
Abstract
Purpose
This review provides a current perspective on the mechanism of vitamin D on skeletal muscle function with the emphasis on oxidative stress, muscle anabolic state and muscle energy metabolism. It focuses on several aspects related to cellular and molecular physiology such as VDR as the trigger point of vitamin D action, oxidative stress as a consequence of vitamin D deficiency.
Method
The interaction between vitamin D deficiency and mitochondrial function as well as skeletal muscle atrophy signalling pathways have been studied and clarified in the last years. To the best of our knowledge, we summarize key knowledge and knowledge gaps regarding the mechanism(s) of action of vitamin D in skeletal muscle.
Result
Vitamin D deficiency is associated with oxidative stress in skeletal muscle that influences the mitochondrial function and affects the development of skeletal muscle atrophy. Namely, vitamin D deficiency decreases oxygen consumption rate and induces disruption of mitochondrial function. These deleterious consequences on muscle may be associated through the vitamin D receptor (VDR) action. Moreover, vitamin D deficiency may contribute to the development of muscle atrophy. The possible signalling pathway triggering the expression of Atrogin-1 involves Src-ERK1/2-Akt- FOXO causing protein degradation.
Conclusion
Based on the current knowledge we propose that vitamin D deficiency results from the loss of VDR function and it could be partly responsible for the development of neurodegenerative diseases in human beings.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
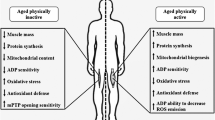
The last decade brought a tremendous number of studies on vitamin D function in human body. In fact, those studies have begun in 1822 when the Polish physician Dr. Jedrzej Sniadecki discovered that the lack of sunlight exposure directly contributes to the onset of rickets (Mozolowski 1939). Later, in 1918 Sir Edward Mellanby showed that nutritional intervention with cod liver oil may replace sunlight in the cure and prevention of rickets (Mellanby 1918). Next, Dr. Elmer McCollum et al. officially termed this nutritional factor as vitamin D (McCollum et al. 1922). The discovery of vitamin D receptor (VDR) (Haussler et al. 1969), and confirming its presence in various tissues has opened the mechanistic link between vitamin D and the occurrence of many diseases and disorders such as: obesity, a chronic, low-grade inflammatory state which aids in the pathogenesis of insulin resistance, metabolic syndrome, and type II diabetes mellitus, (McGill et al. 2008), cardiovascular risk (Kunadian et al. 2014), Alzheimer’s disease (Littlejohns et al. 2014), depression (Jhee et al. 2017) and cancer (Garland et al. 2006). The presence of VDR was also confirmed in skeletal muscle tissue (Simpson et al. 1985; Bischoff et al. 2001), thereby the studies on musculoskeletal disorders gained the potential to evaluate the mechanistic properties of vitamin D function (Fig. 1). The aim of this review is to present the latest reports on skeletal muscle function and vitamin D status. The current review provides the evidence that deficiency of vitamin D through oxidative stress and disruption of mitochondrial function may affect the development of skeletal muscle atrophy.

Overview of biological functions of vitamin D with the emphasis on skeletal muscle
Vitamin D deficiency
Over the past 2 decades, interest in vitamin D has increased significantly. Requests for serum vitamin D concentration measurements increased between the year 2000 and 2010 by over 80-fold and the number of vitamin D supplements sales has risen several times (Shahangian et al. 2014). Although vitamin D intoxication reports are rare (Galior et al. 2018), it does occur and patients and prescribers should be more cognizant of the proper vitamin D treatment and potential dangers of vitamin D overdose. There has been controversy about what exact 25(OH)D (the sum of 25[OH]D2 and 25[OH]D3 concentrations) define vitamin D deficiency and sufficiency. The latest guidelines indicate the concentration lower than 20 ng/mL (50 nmol/L) as vitamin D deficiency. The majority of studies that included 25(OH)D concentrations to analyze relations between health and the risk of diseases pointed on higher 25(OH)D concentrations, i.e., in the range of 30–50 ng/mL (75–125 nmol/L) or 40–60 ng/mL (100–150 nmol/L), not on 20 ng/mL (50 nmol/L) as the necessary minimal concentration for human well-being (Pludowski et al. 2018). However, the true vitamin D deficiency should consider its bioavailability and, therefore, its binding to vitamin D binding protein (VDBP). VDBP is the primary vitamin D carrier, binding 85–90% of circulating 25(OH)D and 1,25-dihydroxyvitamin D3 and the remaining unbound 25(OH)D is considered bioavailable (either free or bound to albumin). About 10–15% of total 25(OH)D is bound to albumin, in contrast to free 25(OH)D, which accounts for 1% of total circulating vitamin D (Bikle et al. 1986). Since the affinity of albumin to 25(OH)D or 1,25(OH)2D3 is weaker than that of VDBP, the loosely bound fraction and the free fraction comprise bioavailable 25(OH)D (Brown et al. 2012). There might be tremendous personal differences in bioavailable vitamin D for humans with a genetic mutation for VDBP or in VDBP-KD mouse models were shown to have lower 25(OH)D blood level (Fu et al. 2009; Jones et al. 2014). There are also big racial differences in VDBP in the general population to some degree. A recent study demonstrated that although Black people had lower levels of VDBP and serum 25(OH)D (38.9 ± 0.5 nmol/L) than White people (64.4 ± 0.9 nmol/L) the levels of bioavailable 25(OH)D of Black people was similar to those of White people (2.9 ± 0.1 and 3.1 ± 0.1 ng/mL, respectively) (Powe et al. 2013). Vitamin D overdose may lead to vitamin D toxicity. The clinical manifestations of vitamin D toxicity reported in these cases were a consequence of the hypercalcemia and included nausea, vomiting, muscle weakness, polyuria, nephrocalcinosis, and renal failure (Galior et al. 2018).
Vitamin D physiology
To be fully active, 25(OH) D3 (calcidiol, 25-hydroxycholecalciferol) must be hydroxylated in the C-1 position, producing 1α,25(OH)2 D3 (calcitriol, 1,25-dihydroxycholecalciferol). It is known that 1α,25(OH)2 D3 concentration is regulated by two vitamin D3 regulating enzymes, CYP24A1 (cytochrome P450 family 24 subfamily A member 1), and CYP27B1 (25-hydroxyvitamin D-1-α-hydroxylase). In general, excess 1α,25(OH)2 D3 can be converted to its catabolite form 1α,24,25(OH)3 D3 via the CYP24A1 enzyme in which 1α,24,25(OH)3 D3 is less active. In contrast, 25-hydroxyvitamin D-1α-hydroxylase functions to increase 1α,25(OH)2 D3 synthesis from 25(OH) D3 (Srikuea et al. 2016). This enzymatic reactions take place mainly in the kidney, although other cells/tissues express the CYP27B1 and CYP24A1 enzymes as well, particularly in C2C12 cells and mouse primary myotubes (Girgis et al. 2014). Therefore, the expression of the vitamin D3 metabolizing enzymes in skeletal muscle suggests the possible local regulation of vitamin D3 in this extrarenal tissue (Srikuea et al. 2016). In addition to the regulation of the concentration of the active vitamin D3, CYP27B1 is a central regulatory axis of the calcium and phosphate homeostatic systems. CYP27B1 is upregulated by parathyroid hormone (PTH), low Ca2+, and low PO43− levels (Omdahl et al. 1972; DeLuca 1974). Therefore, a negative relationship exists between serum 25(OH) D3 and serum PTH. The threshold of serum 25(OH) D3, where serum PTH starts to rise is about 75 nmol/l according to most studies (Lips 2006).
The study of Abboud and coworkers (Abboud et al. 2013), which examined the concentration and time-dependent effects of calcitriol on the capacity of muscle cells to take up and release 25(OH) D3, showed an evidence that skeletal muscle cells indeed contain a mobile pool of 25(OH) D3 which accumulates from and returns to the extracellular environment. 25(OH) D3 is taken up and retained in the muscle cells by binding to VDBP, which is internalized via membrane megalin and then attached to actin in the cytoplasm, that provide high affinity binding for its specific ligand, 25(OH) D3. Interestingly, the early increase in net uptake of 25(OH)D3 after a short pre-incubation (3 h) and short further incubation (4 h) with calcitriol was associated with a significant increase in VDBP protein in the C2 myotubes, perhaps providing more intracellular binding sites for 25(OH)D3. It is possible that this increase in VDBP might be due to reduced degradation in the cell since the authors observed little VDBP in the incubation medium (Abboud et al. 2018). The study also reports that when C2 cells are differentiated into myotubes, the time-dependent uptake of labelled 25(OH) D3 is 2–3 times higher than in undifferentiated myoblasts and osteoblasts. Additionally, they showed that C2 myotubes released only 32% of the previously accumulated 25(OH) D3 after 4 h as compared to 60% for osteoblasts, and that muscle uptake and retention of 25(OH) D3 are modulated by PTH (Abboud et al. 2017). The authors postulated that if the capacity to hold 25(OH) D3 out of the circulation in skeletal muscle is high when vitamin D status is falling in winter, 25(OH) D3 might be protected from inactivating activity of CYP24A1 in the liver. Furthermore, they hypothesize that-storage and gradual release from muscles would increase the level of circulating 25(OH) D3. This would maintain adequate status during the months when vitamin D supply was low giving skeletal muscles a pivotal role in the maintenance of vitamin D status (Abboud et al. 2013, 2017, 2018). It is important to emphasize that also in this hypothesis VDBP plays an important role in regulating bioavailability of vitamin D yet this time in skeletal muscle cells.
