
Abstract
A full mechanistic understanding of how secretory cargo proteins are exported from the endoplasmic reticulum for passage through the early secretory pathway is essential for us to comprehend how cells are organized, maintain compartment identity, as well as how they selectively secrete proteins and other macromolecules to the extracellular space. This process depends on the function of a multi-subunit complex, the COPII coat. Here we describe progress towards a full mechanistic understanding of COPII coat function, including the latest findings in this area. Much of our understanding of how COPII functions and is regulated comes from studies of yeast genetics, biochemical reconstitution and single cell microscopy. New developments arising from clinical cases and model organism biology and genetics enable us to gain far greater insight in to the role of membrane traffic in the context of a whole organism as well as during embryogenesis and development. A significant outcome of such a full understanding is to reveal how the machinery and processes of membrane trafficking through the early secretory pathway fail in disease states.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
The early secretory pathway is made up of the endoplasmic reticulum (ER), Golgi apparatus and intermediate compartments between these two major stations (including transport vesicles and tubules) and the ER-Golgi intermediate compartment itself (Palade 1975). These intracellular compartments are in constant communication, with molecules being passed from one donor compartment to a target compartment via discrete vesicles or convoluted networks of tubules and vesicles (Bannykh and Balch 1997; Martinez-Menarguez et al. 1999). Evidence also exists for direct connections between compartments (e.g. Stinchcombe et al. 1995) that would significantly complicate sorting and segregation processes. Membrane trafficking must be a highly organized process with each outward movement counterbalanced by a retrieval step whereby membrane and selected proteins are returned to their original compartment of origin (Bonifacino and Glick 2004; Salama and Schekman 1995). Each transport vesicle must be highly selective for the proteins that make up vesicle cargo, and for the fusion proteins specifying the target membrane, so that trafficking within the cell is kept well-organized and efficient (Bonifacino and Glick 2004; Salama and Schekman 1995). Here, we try to provide a perspective on the formation of transport vesicles from the ER. For further mechanistic detail, including subtle differences between the animal systems primarily described here and those in plants (Hanton et al. 2005), the reader is referred to other excellent recent reviews in this area (Gurkan et al. 2006; Kirk and Ward 2007; Lee and Miller 2007; Mancias and Goldberg 2005).
ER exit sites
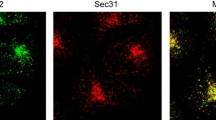
The export of proteins from the ER has been well defined in yeast (both Saccharomyces cerevisiae (Schekman and Novick 2004) and Pichia pastoris (Rossanese et al. 1999)) and mammalian cells (Bannykh and Balch 1997). In S. cerevisiae, budding appears to occur stochastically across the entire ER membrane. In P. pastoris and metazoans, the budding event is highly organized, occurring at discrete sites on the ribosome-free ER, called transitional ER (tER) or ER exit sites (ERES) (Bannykh et al. 1996; Orci et al. 1991; Palade 1975; Tang et al. 2005). These sites are defined by the presence of COPII proteins (Barlowe et al. 1994), the core machinery that directs the budding event. Figure 1a shows the localization of ERES (using a COPII marker, Sec24C) and Golgi membranes in a HeLa cell spread on a glass coverslip. COPII is a multi-subunit complex that mediates the accumulation of secretory cargo, the deformation of the membrane and generation of subsequent transport vesicles. The localization of ERES within the ER membrane is shown in Fig. 1b. The mechanism of assembly and function of this complex forms the core of this review.

Localization of COPII in mammalian cells. a Sec24C (COPII, green) and giantin (a marker of the cis-/medial-Golgi, red) in HeLa cells. Note the close proximity of the two structures in the juxtanuclear region results in apparent colocalization and yellow colouring in the merge. Bar 10 μm. b Sec24C (red) and calnexin (green) show the presence of ERES on ER membranes. Bar 10 μm. c Immunofluorescence labelling of methanol fixed HeLa cell shows the close apposition but distinct localization of Sec24C (COPII, green) and ERGIC-53 (red, a marker of the ER-Golgi intermediate compartment). Arrowheads highlight the clear separation of the two structures. Bar 5 μm. Individual green and red channels are shown alongside the merge for clarity
ERES are relatively immobile structures (Stephens et al. 2000) and face out towards assemblies of vesicular-tubular structures (VTCs), also known as ERGIC clusters (ER-Golgi-intermediate compartment) (Bannykh and Balch 1997; Scales et al. 1997; Schweizer et al. 1990). These ERGIC clusters are COPI positive, cargo-rich compartments that mediate the trafficking of secretory cargo between the ER and the Golgi (Appenzeller-Herzog and Hauri 2006; Schweizer et al. 1990). Figure 1c shows the spatial distribution of ERES and ERGIC at high magnification; one can clearly observe the close juxtaposition but clear offset between the two compartments. ERES are distributed throughout the cytoplasm but also cluster in the juxtanuclear area of cell types with a classic juxtanuclear Golgi (Bannykh et al. 1996; Hammond and Glick 2000; Orci et al. 1991; Stephens et al. 2000). Over half the ERES present cluster at the ER-Golgi interface within this region and data exist that give intriguing glimpses of the possibility of membrane connectivity between ERES and Golgi structures (Ladinsky et al. 1999; Sesso et al. 1992; Stinchcombe et al. 1995).
