but they may be located within different cell domains and/or be associated with distinct regulatory networks. An inactive (non-pumping pool) Na+,K+-ATPase appears to interact with caveolin-1 and regulate its availability at the plasma membrane by affecting caveolin-1 internalization [5, 48]. Such association could serve only as a structural organization governing caveolin trafficking, or it could be part of a more sophisticated signaling network not yet fully elucidated. A possible molecular identity of such a signaling complex has been presented, and in the proposed complex, Src kinase within the caveolin-1/Na+,K+-ATPase microdomains acts as the leading effector of ouabain signals controlling transcription activation after its binding to the Na+,K+-ATPase [47].
In terms of cell physiology, increases in intracellular sodium in mammalian cells may not only increase Na+,K+-ATPase activity and thereby active Na+ transport outside the cells via the SIK1 network, but also may concomitantly reduce cell permeability. Thus, in addition to triggering the activation of SIK1, rises in intracellular sodium are also associated with down-regulation of epithelial Na+ channels (ENaC) [22]. Increases in intracellular sodium affect the proteolytic cleavage of ENaC and thereby their activity at the plasma membrane. As with the regulation of Na+,K+-ATPase activity, it is unlikely that Na+ by itself is directly responsible for regulating the enzymes that influence ENaC cleavage; instead, Na+ probably triggers a series of intracellular signals necessary for achieving the effect. Whether SIK1 is part of such a regulatory network remains to be examined.
Besides being part of a cell survival kit responsible for controlling the clearance of excessive intracellular sodium, the SIK1 network could be highjacked under physiological conditions by hormones, such as angiotensin, that increase sodium reabsorption in the renal tubule and intestinal epithelia. Similarly, it is tempting to speculate that pathophysiological conditions associated with increased renal sodium reabsorption, and possibly resulting in high blood pressure, could be the result of elevated SIK1 activity.
Perspective
The existence of an evolutionarily conserved mechanism in animal and plant cells for controlling intracellular sodium appears to be relevant for cell survival. Sodium, a monovalent cation, can initiate its own homeostasis via a calcium-dependent protein kinase pathway. Although details of the pathway are different in plants and animals, the regulatory principles are conserved. It appears that the signaling network triggered by sodium can also influence gene expression and be the target for regulatory hormones. In addition, there is cross-talk with oxidative stress response pathways [21, 55]. Continued improvement in the understanding of these sodium-related pathways is important for finding better strategies to improve crop performance in saline environments and to improve the diagnosis and treatment of sodium-related pathological conditions in humans.
References
Amtmann A, Sanders D (1998) Mechanisms of Na+ uptake by plant cells. Adv Bot Res 29:76–112
Batelli G, Verslues PE, Agius F, Qiu Q, Fujii H, Pan S, Schumaker KS, Grillo S, Zhu JK (2007) SOS2 promotes salt tolerance in part by interacting with the vaculoar H+-ATPase and upregulating its transport activity. Mol Cell Biol 27:7781–7790
Bertorello AM, Sznajder JI (2005) The dopamine paradox in lung and kidney epithelia: sharing the same target but operating different signaling networks. Am J Respir Cell Mol Biol 33:432–437