VDR in musculoskeletal system
Many biological functions of the active form of vitamin D3 are mediated by VDR, which is a protein that binds 1α,25(OH)2 D3 effectively at sub-nanomolar concentrations (Haussler et al. 1997; Dusso et al. 2005). 1α,25(OH)2 D3 binds to VDR what leads to the conformational changes that allow VDR to interact with its heterodimeric partner, retinoid X receptor (RXR) (Smith et al. 2004). The complex (i.e.1,25D-VDR-RXR) is translocated to the nucleus and binds to vitamin D response elements (VDRE), which ultimately results in activation of transcription (Haussler et al. 1998). Classic VDRE consist of two hexameric direct repeats with a three-nucleotide linker (Umesono et al. 1991; Carlberg et al. 1993). The cell specificity of the actions of VDR and its ligand 1α,25(OH)2 D3 can be explained in part by VDR’s recognition mode for its genomic binding sites and the tissue-specific differences in the expression of VDR and its key co-factors. Moreover, in contrast to other nuclear receptors such as receptors of cortisol or testosterone, the VDR can bind its genomic targets also in the absence of ligand, i.e. in this respect the functional profile of the VDR is larger than that of its ligand (Polly et al. 2000).
Study on chick myoblasts treated with 1α,25(OH)2 D3 revealed rapid translocation of VDR from the nucleus to the plasma membrane within 5 min after the addition of 1α,25(OH)2 D3 (Capiati et al. 2002). The 1α,25(OH)2 D3-dependent intracellular redistribution of the VDR can be blocked by genistein, herbimycin or colchicine, suggesting the involvement of tyrosine kinase/s and microtubular transport in the relocation of the receptor (Capiati et al. 2002). Studies using a VDR knockout (VDRKO) mouse (Zanello et al. 2004) and a naturally occurring human VDR mutation (Nguyen et al. 2004) unquestionably showed that 1α,25(OH)2 D3-mediated rapid responses require a functional VDR. Despite that, the VDR has been found also in the plasma membrane in caveolae (Norman et al. 2002); therefore, it has been proposed that the VDR activates nongenomic signalling. Interestingly, the identification of an alternative ligand-binding pocket in the nuclear VDR has allowed to generate by computer docking a receptor conformational ensemble model providing an explanation for the VDR genomic and non-genomic functions (Mizwicki et al. 2004).
The VDR gene shows highest expression in metabolic tissues, such as kidneys, bone and intestine, but at least low to moderate expression is found in nearly all other of the approximately 250 human tissues and cell-types (Verstuyf et al. 2010). In situ studies on human skeletal muscles confirm the presence of VDR in this tissue (Bischoff et al. 2001) and documented that expression of VDR is essential for effective uptake of vitamin D by muscle cells (Girgis et al. 2014). Additionally, recent study in VDRKO mouse muscle fibers exposed to calcitriol confirmed that VDR is essential for an uptake of labelled 25(OH) D3 (Abboud et al. 2018).
Tanaka and coworkers (Tanaka et al. 2014) using C2C12 and G58 cells demonstrated that myoblasts require downstream signalling from VDR for differentiation into myocytes and that VDR expression is necessary in skeletal muscles for maintaining muscle volume. In addition, it has been presented that VDRKO mice exhibit abnormal skeletal muscle development (Endo et al. 2003). Moreover, serum 25(OH) D3 levels and the expression of VDR in muscle cells, as well as testosterone, levels, decline with age (Bischoff-Ferrari et al. 2004), which contribute to developing sarcopenia and muscle weakness (Lips et al. 2010). VDR is located predominantly on the fast-twitch muscle fibers, which respond first in rapid actions, thus it is not surprising that vitamin D sufficiency increases muscle strength and coordination, enabling prevention of falls (Suzuki et al. 2008; Holick et al. 2011).
Ceglia and coworkers (Ceglia et al. 2013) showed that 4-month vitamin D supplementation increased intramyonuclear VDR concentration by 30% in nonexercised vastus lateralis muscle in the older, mobility-limited, vitamin D-insufficient women. Although, as mentioned before, VDR is predominantly expressed in fast twitch muscles, a study on human paraspinal, slow twice muscle shows that vitamin D deficiency induces its atrophy and decreases the concentration of intramyonucelar VDR and VDR gene expression level (Bang et al. 2018). Also, the study on chronic obstructive pulmonary disease mice model shows that VDR expression in both EDL (extensor digitorum longus) and soleus muscles was reduced in vitamin D-deficient mice as compared with mice with normal vitamin D levels and that the reduction in VDR expression with vitamin D deficiency was more pronounced in the soleus muscle (− 57%) compared with the EDL muscle (− 37%) (Cielen et al. 2016). This data confirms the relationship between serum vitamin D concentration and intramyonuclear VDR concentration, regardless the type of muscle. However, when the disturbed signalling of 1,25(OH)2D3 is explored, it must be considered that the deficiency of vitamin D and the loss of the VDR have some similar but partly meaningful consequences.
Although in many studies VDR has been shown to be necessary for vitamin D function, numerous non classic sites have been proven to act as VDRE (Girgis et al. 2013). Also, non-genomic effects of vitamin D, characterized by rapid activation followed by other complex pathways of intracellular signal transduction after binding of 1,25(OH)D3 to its non-nuclear receptor (Losel et al. 2003; Girgis et al. 2013; Owens et al. 2015) have been reported. Interestingly not only vitamin D itself, but also essential oils (caraway, coriander, dill, ginger, lemongrass, oregano, spearmint, thyme, turmeric and verveine) exhibit the ability to modulate VDR activity (Bartonkova et al. 2018). Intriguingly, essential oils of turmeric, oregano, dill, caraway, verveine and spearmint augmented the activity of both VDR and glucocorticoid receptor (GR) (Bartonkova et al. 2018). The concentrations of essential oils used in this study are naturally occurring in foods and drinks (Usjak et al. 2017). Non-genomic action of vitamin D, diversity in VDR regulation and the presence of numerous VDRE sites widens the range of possible explanations for the mechanism of vitamin D function in the human body and skeletal muscle.
VDR knockout and vitamin D deficiency conditions seem to clearly indicate negative consequences for skeletal muscle homeostasis. Notwithstanding, the overexpression of VDR seems to have damaging consequences on skeletal muscle as well. The FokI polymorphism of VDR gene is a T/C transition in the second exon, resulting in a truncated protein (424aa instead of 427aa) with enhanced transactivation capacity (Whitfield et al. 2001). Two studies in humans suggest that FokI polymorphism is associated with decreased skeletal muscle mass and strength. In particular, Roth and coworkers showed that FokI homozygous men display a low fat-free muscle mass and risk of sarcopenia 2.2-fold higher than controls (Roth et al. 2004). The other study demonstrates that homozygosity for the FokI polymorphism is associated with reduced quadriceps strength as compared with heterozygosity or control patients (Hopkinson et al. 2008). Latest reports show that VDR expression in C2C12 cells is high at the beginning of the differentiation process and is progressively reduced until the cells complete their maturation into myotubes. This observation is consistent with previous data reported that mean intracellular VDR content is higher in undifferentiated than in differentiated cells (Kong et al. 2006). In this regard, VDR down-regulation may represent a condition required to achieve complete myogenic differentiation. The presence of several VDRE in the promoter region of the myogenin gene and the demonstration that VDR may directly bind (in absence of the administration of vitamin D) the myogenin promoter support the proposed mechanism of regulation: the hypothesis of a ligand-independent, VDR-mediated, negative regulation of myogenin transcription. This hypothesis is supported by the results showing that animals administered overdosed of vitamin D display an impaired muscle regeneration that is associated with increased VDR expression (Camperi et al. 2017). Therefore, taking into account differences in undifferentiated and matured myotubes, as well as in recovering muscle cells in the manner of VDR requirements it seems that the solution for the skeletal muscle maintenance lays between vitamin D/VDR deficiency and its overexpression. Moreover, there should be a different approach towards vitamin D supplementation for children and adolescents whose muscles are in the development stage, for athletes requiring recovery, and for elderly people.
Vitamin D relationships to oxidative stress and cellular metabolism in skeletal muscle: data from observational studies
Among many newly discovered functions of vitamin D its involvement in calcium (Ca2+) homeostasis seems to be undeniable. Vitamin D regulates calcium absorption in the gut and maintenance of serum calcium and phosphate concentrations (Gil et al. 2018). Vitamin D was shown to be also involved in cellular metabolism of skeletal muscle, yet precise basis for the molecular mechanisms activated by vitamin D in muscles is unclear. Vitamin D action in skeletal muscle affects calcium (Ca2+) homeostasis which is an important factor in interplay between cytosol and mitochondria which is involved in muscle energy metabolism (Glancy et al. 2012). Vitamin D, through the activity of its active metabolite, 1α,25(OH)2 D3, is essential for normal calcium (Ca2+) and phosphorus balance and the maintenance of skeletal health (DeLuca 2004; Haussler et al. 2008). It has been shown to play an important role in the regulation of skeletal muscle tone and contraction (Li et al. 2018). Vitamin D deficiency is known to alter muscle contraction kinetics by reducing Ca2+ reuptake into the sarcoplasmic reticulum, thereby leading to a prolongation of the relaxation phase of muscle contraction (Rodman et al. 1978; Zittermann 2003). Under physiological conditions mitochondria in skeletal muscle fibers uptake cytoplasmic Ca2+ released from the sarcoplasmic reticulum during twitch and tetanic responses (Rudolf et al. 2004). Thus, the experiments on vitamin D-deficient chick muscles demonstrated the alterations in oxidative phosphorylation and an inability of muscle mitochondria to retain Ca2+ (Mukherjee et al. 1981). Therefore, vitamin D deficiency may be responsible for inadequate Ca2+ uptake by the mitochondria which results in the perturbations of cellular metabolic homeostasis (Sinha et al. 2013).