Studies in S. cerevisiae show well-organized ERES to be absent. Instead vesicular budding is not confined to a certain sub-domain within the membrane but occurs throughout the ER (Rossanese et al 1999). This structural difference between yeast strains has brought about the theory that defined ERES evolved to produce a higher order of COPII budding. This level of organization might reflect some developmental requirement based on multicellularity, or could arise from the need for ERES to take on higher order organization in specific cell types such as muscle cells (Ralston et al. 2001) and neurons (Horton and Ehlers 2004). COPII coated ERES are able to undergo fission and fusion events that appear to control ERES size (Stephens 2003), as well as being able to form de novo (Bevis et al. 2002; Stephens 2003). The core COPII components are peripheral membrane proteins; their constant cycling on and off membranes (Stephens et al. 2000) makes such control possible. Further examination of the mechanism underlying ERES organization requires a discussion of the components of the COPII coat. Here, we attempt to provide some historical background to our understanding of COPII assembly and function, but from the start we attempt to incorporate the latest findings in this area.
An overview of the COPII coat
COPII complex components undergo several rounds of export from the ER. COPII recruitment is initiated by the activation of the small GTPase Sar1 (Nakano et al. 1989) by its ER-localized GEF Sec12 (Nakano et al. 1988). Upon exchange of GDP for GTP, Sar1 exposes an N-terminal amphipathic tail, which inserts into the lipid bilayer (Bielli et al. 2005; Lee et al. 2005). Once tightly associated with the membrane, Sar1 recruits a heterodimeric complex comprising of Sec23/24 (Barlowe et al. 1994; Hicke and Schekman 1989; Hicke et al. 1992; Kaiser and Schekman 1990). Sec23 acts as a GAP for Sar1 (Yoshihisa et al. 1993) increasing the very low rate of hydrolysis by Sar1. Sec24 is required for cargo binding, and together with cargo, Sec23, and Sar1 forms the prebudding complex (Aridor et al. 1998). COPII budding can be reproduced in vitro using synthetic liposomes with which the five core COPII components alone (Sar1, Sec23/24 and Sec13/31) are sufficient to deform the membrane and generated coated vesicles (Matsuoka et al. 1998). Inner coat subunits Sec23 and Sec24 are structurally similar and form a “bow-tie” shape (Lederkremer et al. 2001). The presence of basic residues on the concave inner side of the heterodimer potentially enhances membrane association (Bi et al. 2002). Subsequent recruitment of the heterotetramer Sec13/31, consisting of two Sec13 and two Sec31 subunits, permits minimal cage formation acting as a structural scaffold for the outer layer of the COPII coat (Fath et al. 2007; Stagg et al. 2006).
In mammals, multiple isoforms exist of nearly all COPII subunits, each encoded by a different gene. These are generally denoted with an alphabetical suffix and we use this nomenclature here. Database searching reveals two isoforms of Sar1 (Sar1A and Sar1B), two Sec16 isoforms (which we denote A and B, see below), two of Sec23, (Sec23A and Sec23B) four Sec24 isoforms (A-D), one bona fide Sec13 isoform, and two of Sec31 (A and B). This has important implications for coat assembly and cargo selection, and, as we shall discuss below, has great relevance to COPII function in certain disease states. Further complexity arises from splicing and posttranslational modification of some components (e.g. see Dudognon et al. 2004; Salama et al. 1997; Stankewich et al. 2006).
Organization of ERES: a platform for COPII assembly
Given the differences in organization of COPII budding between species, an obvious question arises—how this organization is achieved? Sec16 was identified in the original screen for secretory pathway mutants in yeast (Novick et al. 1980; for an overview see Schekman and Novick 2004). Upon mutation of the Sec16 gene it was observed that secretory protein precursors rapidly accumulated in their core-glycosylated forms suggesting COPII transport to the Golgi had been blocked (Novick et al. 1981). Electron microscopy of Sec16 mutants has also shown an absence of the distinct 40–80 nm COPII transport vesicles (Kaiser and Schekman 1990). Sec16 encodes a large (∼240 kDa) protein proposed as having three functional domains separated by clusters of proline residues acting as structural spacers (Espenshade et al. 1995). Overexpression of Sec16 in yeast is lethal, a function that maps to the N-terminus of the protein defining one functional domain (Espenshade et al. 1995); two other domains can be defined by a ∼250 amino acid central conserved domain and a smaller C-terminal domain that interacts directly with COPII coat protein subunits Sec23, Sec24 and Sec13/31 (Espenshade et al. 1995; Gimeno et al. 1996). Yeast two hybrid and direct binding experiments have identified that both inner and outer COPII subunits, Sec24 and Sec13/31, bind to the central conserved domain of Sec16. Sec23 interacts with the C-terminal domain, to which Sec24 also binds weakly (Gimeno et al. 1996; Shaywitz et al. 1997). Therefore, Sec16 probably serves as a scaffold onto which COPII subunits assemble (Supek et al. 2002).