Blanco G (2005) Na, K-ATPase subunit heterogeneity as a mechanism for tissue-specific ion regulation. Semin Nephrol 25:292–330
Cai T, Wang H, Chen Y, Liu L, Gunning WT, Quintas LE, Xie ZJ (2008) Regulation of caveolin-1 membrane trafficking by the Na/K-ATPase. J Cell Biol 182:1153–1169
Cheng NH, Pittman JK, Zhu JK, Hirschi KD (2004) The protein kinase SOS2 activates the Arabidopsis H(+)/Ca(2+) antiporter CAX1 to integrate calcium transport and salt tolerance. J Biol Chem 279:2922–2926
Efendiev R, Chen Z, Krmar RT, Uhles S, Katz AI, Pedemonte CH, Bertorello AM (2005) The 14-3-3 protein translates the NA+, K+-ATPase α1-subunit phosphorylation signal into binding and activation of phosphoinositide 3-kinase during endocytosis. J Biol Chem 280:16272–16277
Efendiev R, Yudowski GA, Zwiller J, Leibiger B, Katz AI, Berggren PO, Pedemonte CH, Leibiger IB, Bertorello AM (2002) Relevance of dopamine signals anchoring dynamin-2 to the plasma membrane during Na+, K+-ATPase endocytosis. J Biol Chem 277:44108–44114
Feldman JD, Vician L, Crispino M, Hoe W, Baudry M, Herschman HR (2000) The salt-inducible kinase, SIK, is induced by depolarization in brain. J Neurochem 74:2227–2238
Gaxiola RA, Rao R, Sherman A, Grisafi P, Alper SL, Fink GR (1999) The Arabidopsis thaliana proton transporters, AtNhx1 and Avp1, can function in cation detoxification in yeast. Proc Natl Acad Sci U S A 96:1480–1485
Gong D, Guo Y, Schumaker KS, Zhu JK (2004) The SOS3 family of calcium sensors and SOS2 family of protein kinases in Arabidopsis. Plant Physiol 134:919–926
Guo Y, Halfter U, Ishitani M, Zhu JK (2001) Molecular characterization of functional domains in the protein kinase SOS2 that is required for plant salt tolerance. Plant Cell 13:1383–1400
Halfter U, Ishitani M, Zhu JK (2000) The Arabidopsis SOS2 protein kinase physically interacts with and is activated by the calcium-binding protein SOS3. Proc Natl Acad Sci U S A 97:3735–3740
Hardie DG, Carling D, Halford N (1994) Roles of the Snf1/Rkin1/AMP-activated protein kinase family in the response to environmental and nutritional stress. Semin Cell Biol 5:409–416
Hrabak EM, Chan CW, Gribskov M, Harper JF, Choi JH, Halford N, Kudla J, Luan S, Nimmo HG, Sussman MR, Thomas M, Walker-Simmons K, Zhu JK, Harmon AC (2003) The Arabidopsis CDPK–SnRK superfamily of protein kinases. Plant Physiol 132:666–680
Hunter T (2000) Signaling—2000 and beyond. Cell 100:113–127
Ishitani M, Liu J, Halfter U, Kim CS, Shi W, Zhu JK (2000) SOS3 function in plant salt tolerance requires N-myristoylation and calcium binding. Plant Cell 12:1667–1678
Jaitovich AA, Bertorello AM (2006) Na+, K+-ATPase: an indispensable ion pumping-signaling mechanism across mammalian cell membranes. Semin Nephrol 26:386–392
Jaleel M, McBride A, Lizcano JM, Deak M, Toth R, Morrice NA, Alessi DR (2005) Identification of the sucrose non-fermenting related kinase SNRK, as a novel LKB1 substrate. FEBS Lett 579:1417–1423
Janssens V, Goris J (2001) Protein phosphatase 2A: a highly regulated family of serine/threonine phosphatases implicated in cell growth and signaling. Biochem J 353:417–439
Katiyar-Agarwal S, Zhu J, Kim K, Agarwal M, Fu X, Huang A, Zhu JK (2006) The plasma membrane Na+/H+ antiporter SOS1 interacts with RCD1 and functions in oxidative stress tolerance in Arabidopsis. Proc Natl Acad Sci U S A 103:18816–18821
Knight KK, Wentzlaff DM, Snyder PM (2008) Intracellular sodium regulates proteolytic activation of the epithelial sodium channel. J Biol Chem 283:27477–27482