Latest study of Ryan and coworkers (Ryan et al. 2016) demonstrated increased oxygen consumption rate (OCR) of skeletal muscle cells after treatment with1α,25(OH)2 D3, indicating vitamin D action in the regulation of mitochondrial oxygen consumption and dynamics. In particular, this study showed that respiration coupled to the generation of ATP was increased, which suggests that vitamin D increases the function of mitochondria in muscle. However, direct treatment of isolated mitochondria with 1α,25(OH)2 D3 failed to increase OCR suggesting that the effects of 1α,25(OH)2 D3 on OCR might be VDR-dependent or other extra-mitochondrial biochemical events (Ryan et al. 2016). It is important to mention that the treatment with inactive form of vitamin D, 25(OH) D3, did not influence the OCR in isolated mitochondria, which suggested that vitamin D and 25(OH) D3 will not be useful in the treatment of muscle weakness unless they are metabolized to 1α,25(OH)2 D3. In fact, that is operative in the context of vitamin D deficiency where high PTH levels drive the rapid metabolism of 25(OH)D3 to 1α,25(OH)2 D3.
The vitamin D influence on mitochondria was also reported by Sinha and coauthors who showed that treatment of vitamin D deficient humans with cholecalciferol improves the maximal mitochondrial oxidative phosphorylation rate (τ1/2PCr and τ1/2ADP recovery times were reduced) measured by 31P-NMR spectroscopy (Sinha et al. 2013). Oxidative phosphorylation rate is a function that reflects a composite of mechanisms including mitochondrial number, oxidative enzyme content, mitochondrial components, and vascular supply of substrates and oxygen (Kemp et al. 1993). Moreover, it was reported that the eradication of vitamin D deficiency was associated with an improvement in symptoms of myopathy and fatigue in all participants (Sinha et al. 2013). We found that mitochondrial function was improved in skeletal muscle of patients with low back pain (LBP) supplemented with vitamin D (3200 U/day x 5 weeks). The activity of citrate synthase was approximately 40% higher in the paraspinal muscle after supplementation. Also it was observed higher protein content of PGC-1α, a transcriptional coactivator (unpublished data). The interplay between vitamin D-VDR, reactive oxygen species (ROS) signalling, and the antioxidant system is complex. To the best of our knowledge our group was first demonstrated that vitamin deficiency D increased the cytotoxicity mediated by ROS (Dzik et al. 2018). Taken together, it is very likely that vitamin D deficiency in the long run induces VDR ablation, ROS generation and in consequence deleterious effects on the mitochondrial function, which in turn leads to elevated muscle atrophy (Fig. 2).

The graphical abstract of the vitamin D action in the skeletal muscle in vitamin D deficiency conditions. Vitamin D deficiency decreases IGF-1 and PGC-1α via VDR—the nuclear receptor. Src/ERK1/2/Akt/FOXO3a signalling cascade triggers the muscle atrophy through Murf-1 and MaFbx. Vitamin D deficiency increases oxidative stress and attenuates mitochondrial biogenesis and function. Akt serine/threonine-specific protein kinase, ERK 1/2 extracellular signal-regulated kinases 1 and 2, FOXO forkhead box protein, IGF-1 insulin-like growth factor 1, MaFbx muscle atrophy F-box protein, mTOR mammalian target of rapamycin kinase, MuRF1 muscle ring finger protein, OCR oxygen consumption rate, PGC-1α peroxisome proliferator-activated receptor gamma coactivator 1-alpha, ROS reactive oxygen species, RXR retinoid X receptor, Src steroid receptor coactivator complex, VDR vitamin D receptor, VDRE vitamin D response elements
Mitochondria not only play an important role in cellular energy metabolism, but they also are a source of ROS. Although mitochondria are not always considered as the main producer of ROS in the cell (NAD(P)H oxidase or xanthine oxidase being able to produce high levels of ROS), the electron transport chain produces ROS continuously (Panel et al. 2018). Therefore, given the data of vitamin D effect on mitochondrial function, the aspect of oxidative stress in skeletal muscle regarding vitamin D deficiency is of great value. Recent study on patients with chronic LBP showed that vitamin D deficiency increases antioxidative enzymes activities (Cu/ZnSOD and GPx) in paraspinal muscle and leads to elevated lipid and protein peroxidation. Moreover, this data demonstrate that five week vitamin D supplementation increases serum vitamin D concentration in LBP patients and decreases oxidative stress in skeletal muscle (Dzik et al. 2018). Similar findings reported increased protein oxidation and nitrosative stress and reduced activities of the antioxidant enzymes (Bhat et al. 2015) as well as increased lipid peroxidation in the muscles of vitamin D-deficient rats (Cielen et al. 2016). Furthermore, another study indicated that rats treated with vitamin D showed reduced tissue damage and attenuated oxidative stress after exhaustive exercise (Ke et al. 2016). This data supports the thesis that vitamin D is involved not only in calcium homeostasis and mitochondrial function but also is responsible for oxidative stress in skeletal muscle.
The exact mechanism which might explain the regulation of oxidative stress via vitamin D is not yet elucidated. As previously mentioned, vitamin D regulates mitochondrial dynamics and function, therefore, it might directly influence the mitochondrial ROS generation. However, it is still a manner of debate if the observed reduction of oxidative stress in skeletal muscle is a result of altered mitochondrial function or it may possibly involve any other capacity of vitamin D action in human body. There are studies demonstrating that vitamin D is a very effective antioxidant. It was shown that vitamin D has a capacity to inhibit zinc-induced oxidative stress in the central nervous system which is 103 times higher than vitamin E analogues (Lin et al. 2005). In addition, another study shows that a vitamin D analogue exerts antioxidant effects by activating the Nrf2-Keap1 antioxidant pathway (Nakai et al. 2014). Nevertheless, vitamin D or vitamin D analogs are able to limit oxidative stress in animals (Hamden et al. 2009; Husain et al. 2009) and humans (Tanaka et al. 2011). Moreover, serum vitamin D concentration correlates with oxidative stress in asthmatic children (Igde et al. 2018) and is associated with adiposity in schoolchildren, suggesting that vitamin D deficiency potentially increases the risk for diseases caused by higher adiposity and oxidative stress (Zhang et al. 2014). For example, adipose tissue releases pro-inflammatory cytokines, resulting in chronic inflammation, which may induce oxidative stress and in consequence leads to muscle damage.
Despite the fact that oxidative stress undeniably has a devastating effect on human body, the importance of ROS and reactive nitrogen species as signals in the skeletal muscle adaptation to exercise is now evident (Merry et al. 2016). There are numerous studies reporting negative consequences of antioxidant supplementation in regard of skeletal muscle function, particularly it was shown to impair mitochondrial biogenesis (Gomez-Cabrera et al. 2008; Ristow et al. 2009), to reduce post-exercise insulin sensitivity (Trewin et al. 2015), as well as to attenuate performance improvements (Braakhuis et al. 2015). In a similar manner, the supraphysiological dose of 1α,25(OH)2D3 injected into damaged muscle (days 4–7 after BaCl2 treatment) delays the regenerative response in muscle namely, decreases satellite cell differentiation, delays regenerative muscle fiber formation, and increases muscular fibrosis (Srikuea et al. 2016). Moreover, as previously mentioned, the study on C2C12 and primary myoblasts clearly shows that vitamin D treatment at supraphysiological dose causes VDR overexpression and impairs their differentiation into mature myotubes (Camperi et al. 2017). These observations suggest that while vitamin D deficiency enhances oxidative stress, overcorrection of vitamin D status may also have a negative impact on skeletal muscle in the same manner as antioxidants while overdosed.
Vitamin D signalling with anabolic/catabolic pathways
An increasing body of knowledge suggests the involvement of vitamin D in both anabolic and catabolic pathways in skeletal muscle. One of the reasons of muscle wasting results because of an altered balance in the protein degradation and synthesis rates. Thus, there are three major proteolytic pathways described in the skeletal muscle, namely: the ATP-ubiquitin-dependent system, the lysosomal system, and the cytosolic calcium-activated system (Kandarian et al. 2006). Only, the ATP-ubiquitin-dependent system has been shown to be dependent on vitamin D (Bhat et al. 2013). The study on diet-induced vitamin D deficiency in rats reports no alternation in lysosomal and calpain enzyme activities in vitamin D deficiency-induced muscle wasting. However, it shows a significant increase in the enzymes activities of the 20S proteasome catalytic core (Bhat et al. 2013). 20S proteosomal subunit are catalytic part of the 26S proteasome that functions as the key role in nonlysosomal protein degradation (Tawa et al. 1997). Moreover, the study of Bhat and coworkers has been shown an increase in the expression of E2- ubiquitin conjugating enzyme and ubiquitin conjugates in vitamin D deficiency muscle as well as an increase in the expression of 2 muscle-specific E3 ligases. Atrogin-1 also known as MaFbx (muscle atrophy F-box protein) and MuRF1 (muscle ring finger protein) which were increased by twofold in the vitamin D deficient muscle as compared with control (Bhat et al. 2013). It was postulated that Atrogin-1 and MuRF1, which provide substrate specificity in ATP-dependent ubiquitin proteasome pathway (UPP) responsible for intracellular proteolysis are critical for the development of muscle atrophy.
Recent study on mice reports that prolonged, 12 months, vitamin D insufficiency induces characteristics of sarcopenia that include poor anaerobic capacity, lower lean mass, and a trend towards smaller fast twitch fiber cross-sectional area, as well as gait disturbance. Moreover, this study shows that vitamin D insufficient mice also exhibited increased expression of atrophy-associated Atrogin-1 and differential expression of muscle regulation associated miR-26a when compared to mice with normal vitamin D level (Sleeman et al. 2017). This data strongly suggest that vitamin D insufficiency/deficiency is involved in muscular atrophy development, yet the exact mechanism still needs to be explicated.
Hitherto studies postulated that the possible signalling pathway involved in muscle vitamin D function might require steroid receptor coactivator complex (Src), non-receptor tyrosine kinase, which has been shown to activate mitogen-activated protein kinases (MAPK) (Li et al. 2006; Thobe et al. 2006) in various tissues. There are studies that demonstrate fast non-genomic Src activation by vitamin D in various cell types, including skeletal muscle myoblasts (Chappel et al. 1997; Gniadecki 1998; Khare et al. 1999; Buitrago et al. 2001a, b). Furthermore, there is evidence that vitamin D induces rapid association of VDR with Src in skeletal muscle and osteoblastic cells (Buitrago et al. 2000; Vertino et al. 2005). The study of Buitrago and coworkers (Buitrago et al. 2001a, b) reported that the activation of nonreceptor tyrosine kinase Src coincides with a 1,25(OH)2 D3-induced interaction between Src kinase and VDR in chick muscle cells. Src activation is required for vitamin D-dependent activation of extracellular signal-regulated kinases 1 and 2 (ERK 1/2) and p38 MAPK in skeletal muscle myoblasts (Buitrago et al. 2001a, b, 2006). MAPK signalling is necessary for the maintenance of skeletal muscle mass because inhibition of these signalling cascades elicits muscle atrophy in vitro and in vivo. Particularly, the study on cultured myocytes show that inhibition of ERK1/2 signalling induce myotube atrophy and cause the upregulation of atrophic markers Atrogin-1 and MuRF1 and downregulates the phosphorylation of Akt and its downstream kinases (Shi et al. 2009). Akt along with its downstream signal cascades has been identified as pivotal regulators of muscle hypertrophy by enhancing protein synthesis and concomitant repression of protein breakdown (Sandri et al. 2004; Stitt et al. 2004). It has been reported that 1,25(OH)2 D3-induced Akt activation in skeletal muscle myoblasts was mediated by Src (Buitrago et al. 2012).
ERK signalling is also suggested to mediate the hypertrophic effects of IGF-I, a muscle hypertrophy factor (Haddad et al. 2004). The IGF-1 is well described as far as it concerns its circulating level. Wei and coworkers have shown that IGF-1 caused an increase in the blood levels of 1,25(OH)2D3, the hormonally active vitamin D metabolite, by stimulating the expression and activity of the hydroxylase-1α that produces 1α,25(OH)2 D3 in the kidney (Wei et al. 1998). Moreover, when vitamin D was administered to adult human, IGF-1 serum levels were increased (Ameri et al. 2013). On the other hand, another study reported that 1 year of high-dose vitamin D supplementation did not significantly alter serum IGF-1 among women at high risk in breast cancer (Crew et al. 2015) nor in pre-diabetes patients (Sinha-Hikim et al. 2015).
Recently, Hayakawa and coworkers showed that IGF-1 is not directly affected by 1,25(OH)2D3 in skeletal muscle. They suggested that vitamin D stimulated IGF-1 production in tissues other than skeletal muscle and that the induced IGF-1 circulates in the blood, and exert hypertrophic effects on muscle tissue or supportive effects on muscle function (Hayakawa et al. 2015). It was reported that IGF-1 signalling induces anabolism, being upregulated during resistance exercise, and this influences the IGF- 1–Akt–FOXO pathway (Perrini et al. 2010; Banerjee et al. 2013; Bonaldo et al. 2013). The major signalling pathways that regulate the size of myofibres are the IGF-1– Akt– FOXO pathway, myostatin, NFκ B and glucocorticoids (Fielding et al. 2011; Bonaldo et al. 2013). In the IGF-1– Akt– FOXO pathway, Akt controls protein synthesis via mTOR, and protein degradation via transcription factors of the FOXO family. FoxO also signals between protein breakdown and synthesis, with FOXO3 playing a role in suppression of protein synthesis, and Akt plays a role in anabolism by suppressing protein breakdown (Bonaldo et al. 2013). FoxO are major regulators of the ubiquitin proteasome system acting by directly regulating muscle-specific E3 ligases.
Recent study showed that VDR signalling enhanced by vitamin D treatment inhibited FOXO1 expression, nuclear translocation, and activity in C2C12 muscle cells. The vitamin D-dependent suppression of FOXO1 activation disappeared when VDR was knocked down. These results suggest that FOXO1 is a major target mediating VDR-null signalling in skeletal muscle including the progression of muscle atrophy (Chen et al. 2016). Hence, FOXO transcription factors are thought to control half of the genes identified in the molecular “common atrophy blueprint” present in different atrophy types (Sandri et al. 2004). Akt, a protein kinase B, which is important in signalling pathways is involved in the protein synthesis and skeletal muscle growth (Schiaffino et al. 2011). Akt blocks the function of the FOXO3 by phosphorylation of conserved residues, leading to their sequestration in the cytoplasm away from target genes (Brunet et al. 1999). Phosphorylated FOXO3a does not translocate to the nuclei, and consequently the expression of MAFbx and MuRF, both target genes of FOXO, are inhibited. It is important to note that elevated PGC-1α content, besides its function in mitochondrial biogenesis, prevents transcriptional activity of FOXO3a (Sandri et al. 2006), therefore the mitochondria might be involved in the atrophy progression. Besides the involvement in the progression of muscle atrophy Akt may also regulate muscle synthesis via mTOR. The study of Salles and coworkers (Salles et al. 2013) 1α,25(OH)2D3 sensitizes the Akt/mTOR-dependant pathway to the stimulating effect of leucine and insulin, resulting in a further activation of protein synthesis in murine C2C12 skeletal myotubes.
Although vitamin D deficiency has been shown to lead to muscle atrophy both in animals and humans (Endo et al. 2003; Dhesi et al. 2004; Sato et al. 2005; Snijder et al. 2006) the problem of muscle atrophy seems to be more complicated and may not be fully solved with vitamin D alone. While the studies on vitamin D deficiency seem to undoubtedly connect the low level of vitamin D with the progression of muscle atrophy, it is necessary to mention that the propriety to prevent muscle atrophy is not only typical for vitamin D. Catechins and other antioxidants possess the ability to prevent, mitigate, delay, and even treat muscle-related disorders caused by aging and diseases as well (Li et al. 2019). It was reported that epigallocatechin gallate protects the skeletal muscle mitochondria (Oliveira et al. 2016) reduces skeletal muscle oxidative stress in non-obese diabetic rats and restores the content of complex I and voltage-dependent anion selective channel protein 1 (VDAC1) to improve the function of mitochondria (Yan et al. 2012). Epigallocatechin gallate may also decrease the protein degradation rate caused by muscle atrophy, increase the expression of anabolic factors and promote the cross-sectional area of muscle fiber (Mirza et al. 2014; Meador et al. 2015). Catechins were also shown to promote the differentiation of myoblasts (Kim et al. 2017). It is necessary to mention that the changes triggered by catechins involve the same pathways that we previously described to be engaged in vitamin D action in skeletal muscle. Epigallocatechin gallate was shown to promote the phosphorylation of Akt, inhibit the activation of FOXO, prevent nuclear accumulation and reduce the degradation of muscle protein (Bartholome et al. 2010). Epicatechin has been reported to activate Erk1/2 and p38 MAPK, suggesting its potential role in promoting cell survival (Deng et al. 2012). The recent study conducted on human, mice and C2C12 myoblasts demonstrate that the improvement in the glutathione (GSH) status exerts measurable and beneficial effects on both mRNA and protein expression levels of VDBP, VD-25-hydroxylase, VDR as well as PGC-1a/GLUT 4 (Jain et al. 2018). GSH is a major antioxidant and a cofactor of many enzymes in the human body (Franco et al. 2012) and the authors suggest that GSH status positively upregulates the bioavailability of 25(OH)VD. This may explain why consumption of food rich in l-cysteine/methionine and GSH, such as milk and leafy vegetables, can increase the bioavailability of vitamin D and improve the quality of life, while the studies on vitamin D itself are not always consistent.
Intriguingly, glucocorticoids were shown to increase VDR expression, particularly, dexamethasone (Dex) was reported to potentate calcitriol effects by increasing VDR. Treatment of squamous cell carcinoma VII cells with Dex produces an important increase of VDR transcripts. Similar effects have been observed in mouse adipocytes and human breast cancer cell lines. The VDR gene contains a number of putative glucocorticoid response elements. Rapid increase in VDR transcript levels may indicate glucocorticoids directly induce VDR de novo transcription (Hidalgo et al. 2010). Also, the study in human monocytes shows that vitamin D enhances glucocorticoid action (Zhang et al. 2013). However, glucocorticoids are known to cause both muscle wasting and decreased bone formation (Ziegler et al. 1998), so the opposite effect to the vitamin D action in skeletal muscle. Vitamin D is considered a true steroid hormone, and like glucocorticoids and gonadal hormones, may exert several immunomodulatory activities (Cutolo et al. 2014). In fact, vitamin D is recognized as an anti-inflammatory agent (Calton et al. 2015), thus vitamin D may influence bioenergetics through inflammatory mechanisms, whereas glucocorticoids rise in the stress condition (Kudielka et al. 2010). Also, our recent study showed that the elevated plasma corticosterone concentration due to chronic stress response was associated with increased oxidative stress in skeletal muscle (Karnia et al. 2018) and the progression of muscle atrophy (data not shown) in rats. Also, cross-sectional analysis of the National Health and Nutrition Examination Survey 2001–2006 report demonstrated that the odds of having vitamin D deficiency were twice as likely in patients who reported glucocorticoids use compared with those without steroid use (Skversky et al. 2011). The different action of vitamin D and glucocorticoids might be due to different concentration, and the right level of vitamin D deficiency and toxicity should be clearly defined in order to alleviate the possible negative glucocorticoid action in skeletal muscle.