Lecuona E, Dada LA, Sun H, Butti ML, Zhou G, Chew TL, Sznajder JI (2006) Na, K-ATPase a1-subunit dephosphorylation by protein phosphatase 2A is necessary for its recruitment to the plasma membrane. FASEB J 20:2618–2620
Liu J, Ishitani M, Halfter U, Kim CS, Zhu JK (2000) The Arabidopsis thaliana SOS2 gene encodes a protein kinase that is required for salt tolerance. Proc Natl Acad Sci U S A 97:3730–3734
Liu J, Zhu JK (1998) A calcium sensor homolog required for plant salt tolerance. Science 280:1943–1945
Longin S, Jordens J, Martens E, Stevens I, Janssens V, Rondelez E, De Baere I, Derua R, Waelkens E, Goris J, Van Hoof C (2004) An inactive protein phosphatase 2A population is associated with methylesterase and can be re-activated by the phosphotyrosyl phosphatase activator. Biochem J 380:111–119
McDonough AA, Velotta JB, Schwinger RH, Philipson KD, Farley RA (2002) The cardiac sodium pump: structure and function. Basic Res Cardiol 97(Suppl 1):I19–24
Ogimoto G, Yudowski GA, Barker CJ, Köhler M, Katz AI, Féraille E, Pedemonte CH, Berggren PO, Bertorello AM (2000) G protein-coupled receptors regulate Na+, K+-ATPase activity and endocytosis by modulating the recruitment of adaptor protein 2 and clathrin. Proc Natl Acad Sci U S A 97:3242–3247
Ogris E, Du X, Nelson KC, Mak EK, Yu XX, Lane WS, Pallas DC (1999) A protein phosphatase methylesterase (PME-1) is one of several novel proteins stably associating with two inactive mutants of protein phosphatase 2A. J Biol Chem 274:14382–14391
Ohta M, Guo Y, Halfter U, Zhu JK (2003) A novel domain in the protein kinase SOS2 mediates interaction with the protein phosphatase 2C ABI2. Proc Natl Acad Sci U S A 100:11771–11776
Okamoto M, Takemori H, Katoh Y (2004) Salt-inducible kinase in steroidogenesis and adipogenesis. Trends Endocrinol Metab 15:21–26
Pagel P, Zatti A, Kimura T, Duffield A, Chauvet V, Rajendran V, Caplan MJ (2003) Ion pump -interacting proteins: promising new partners. Ann N Y Acad Sci 986:360–368
Panayiotidis MI, Bortner CD, Cidlowski JA (2006) On the mechanisms of ionic regulation of apoptosis: would the Na+/K+-ATPase please stand up? Acta Physiol 187:205–215
Qiu QS, Barkla BJ, Vera-Estrella R, Zhu JK, Schumaker KS (2003) Na+/H+exchange activity in the plasma membrane of Arabidopsis. Plant Physiol 132:1041–1052
Qiu QS, Guo Y, Dietrich MA, Schumaker KS, Zhu JK (2002) Regulation of SOS1, a plasma membrane Na+/H+ exchanger in Arabidopsis thaliana, by SOS2 and SOS3. Proc Natl Acad Sci U S A 99:8436–8441
Qiu QS, Guo Y, Quintero FJ, Pardo JM, Schumaker KS, Zhu JK (2004) Regulation of vacuolar Na+/H+ exchange in Arabidopsis thaliana by the salt-overly-sensitive (SOS) pathway. J Biol Chem 279:207–215
Quan R, Lin H, Mendoza I, Zhang Y, Cao W, Yang Y, Shang M, Chen S, Pardo JM, Guo Y (2007) SCABP8/CBL10, a putative calcium sensor, interacts with the protein kinase SOS2 to protect Arabidopsis shoots from salt stress. Plant Cell 19:1415–1431
Quintero FJ, Ohta M, Shi H, Zhu JK, Pardo JM (2002) Reconstitution in yeast of the Arabidopsis SOS signaling pathway for Na+ homeostasis. Proc Natl Acad Sci U S A 99:9061–9066
Rajasekaran SA, Barwe SP, Rajasekaran AK (2005) Multiple functions of NaK-ATPase in epithelial cells. Semin Nephrol 25:328–334
Rus A, Yokoi S, Sharkhuu A, Reddy M, Lee BH, Matsumoto TK, Koiwa H, Zhu JK, Bressan RA, Hasegawa PM (2001) AtHKT1 is a salt tolerance determinant that controls Na(+) entry into plant roots. Proc Natl Acad Sci U S A 98:14150–14155