Summary
As summarized in Table 1. recently published data indicates that vitamin D deficiency is associated with lower VDR content, increased oxidative stress and altered the activity of antioxidant enzymes in skeletal muscle. Moreover, it is shown that vitamin D deficiency may induce paraspinal muscle atrophy and decreases the concentration of intramyonucelar VDR and gene expression of VDR. In addition, it is also reported that vitamin D regulates mitochondrial oxygen consumption and dynamics. Namely, vitamin D deficiency decreases oxygen consumption rate and induces disruption of mitochondrial function. Taken together, it is very likely that vitamin D deficiency in the long run induces VDR ablation, ROS generation and in consequence deleterious effects on the mitochondrial function, which in turn leads to elevated muscle atrophy. The possible signalling pathway that triggers the expression of Murf1 and MaFbx (markers of muscle atrophy) may involve Src-ERK1/2-Akt- FOXO. In addition, it should be stressed that the dysfunctions of mitochondrial respiratory chain and dangerous ROS generation are crucial factors in human pathologies, especially in neurodegenerative diseases where muscle atrophy is observed. We assume that vitamin D deficiency results from the loss of VDR function and it could be partly responsible for the development of neurodegenerative diseases in human beings. However, the correction of vitamin D deficiency should be done wisely in order to avoid negative consequences of VDR overexpression and vitamin D toxicity. Vitamin D supplementation should be addressed towards bioavailability of vitamin D and towards personal requirements that may differ between children, athletes, adults and elderly people. Also, the changes in diet in regard of antioxidants, GSH precursors and essential oils supply should be considered as the support for vitamin D treatment.
Future directions
Given the important action of vitamin D on skeletal muscle tissue, a better understanding of the mechanisms involved in muscle atrophy is needed. In particular, there is a great need of a new insight into VDR expression and activation, biogenesis and the function of mitochondria as well as signalling pathways associated with progressive muscle atrophy in vitamin D deficiency. On the other hand, beneficial effect of normalized serum vitamin D concentration should be explored in regard to muscle aerobic energy metabolism, oxidative stress and prevention of muscle atrophy. Even more, we suppose that supplementation with vitamin D to sufficient serum vitamin D level will: reduce ROS overproduction, increase VDR gene expression and protein content, improve mitochondrial function and inhibit the atrophy of muscle. Finally, the broadened knowledge about vitamin D mechanism(s), may contribute to the reduced progression of neurodegenerative diseases in humans.
Abbreviations
- Akt:
-
Serine/threonine-specific protein kinase
- Cu/ZnSOD:
-
Copper/zinc-dependent dismutase
- CS:
-
Citrate synthase
- CYP24A1:
-
Cytochrome P450 family 24 subfamily A member 1
- CYP27B1:
-
25-hydroxyvitamin D-1-α-hydroxylase
- ERK 1/2:
-
Extracellular signal-regulated kinases 1 and 2
- FOXO1:
-
Forkhead box protein O1
- FOXO3:
-
Forkhead box protein O3
- GPx:
-
Glutathione peroxidase
- GR:
-
Glucocorticoid receptor
- IGF-1:
-
Insulin-like growth factor 1
- LBP:
-
Low back pain
- MaFbx:
-
Muscle atrophy F-box protein
- MAPK:
-
Mitogen-activated protein kinases
- MnSOD:
-
Manganese-dependent superoxide dismutase
- mTOR:
-
Mammalian target of rapamycin kinase
- MuRF1:
-
Muscle ring finger protein
- OCR:
-
Oxygen consumption rate
- PGC-1α:
-
Peroxisome proliferator-activated receptor gamma coactivator 1-alpha
- PTH:
-
Parathyroid hormone
- PLIF:
-
Posterior lumbar interbody fusion
- ROS:
-
Reactive oxygen species
- RXR:
-
Retinoid X receptor
- Src:
-
Steroid receptor coactivator complex
- VDBP:
-
Vitamin D binding protein
- VDR:
-
Vitamin D receptor
- VDRE:
-
Vitamin D response elements
References
Abboud M, Puglisi DA, Davies BN, Rybchyn M, Whitehead NP, Brock KE et al (2013) “Evidence for a specific uptake and retention mechanism for 25-hydroxyvitamin D (25OHD) in skeletal muscle cells.” Endocrinology 154(9): 3022–3030
Abboud M, Rybchyn MS, Liu J, Ning Y, Gordon-Thomson C, Brennan-Speranza TC et al (2017) “The effect of parathyroid hormone on the uptake and retention of 25-hydroxyvitamin D in skeletal muscle cells”. J Steroid Biochem Molecular Biol 173:173–179
Abboud M, Rybchyn MS, Ning YJ, Brennan-Speranza TC, Girgis CM, Gunton JE et al (2018) “1,25-Dihydroxycholecalciferol (calcitriol) modifies uptake and release of 25-hydroxycholecalciferol in skeletal muscle cells in culture”. J Steroid Biochem Molecular Biol 177:109–115
Ameri P, Giusti A, Boschetti M, Bovio M, Teti C, Leoncini G et al (2013) “Vitamin D increases circulating IGF1 in adults: potential implication for the treatment of GH deficiency”. Eur J Endocrinol 169(6):767–772
Banerjee A, Apponi LH, Pavlath GK, Corbett AH (2013) “PABPN1: molecular function and muscle disease”. FEBS J 280(17):4230–4250
Bang WS, Lee DH, Kim KT, Cho DC, Sung JK, Han IB et al (2018) “Relationships between vitamin D and paraspinal muscle: human data and experimental rat model analysis”. Spine J 18(6):1053–1061
Bartholome A, Kampkotter A, Tanner S, Sies H, Klotz LO (2010) Epigallocatechin gallate-induced modulation of FoxO signaling in mammalian cells and C. elegans: FoxO stimulation is masked via PI3K/Akt activation by hydrogen peroxide formed in cell culture. Arch Biochem Biophys 501(1):58–64
Bartonkova I, Dvorak Z (2018) “Assessment of endocrine disruption potential of essential oils of culinary herbs and spices involving glucocorticoid, androgen and vitamin D receptors”. Food Funct 9(4):2136–2144
Bhat M, Ismail A (2015) Vitamin D treatment protects against and reverses oxidative stress induced muscle proteolysis. J Steroid Biochem Mol Biol 152:171–179
Bhat M, Kalam R, Qadri SS, Madabushi S, Ismail A (2013) Vitamin D deficiency-induced muscle wasting occurs through the ubiquitin proteasome pathway and is partially corrected by calcium in male rats. Endocrinology 154(11): 4018–4029
Bikle DD, Gee E, Halloran B, Kowalski MA, Ryzen E, Haddad JG (1986) Assessment of the free fraction of 25-hydroxyvitamin D in serum and its regulation by albumin and the vitamin D-binding protein. J Clin Endocrinol Metabol 63(4):954–959
Bischoff HA, Borchers M, Gudat F, Duermueller U, Theiler R, Stahelin HB et al (2001) “In situ detection of 1,25-dihydroxyvitamin D3 receptor in human skeletal muscle tissue”. Histochem J 33(1):19–24
Bischoff-Ferrari HA, Borchers M, Gudat F, Durmuller U, Stahelin HB, Dick W (2004) Vitamin D receptor expression in human muscle tissue decreases with age. J Bone Miner Res 19(2):265–269
Bonaldo P, Sandri M (2013) Cellular and molecular mechanisms of muscle atrophy. Dis Model Mech 6(1):25–39
Braakhuis AJ, Hopkins WG (2015) “Impact of dietary antioxidants on sport performance. A review. Sports Med 45(7):939–955
Brown AJ, Coyne DW (2012) Bioavailable vitamin D in chronic kidney disease. Kidney Int 82(1):5–7
Brunet A, Bonni A, Zigmond MJ, Lin MZ, Juo P, Hu LS et al (1999) Akt promotes cell survival by phosphorylating and inhibiting a Forkhead transcription factor. Cell 96(6):857–868
Buitrago C, Vazquez G, De Boland AR, Boland RL (2000) Activation of Src kinase in skeletal muscle cells by 1, 1,25-(OH(2))-vitamin D(3) correlates with tyrosine phosphorylation of the vitamin D receptor (VDR) and VDR-Src interaction. J Cell Biochem 79(2):274–281
Buitrago C, Boland R, de Boland AR (2001a) The tyrosine kinase c-Src is required for 1,25(OH)2-vitamin D3 signalling to the nucleus in muscle cells. Biochimica et biophysica acta 1541(3):179–187
Buitrago C, Vazquez G, De Boland AR, Boland R (2001b) “The vitamin D receptor mediates rapid changes in muscle protein tyrosine phosphorylation induced by 1,25(OH)(2)D(3). Biochem Biophys Res Commun 289(5): 1150–1156