Sanchez-Barrena MJ, Fujii H, Angulo I, Martinez-Ripoll M, Zhu JK, Albert A (2007) The structure of the c-terminal domain of the protein kinase AtSOS2 bound to the calcium sensor AtSOS3. Mol Cell 26:427–435
Sanchez-Barrena MJ, Martinez-Ripoll M, Zhu JK, Albert A (2005) The structure of the Arabidopsis thaliana SOS3: molecular mechanism of sensing calcium for salt stress response. J Mol Biol 345:1253–1264
Shi H, Ishitani M, Kim C, Zhu JK (2000) The Arabidopsis thaliana salt tolerance gene SOS1 encodes a putative Na+/H+ antiporter. Proc Natl Acad Sci U S A 97:6896–6901
Sjöström M, Stenström K, Eneling K, Zwiller J, Katz AI, Takemori H, Bertorello AM (2007) SIK1 is part of a cell sodium-sensing network that regulates active sodium transport through a calcium-dependent process. Proc Natl Acad Sci U S A 104:16922–16927
Skou JC (1988) The Na, K-pump. Methods Enzymol 156:1–25
Sweeney G, Klip A (2001) Mechanisms and consequences of Na+, K+-pump regulation by insulin and leptin. Cell Mol Biol 47:363–372
Tian J, Cai T, Yuan Z, Wang H, Liu L, Haas M, Maksimova E, Huang XY, Xie ZJ (2006) Binding of Src to Na+/K+-ATPase forms a functional signaling complex. Mol Biol Cell 17:317–326
Wang H, Haas M, Liang M, Cai T, Tian J, Li S, Xie Z (2004) Ouabain assembles signaling cascades through the caveolar Na+/K+-ATPase. J Biol Chem 279:17250–17259
Wang Z, Takemori H, Halder SK, Nonaka Y, Okamoto M (1999) Cloning of a novel kinase (SIK) of the SNF1/AMPK family from high salt diet-treated rat adrenal. FEBS Lett 453:135–139
Wu SJ, Ding L, Zhu JK (1996) SOS1, a genetic locus essential for salt tolerance and potassium acquisition. Plant Cell 8:617–627
Yudowski GA, Efendiev R, Pedemonte CH, Katz AI, Berggren PO, Bertorello AM (2000) Phosphoinositide-3 kinase binds to a proline-rich motif in the Na+, K+-ATPase alpha subunit and regulates its trafficking. Proc Natl Acad Sci U S A 97:6556–6561
Zhu JK, Liu J, Xiong L (1998) Genetic analysis of salt tolerance in Arabidopsis. Evidence for a critical role of potassium nutrition. Plant Cell 10:1181–1191
Zhu JK (2000) Genetic analysis of plant salt tolerance using Arabidopsis. Plant Physiol 124:941–948
Zhu JK (2001) Plant salt tolerance. Trends Plant Sci 6:66–71
Verslues PE, Batelli G, Grillo S, Agius F, Kim YS, ZhuJ Agarwal M, Katiyar-Agarwal S, Zhu JK (2007) Interaction of SOS2 with nucleoside diphosphate kinase 2 and catalases reveals a point of connection between salt stress and H2O2 signaling in Arabidopsis thaliana. Mol Cell Biol 27:7771–7780
Efendiev R, Bertorello AM, Zandomeni R, Cinelli AR, Pedemonte CH (2002) Agonist-dependent regulation of renal Na+, K+-ATPase activity is modulated by intracellular sodium concentration. J Biol Chem 277:11489–96
Acknowledgments
The critical reading and suggestions of Dr A. Jaitovich are greatly appreciated. Work in the Bertorello lab has been supported in part by funds from the Swedish Research Council (32X-10860, 65X-10860, and 32P-14879), the Swedish Heart and Lung Foundation, and the Swedish Kidney Research Fund. Work in the Zhu lab has been supported by USDA, NSF, and NIH.
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License (https://creativecommons.org/licenses/by-nc/2.0), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
Bertorello, A.M., Zhu, JK. SIK1/SOS2 networks: decoding sodium signals via calcium-responsive protein kinase pathways. Pflugers Arch - Eur J Physiol 458, 613–619 (2009). https://doi.org/10.1007/s00424-009-0646-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00424-009-0646-2