Buitrago CG, Ronda AC, de Boland AR, Boland R (2006) “MAP kinases p38 and JNK are activated by the steroid hormone 1alpha,25(OH)2-vitamin D3 in the C2C12 muscle cell line”. J Cell Biochem 97(4):698–708
Buitrago CG, Arango NS, Boland RL (2012) “1alpha,25(OH)2D3-dependent modulation of Akt in proliferating and differentiating C2C12 skeletal muscle cells”. J Cell Biochem 113(4):1170–1181
Calton EK, Keane KN, Newsholme P, Soares MJ (2015) The impact of vitamin D levels on inflammatory status: a systematic review of immune cell studies. PLoS One 10(11):e0141770
Camperi A, Pin F, Costamagna D, Penna F, Menduina ML, Aversa Z et al (2017) Vitamin D and VDR in cancer cachexia and muscle regeneration. Oncotarget 8(13):21778–21793
Capiati D, Benassati S, Boland RL (2002) 1,25(OH)2-vitamin D3 induces translocation of the vitamin D receptor (VDR) to the plasma membrane in skeletal muscle cells. J Cell Biochem 86(1):128–135
Carlberg C, Bendik I, Wyss A, Meier E, Sturzenbecker LJ, Grippo JF et al (1993) Two nuclear signalling pathways for vitamin D. Nature 361(6413):657–660
Ceglia L, Niramitmahapanya S, da Silva Morais M, Rivas DA, Harris SS, Bischoff-Ferrari H et al (2013) A randomized study on the effect of vitamin D(3) supplementation on skeletal muscle morphology and vitamin D receptor concentration in older women. J Clin Endocrinol Metabol 98(12):E1927–E1935
Chappel J, Ross FP, Abu-Amer Y, Shaw A, Teitelbaum SL (1997) “1,25-dihydroxyvitamin D3 regulates pp60c-src activity and expression of a pp60c-src activating phosphatase”. J Cell Biochem 67(4):432–438
Chen S, Villalta SA, Agrawal DK (2016) FOXO1 mediates vitamin D deficiency-induced insulin resistance in skeletal muscle. J Bone Miner Res 31(3):585–595
Cielen N, Heulens N, Maes K, Carmeliet G, Mathieu C, Janssens W et al (2016) Vitamin D deficiency impairs skeletal muscle function in a smoking mouse model. J Endocrinol 229(2):97–108
Crew KD, Xiao T, Thomas PS, Terry MB, Maurer M, Kalinsky K et al (2015) Safety, feasibility, and biomarker effects of high-dose vitamin D supplementation among women at high risk for breast cancer. Int J Food Sci Nutr Diet 2015(Suppl 1): 1–16
Cutolo M, Paolino S, Sulli A, Smith V, Pizzorni C, Seriolo B (2014) Vitamin D, steroid hormones, and autoimmunity. Ann N Y Acad Sci 1317:39–46
DeLuca HF (1974) Vitamin D: the vitamin and the hormone. Fed Proc 33(11):2211–2219
DeLuca HF (2004) “Overview of general physiologic features and functions of vitamin D”. Am J Clin Nutr 80(6 Suppl):1689S–1696S
Deng YT, Chang TW, Lee MS, Lin JK (2012) Suppression of free fatty acid-induced insulin resistance by phytopolyphenols in C2C12 mouse skeletal muscle cells. J Agric Food Chem 60(4):1059–1066
Dhesi JK, Jackson SH, Bearne LM, Moniz C, Hurley MV, Swift CG et al (2004) Vitamin D supplementation improves neuromuscular function in older people who fall. Age Ageing 33(6):589–595
Dusso AS, Brown AJ, Slatopolsky E (2005) Vitamin D. Am J Physiol. Renal Physiol 289(1):F8–F28
Dzik K, Skrobot W, Flis DJ, Karnia M, Libionka W, Kloc W et al (2018) “Vitamin D supplementation attenuates oxidative stress in paraspinal skeletal muscles in patients with low back pain”. Eur J Appl Physiol 118(1):143–151
Endo I, Inoue D, Mitsui T, Umaki Y, Akaike M, Yoshizawa T et al (2003) Deletion of vitamin D receptor gene in mice results in abnormal skeletal muscle development with deregulated expression of myoregulatory transcription factors. Endocrinology 144(12):5138–5144
Fielding RA, Vellas B, Evans WJ, Bhasin S, Morley JE, Newman AB et al (2011) “Sarcopenia: an undiagnosed condition in older adults. Current consensus definition: prevalence, etiology, and consequences. International working group on sarcopenia”. J Am Med Dir Assoc 12(4):249–256
Franco R, Cidlowski JA (2012) Glutathione efflux and cell death. Antioxid Redox Signal 17(12):1694–1713
Fu L, Yun F, Oczak M, Wong BY, Vieth R, Cole DE (2009) “Common genetic variants of the vitamin D binding protein (DBP) predict differences in response of serum 25-hydroxyvitamin D [25(OH)D] to vitamin D supplementation”. Clin Biochem 42(10–11):1174–1177
Galior K, Grebe S, Singh R (2018) Development of vitamin D toxicity from overcorrection of vitamin D deficiency: a review of case reports. Nutrients 10(8):953
Garland CF, Garland FC, Gorham ED, Lipkin M, Newmark H, Mohr SB et al (2006) “The role of vitamin D in cancer prevention”. Am J Public Health 96(2):252–261
Gil A, Plaza-Diaz J, Mesa MD (2018) “Vitamin D: classic and novel actions”. Ann Nutr Metab 72(2):87–95
Girgis CM, Clifton-Bligh RJ, Hamrick MW, Holick MF, Gunton JE (2013) The roles of vitamin D in skeletal muscle: form, function, and metabolism. Endocr Rev 34(1):33–83
Girgis CM, Mokbel N, Cha KM, Houweling PJ, Abboud M, Fraser DR et al (2014) The vitamin D receptor (VDR) is expressed in skeletal muscle of male mice and modulates 25-hydroxyvitamin D (25OHD) uptake in myofibers. Endocrinology 155(9):3227–3237
Glancy B, Balaban RS (2012) Role of mitochondrial Ca2 + in the regulation of cellular energetics. Biochemistry 51(14):2959–2973
Gniadecki R (1998) Involvement of Src in the vitamin D signaling in human keratinocytes. Biochemical pharmacology 55(4):499–503
Gomez-Cabrera MC, Domenech E, Romagnoli M, Arduini A, Borras C, Pallardo FV et al (2008) “Oral administration of vitamin C decreases muscle mitochondrial biogenesis and hampers training-induced adaptations in endurance performance”. Am J Clin Nutr 87(1):142–149
Haddad F, Adams GR (2004) Inhibition of MAP/ERK kinase prevents IGF-I-induced hypertrophy in rat muscles. J Appl Physiol 96(1):203–210
Hamden K, Carreau S, Jamoussi K, Miladi S, Lajmi S, Aloulou D et al (2009) “1Alpha,25 dihydroxyvitamin D3: therapeutic and preventive effects against oxidative stress, hepatic, pancreatic and renal injury in alloxan-induced diabetes in rats”. J Nutr Sci Vitaminol (Tokyo) 55(3):215–222
Haussler MR, Norman AW (1969) Chromosomal receptor for a vitamin D metabolite. Proc Natl Acad Sci USA 62(1):155–162
Haussler MR, Haussler CA, Jurutka PW, Thompson PD, Hsieh JC, Remus LS et al (1997) The vitamin D hormone and its nuclear receptor: molecular actions and disease states. J Endocrinol 154:S57–S73
Haussler MR, Whitfield GK, Haussler CA, Hsieh JC, Thompson PD, Selznick SH et al (1998) The nuclear vitamin D receptor: biological and molecular regulatory properties revealed. J Bone Miner Res 13(3):325–349
Haussler MR, Haussler CA, Bartik L, Whitfield GK, Hsieh JC, Slater S et al (2008) “Vitamin D receptor: molecular signaling and actions of nutritional ligands in disease prevention”. Nutr Rev 66(10 Suppl 2):S98–S112
Hayakawa N, Fukumura J, Yasuno H, Fujimoto-Ouchi K, Kitamura H (2015) “1alpha,25(OH)2D3 downregulates gene expression levels of muscle ubiquitin ligases MAFbx and MuRF1 in human myotubes”. Biomed Res 36(2):71–80
Hidalgo AA, Trump DL, Johnson CS (2010) “Glucocorticoid regulation of the vitamin D receptor”. J Steroid Biochem Mol Biol 121(1–2):372–375
Holick MF, Binkley NC, Bischoff-Ferrari HA, Gordon CM, Hanley DA, Heaney RP et al (2011) “Evaluation, treatment, and prevention of vitamin D deficiency: an Endocrine Society clinical practice guideline”. J Clin Endocrinol Metabol 96(7):1911–1930
Hopkinson NS, Li KW, Kehoe A, Humphries SE, Roughton M, Moxham J et al (2008) “Vitamin D receptor genotypes influence quadriceps strength in chronic obstructive pulmonary disease”. Am J Clin Nutr 87(2):385–390
Husain K, Ferder L, Mizobuchi M, Finch J, Slatopolsky E (2009) “Combination therapy with paricalcitol and enalapril ameliorates cardiac oxidative injury in uremic rats”. Am J Nephrol 29(5):465–472
Igde M, Baran P, Oksuz BG, Topcuoglu S, Karatekin G (2018) “Association between the oxidative status, vitamin D levels and respiratory function in asthmatic children”. Niger J Clin Pract 21(1):63–68
Jain SK, Parsanathan R, Achari AE, Kanikarla-Marie P, Bocchini JA Jr (2018) Glutathione stimulates vitamin D regulatory and glucose-metabolism genes, lowers oxidative stress and inflammation, and increases 25-hydroxy-vitamin D levels in blood: a novel approach to treat 25-hydroxyvitamin D Deficiency. Antioxid Redox Signal 29(17):1792–1807
Jhee JH, Kim H, Park S, Yun HR, Jung SY, Kee YK et al (2017) Vitamin D deficiency is significantly associated with depression in patients with chronic kidney disease. PLoS One 12(2):e0171009
Jones KS, Assar S, Harnpanich D, Bouillon R, Lambrechts D, Prentice A et al (2014) “25(OH)D2 half-life is shorter than 25(OH)D3 half-life and is influenced by DBP concentration and genotype”. J Clin Endocrinol Metab 99(9):3373–3381
Kandarian SC, Jackman RW (2006) Intracellular signaling during skeletal muscle atrophy. Muscle Nerve 33(2):155–165
Karnia MJ, Myslinska D, Dzik KP, Flis DJ, Ciepielewski ZM, Podlacha M et al (2018) The electrical stimulation of the bed nucleus of the stria terminalis causes oxidative stress in skeletal muscle of rats. Oxidative med Cell Longev 2018:4671213
Ke CY, Yang FL, Wu WT, Chung CH, Lee RP, Yang WT et al (2016) “Vitamin D3 reduces tissue damage and oxidative stress caused by exhaustive exercise”. Int J Med Sci 13(2):147–153
Kemp GJ, Taylor DJ, Thompson CH, Hands LJ, Rajagopalan B, Styles P et al (1993) Quantitative analysis by 31P magnetic resonance spectroscopy of abnormal mitochondrial oxidation in skeletal muscle during recovery from exercise. NMR Biomed 6(5): 302–310
Khare S, Bissonnette M, Wali R, Skarosi S, Boss GR, von Lintig FC et al (1999) “1,25-dihydroxyvitamin D3 but not TPA activates PLD in Caco-2 cells via pp60(c-src) and RhoA”. Am J Physiol 276(4 Pt 1):G1005–G1015
Kim AR, Kim KM, Byun MR, Hwang JH, Park JI, Oh HT et al (2017) Catechins activate muscle stem cells by Myf5 induction and stimulate muscle regeneration. Biochem Biophys Res Commun 489(2):142–148
Kong J, Li YC (2006) Molecular mechanism of 1,25-dihydroxyvitamin D3 inhibition of adipogenesis in 3T3-L1 cells. Am J Physiol. Endocrinol Metab 290(5):E916–E924
Kudielka BM, Wust S (2010) Human models in acute and chronic stress: assessing determinants of individual hypothalamus-pituitary-adrenal axis activity and reactivity. Stress 13(1):1–14
Kunadian V, Ford GA, Bawamia B, Qiu W, Manson JE (2014) “Vitamin D deficiency and coronary artery disease: a review of the evidence”. Am Heart J 167(3):283–291
Li Z, Hosoi Y, Cai K, Tanno Y, Matsumoto Y, Enomoto A et al (2006) “Src tyrosine kinase inhibitor PP2 suppresses ERK1/2 activation and epidermal growth factor receptor transactivation by X-irradiation”. Biochem Biophys Res Commun 341(2):363–368
Li J, Mihalcioiu M, Li L, Zakikhani M, Camirand A, Kremer R (2018) Vitamin D prevents lipid accumulation in murine muscle through regulation of PPARgamma and perilipin-2 expression. J Steroid Biochem Mol Biol 177:116–124
Li P, Liu A, Xiong W, Lin H, Xiao W, Huang J et al (2019) Catechins enhance skeletal muscle performance. Crit Rev Food Sci Nutr: 1–14
Lin AM, Chen KB, Chao PL (2005) Antioxidative effect of vitamin D3 on zinc-induced oxidative stress in CNS. Ann N Y Acad Sci 1053:319–329
Lips P (2006) “Vitamin D physiology”. Prog Biophys Mol Biol 92(1):4–8
Lips P, Binkley N, Pfeifer M, Recker R, Samanta S, Cohn DA et al (2010) “Once-weekly dose of 8400 IU vitamin D(3) compared with placebo: effects on neuromuscular function and tolerability in older adults with vitamin D insufficiency”. Am J Clin Nutr 91(4):985–991
Littlejohns TJ, Henley WE, Lang IA, Annweiler C, Beauchet O, Chaves PH et al (2014) Vitamin D and the risk of dementia and Alzheimer disease. Neurology 83(10):920–928
Losel R, Wehling M (2003) “Nongenomic actions of steroid hormones”. Nat Rev Mol Cell Biol 4(1):46–56
McCollum EF, Simmonds N, Becker JE, Shipley PG (1922) Studies on experimental rickets: and experimental demonstration of the existence of a vitamin which promotes calcium deposition. J Biol Chem 53:293–312
McGill AT, Stewart JM, Lithander FE, Strik CM, Poppitt SD (2008) “Relationships of low serum vitamin D3 with anthropometry and markers of the metabolic syndrome and diabetes in overweight and obesity”. Nutr J 7:4
Meador BM, Mirza KA, Tian M, Skelding MB, Reaves LA, Edens NK et al (2015) “The Green Tea Polyphenol Epigallocatechin-3-Gallate (EGCg) Attenuates Skeletal Muscle Atrophy in a Rat Model of Sarcopenia”. J Frailty Aging 4(4):209–215
Mellanby T (1918) The part played by an “accessory factor” in the production of experimental rickets. J Physiol 52:11–14
Merry TL, Ristow M (2016) “Do antioxidant supplements interfere with skeletal muscle adaptation to exercise training?”. J Physiol 594(18):5135–5147
Mirza KA, Pereira SL, Edens NK, Tisdale MJ (2014) “Attenuation of muscle wasting in murine C2C 12 myotubes by epigallocatechin-3-gallate”. J Cachexia Sarcopenia Muscle 5(4):339–345
Mizwicki MT, Keidel D, Bula CM, Bishop JE, Zanello LP, Wurtz JM et al (2004) Identification of an alternative ligand-binding pocket in the nuclear vitamin D receptor and its functional importance in 1alpha,25(OH)2-vitamin D3 signaling. Proc Natl Acad Sci USA 101(35):12876–12881
Mozolowski W (1939) “Jedrzej Sniadecki (1768–1838) on the cure of rickets.” 143: 121–124
Mukherjee A, Zerwekh JE, Nicar MJ, McCoy K, Buja LM (1981) Effect of chronic vitamin D deficiency on chick heart mitochondrial oxidative phosphorylation. J Mol Cell Cardiol 13(2):171–183
Nakai K, Fujii H, Kono K, Goto S, Kitazawa R, Kitazawa S et al (2014) “Vitamin D activates the Nrf2-Keap1 antioxidant pathway and ameliorates nephropathy in diabetic rats”. Am J Hypertens 27(4):586–595
Nguyen TM, Lieberherr M, Fritsch J, Guillozo H, Alvarez ML, Fitouri Z et al (2004) “The rapid effects of 1,25-dihydroxyvitamin D3 require the vitamin D receptor and influence 24-hydroxylase activity: studies in human skin fibroblasts bearing vitamin D receptor mutations”. J Biol Chem 279(9):7591–7597
Norman AW, Olivera CJ, Barreto Silva FR, Bishop JE (2002) A specific binding protein/receptor for 1alpha,25-dihydroxyvitamin D(3) is present in an intestinal caveolae membrane fraction. Biochem Biophys Res Commun 298(3):414–419
Oliveira MR, Nabavi SF, Daglia M, Rastrelli L, Nabavi SM (2016) Epigallocatechin gallate and mitochondria—a story of life and death. Pharmacol Res 104:70–85
Omdahl JL, Gray RW, Boyle IT, Knutson J, DeLuca HF (1972) “Regulation of metabolism of 25-hydroxycholecalciferol by kidney tissue in vitro by dietary calcium”. Nat New Biol 237(71):63–64
Owens DJ, Sharples AP, Polydorou I, Alwan N, Donovan T, Tang J et al (2015) A systems-based investigation into vitamin D and skeletal muscle repair, regeneration, and hypertrophy. Am J Physiol. Endocrinol Metab 309(12):E1019–E1031
Panel M, Ghaleh B, Morin D (2018) Mitochondria and aging: a role for the mitochondrial transition pore? Aging Cell: e12793
Perrini S, Laviola L, Carreira MC, Cignarelli A, Natalicchio A, Giorgino F (2010) “The GH/IGF1 axis and signaling pathways in the muscle and bone: mechanisms underlying age-related skeletal muscle wasting and osteoporosis”. J Endocrinol 205(3):201–210
Pludowski P, Holick MF, Grant WB, Konstantynowicz J, Mascarenhas MR, Haq A et al (2018) “Vitamin D supplementation guidelines”. J Steroid Biochem Mol Biol 175:125–135
Polly P, Herdick M, Moehren U, Baniahmad A, Heinzel T, Carlberg C (2000) VDR-Alien: a novel, DNA-selective vitamin D(3) receptor-corepressor partnership. FASEB J 14(10):1455–1463
Powe CE, Evans MK, Wenger J, Zonderman AB, Berg AH, Nalls M et al (2013) “Vitamin D-binding protein and vitamin D status of black Americans and white Americans”. New Engl J Med 369(21):1991–2000
Ristow M, Zarse K, Oberbach A, Kloting N, Birringer M, Kiehntopf M et al (2009) Antioxidants prevent health-promoting effects of physical exercise in humans. Proc Natl Acad Sci USA 106(21):8665–8670
Rodman JS, Baker T (1978) Changes in the kinetics of muscle contraction in vitamin D-depleted rats. Kidney Int 13(3):189–193
Roth SM, Zmuda JM, Cauley JA, Shea PR, Ferrell RE (2004) Vitamin D receptor genotype is associated with fat-free mass and sarcopenia in elderly men. J Gerontol. Series A, Biol Sci Med Sci 59(1): 10–15
Rudolf R, Mongillo M, Magalhaes PJ, Pozzan T (2004) “In vivo monitoring of Ca(2+) uptake into mitochondria of mouse skeletal muscle during contraction”. J Cell Biol 166(4):527–536
Ryan ZC, Craig TA, Folmes CD, Wang X, Lanza IR, Schaible NS et al (2016) “1alpha,25-dihydroxyvitamin D3 regulates mitochondrial oxygen consumption and dynamics in human skeletal muscle cells”. J Biol Chem 291(3):1514–1528
Salles J, Chanet A, Giraudet C, Patrac V, Pierre P, Jourdan M et al (2013) 1,25(OH)2-vitamin D3 enhances the stimulating effect of leucine and insulin on protein synthesis rate through Akt/PKB and mTOR mediated pathways in murine C2C12 skeletal myotubes. Mol Nutr Food Res 57(12):2137–2146
Sandri M, Sandri C, Gilbert A, Skurk C, Calabria E, Picard A et al (2004) Foxo transcription factors induce the atrophy-related ubiquitin ligase atrogin-1 and cause skeletal muscle atrophy. Cell 117(3): 399–412
Sandri M, Lin J, Handschin C, Yang W, Arany ZP, Lecker SH et al (2006) PGC-1alpha protects skeletal muscle from atrophy by suppressing FoxO3 action and atrophy-specific gene transcription. Proc Natl Acad Sci USA 103(44):16260–16265
Sato Y, Iwamoto J, Kanoko T, Satoh K (2005) Low-dose vitamin D prevents muscular atrophy and reduces falls and hip fractures in women after stroke: a randomized controlled trial. Cerebrovasc Dis 20(3):187–192
Schiaffino S, Mammucari C (2011) Regulation of skeletal muscle growth by the IGF1-Akt/PKB pathway: insights from genetic models. Skelet Muscle 1(1):4
Shahangian S, Alspach TD, Astles JR, Yesupriya A, Dettwyler WK (2014) “Trends in laboratory test volumes for Medicare Part B reimbursements, 2000–2010”. Arch Pathol Lab Med 138(2):189–203
Shi H, Scheffler JM, Zeng C, Pleitner JM, Hannon KM, Grant AL et al (2009) Mitogen-activated protein kinase signaling is necessary for the maintenance of skeletal muscle mass. Am J Physiol. Cell Physiol 296(5):C1040–C1048
Simpson RU, Thomas GA, Arnold AJ (1985) Identification of 1,25-dihydroxyvitamin D3 receptors and activities in muscle. J Biol Chem 260(15):8882–8891
Sinha A, Hollingsworth KG, Ball S, Cheetham T (2013) Improving the vitamin D status of vitamin D deficient adults is associated with improved mitochondrial oxidative function in skeletal muscle. J Clin Endocrinol Metab 98(3):E509–E513
Sinha-Hikim I, Duran P, Shen R, Lee M, Friedman TC, Davidson MB (2015) “Effect of long term vitamin D supplementation on biomarkers of inflammation in Latino and African-American subjects with pre-diabetes and hypovitaminosis D.” Hormone and metabolic research = Hormon- und Stoffwechselforschung = Hormones et metabolisme 47(4): 280–283
Skversky AL, Kumar J, Abramowitz MK, Kaskel FJ, Melamed ML (2011) Association of glucocorticoid use and low 25-hydroxyvitamin D levels: results from the National Health and Nutrition Examination Survey (NHANES): 2001–2006. J Clin Endocrinol Metab 96(12): 3838–3845
Sleeman I, Aspray T, Lawson R, Coleman S, Duncan G, Khoo TK et al (2017) “The role of vitamin D in disease progression in early Parkinson’s disease. J Parkinsons Dis 7(4):669–675
Smith CL, O’Malley BW (2004) Coregulator function: a key to understanding tissue specificity of selective receptor modulators. Endocr Rev 25(1):45–71
Snijder MB, van Schoor NM, Pluijm SM, van Dam RM, Visser M, Lips P (2006) Vitamin D status in relation to one-year risk of recurrent falling in older men and women. J Clin Endocrinol Metab 91(8):2980–2985
Srikuea R, Hirunsai M (2016) Effects of intramuscular administration of 1alpha,25(OH)2D3 during skeletal muscle regeneration on regenerative capacity, muscular fibrosis, and angiogenesis. J Appl Physiol 120(12):1381–1393
Stitt TN, Drujan D, Clarke BA, Panaro F, Timofeyva Y, Kline WO et al (2004) “The IGF-1/PI3K/Akt pathway prevents expression of muscle atrophy-induced ubiquitin ligases by inhibiting FOXO transcription factors”. Mol Cell 14(3):395–403
Suzuki T, Kwon J, Kim H, Shimada H, Yoshida Y, Iwasa H et al (2008) Low serum 25-hydroxyvitamin D levels associated with falls among Japanese community-dwelling elderly. J Bone Miner Res 23(8):1309–1317
Tanaka M, Tokunaga K, Komaba H, Itoh K, Matsushita K, Watanabe H et al (2011) “Vitamin D receptor activator reduces oxidative stress in hemodialysis patients with secondary hyperparathyroidism”. Ther Apher Dial 15(2):161–168
Tanaka M, Kishimoto KN, Okuno H, Saito H, Itoi E (2014) Vitamin D receptor gene silencing effects on differentiation of myogenic cell lines. Muscle Nerve 49(5):700–708
Tawa NE Jr, Odessey R, Goldberg AL (1997) “Inhibitors of the proteasome reduce the accelerated proteolysis in atrophying rat skeletal muscles”. J Clin Investig 100(1):197–203
Thobe BM, Frink M, Choudhry MA, Schwacha MG, Bland KI, Chaudry IH (2006) Src family kinases regulate p38 MAPK-mediated IL-6 production in Kupffer cells following hypoxia. Am J Physiol. Cell Physiol 291(3):C476–C482
Trewin AJ, Lundell LS, Perry BD, Patil KV, Chibalin AV, Levinger I et al (2015) Effect of N-acetylcysteine infusion on exercise-induced modulation of insulin sensitivity and signaling pathways in human skeletal muscle. Am J Physiol Endocrinol Metab 309(4):E388–E397
Umesono K, Murakami KK, Thompson CC, Evans RM (1991) Direct repeats as selective response elements for the thyroid hormone, retinoic acid, and vitamin D3 receptors. Cell 65(7):1255–1266
Usjak L, Petrovic S, Drobac M, Sokovic M, Stanojkovic T, Ciric A et al (2017) Essential oils of three cow parsnips—composition and activity against nosocomial and foodborne pathogens and food contaminants. Food Funct 8(1):278–290
Verstuyf A, Carmeliet G, Bouillon R, Mathieu C (2010) “Vitamin D: a pleiotropic hormone”. Kidney Int 78(2):140–145
Vertino AM, Bula CM, Chen JR, Almeida M, Han L, Bellido T et al (2005) “Nongenotropic, anti-apoptotic signaling of 1alpha,25(OH)2-vitamin D3 and analogs through the ligand binding domain of the vitamin D receptor in osteoblasts and osteocytes. Mediation by Src, phosphatidylinositol 3-, and JNK kinases”. J Biol Chem 280(14):14130–14137
Wei S, Tanaka H, Seino Y (1998) “Local action of exogenous growth hormone and insulin-like growth factor-I on dihydroxyvitamin D production in LLC-PK1 cells”. Eur J Endocrinol 139(4):454–460
Whitfield GK, Remus LS, Jurutka PW, Zitzer H, Oza AK, Dang HT et al (2001) “Functionally relevant polymorphisms in the human nuclear vitamin D receptor gene”. Mol Cell Endocrinol 177(1–2):145–159
Yan J, Feng Z, Liu J, Shen W, Wang Y, Wertz K et al (2012) “Enhanced autophagy plays a cardinal role in mitochondrial dysfunction in type 2 diabetic Goto-Kakizaki (GK) rats: ameliorating effects of (-)-epigallocatechin-3-gallate”. J Nutr Biochem 23(7):716–724
Zanello LP, Norman AW (2004) Rapid modulation of osteoblast ion channel responses by 1alpha,25(OH)2-vitamin D3 requires the presence of a functional vitamin D nuclear receptor. Proc Natl Acad Sci USA 101(6):1589–1594
Zhang Y, Leung DY, Goleva E (2013) “Vitamin D enhances glucocorticoid action in human monocytes: involvement of granulocyte-macrophage colony-stimulating factor and mediator complex subunit 14”. J Biol Chem 288(20):14544–14553
Zhang HQ, Teng JH, Li Y, Li XX, He YH, He X et al (2014) Vitamin D status and its association with adiposity and oxidative stress in schoolchildren. Nutrition 30(9):1040–1044
Ziegler R, Kasperk C (1998) “Glucocorticoid-induced osteoporosis: prevention and treatment”. Steroids 63(5–6):344–348
Zittermann A (2003) “Vitamin D in preventive medicine: are we ignoring the evidence?”. Br J Nutr 89(5):552–572
Acknowledgements
We would like to thank Prof. Michal Zmijewski for revise and dedicated comments that helped to improve our manuscript.
Funding
This study was partially supported by NCN UMO-2012/05/B/NZ7/02493.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that there is no conflict of interests regarding the publication of this paper.
Additional information
Communicated by Michael Lindinger.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
OpenAccess This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Dzik, K.P., Kaczor, J.J. Mechanisms of vitamin D on skeletal muscle function: oxidative stress, energy metabolism and anabolic state. Eur J Appl Physiol 119, 825–839 (2019). https://doi.org/10.1007/s00421-019-04104-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00421-019-04104-x