Further information relating to the function of Sec16 came with the identification and characterization of Sec16 from other species. Initial key work in this area arose from the use of P. pastoris as a model for the study of membrane trafficking. This system has proven so useful as it shows a level of organization above that of S. cerevisiae but is still genetically tractable (Gould et al. 1992; Payne et al. 2000). P. pastoris Sec16 localizes to ERES (transitional ER) (Connerly et al. 2005) and is specifically involved in the determination of these sites and potential stabilization of the COPII coat. A screen for temperature sensitive mutants, generated from chemical mutagenesis, identified one particular missense mutation shown to be significant to ER export (Connerly et al. 2005). This mutant, termed dot1, lies within the central conserved domain region of Sec16 consequently causing disruption of the typical ERES phenotype (Connerly et al. 2005). Conversely, upon overexpression of a GDP-restricted Sar1 mutant Sar1T34N, known to block COPII vesicle formation, ERES remain intact (Connerly et al. 2005; Kuge et al. 1994). Furthermore, the membrane-bound GEF Sec12 is not responsible for defining ERES formation as when it is delocalized to the general ER, ERES remain intact (Connerly et al. 2005). However, one should note that mammalian Sec12 is localized through the entire ER anyway (Weissman et al. 2001 and DJS unpublished observations). This fragmentation of ERES resulting from the dot1 mutation, suggests a specific role for Sec16 in coordinating COPII subunit assembly. It is speculated that Sec16 prevents premature COPII subunit disassembly upon Sar1-GTP hydrolysis with by linking subunits via Sec16 self-association (Connerly et al. 2005). This self-association mechanism results in the accumulation of COPII subunits at specific domains of fixed locations within the cell, in proximity to the cis-Golgi.
More recently, metazoan orthologues of Sec16 have been identified, the best characterized of which is now that from human. Sec16 sequences from yeast species S. cerevisiae and P. pastoris show highest homology within the central conserved domain; homology searching with this central domain of yeast Sec16 led to the identification of the human orthologue (Bhattacharyya and Glick 2007; Iinuma et al. 2007; Watson et al. 2006), which was originally annotated as KIAA0310 (by the Kazusa consortium who identified this gene in a collection of large human cDNA clones, Kikuno et al. 2004). The large (∼250 kDa) protein, KIAA0310 shows only 19.8% sequence similarity to S. cerevisiae Sec16 protein with the majority lying within the central conserved domain. Both endogenous (Watson et al. 2006) and GFP-tagged Sec16 (Bhattacharyya and Glick 2007; Iinuma et al. 2007; Watson et al. 2006) localize to ERES. Sec16 clusters in the juxtanuclear area, aligned with yet distinct from Golgi membrane (Watson et al. 2006). Peripheral punctate spots colocalize with ERES markers Sec24C and Sec31A and are both long-lived and stable (Watson et al. 2006). The localization of Sec16 to ERES is dependent upon Sar1-GTP as Sec16 mutations are shown to be partially suppressed by the overexpression of Sar1 (Saito-Nakano and Nakano 2000). In addition, when a GDP-restricted form of Sar1 was expressed in mammalian cells Sec16, Sec24C and Sec31A were almost completely lost from punctate structures (Iinuma et al. 2007; Watson et al. 2006).
The distribution of Sec16 throughout the cell has been analysed by subcellular fractionation experiments, and although immunofluorescence shows a large presence of Sec16 at ERES throughout the cell, Sec16 is predominantly cytosolic and is thought to cycle on and off the ER membrane in a Sar1 dependent fashion (Iinuma et al. 2007; Watson et al. 2006). The S. cerevisiae protein becomes incorporated in to budding vesicles (Espenshade et al. 1995). It remains to be determined whether this is also true of the metazoan orthologues.
The depletion (using RNA interference) or overexpression of Sec16 gives rise to phenotypes that support a role for Sec16 in the organization of COPII at ERES. Following depletion of Sec16 expression, the number of ERES in a cell decreases and protein transport from the ER is delayed, but significantly, it is not abolished (Bhattacharyya and Glick 2007; Iinuma et al. 2007; Watson et al. 2006). In contrast, overexpression of Sec16 causes displacement of other COPII subunits (Sec24 and Sec31) from ERES (Watson et al. 2006), presumably due to sequestration of other COPII subunits by recombinant Sec16. As seen in P. pastoris, Sec16 is present at ERES at a concentration that is an order of magnitude lower than that of COPII subunits, as a result of this Sec16 is suggested to be a modulator of COPII function rather than a stoichiometric subunit of the COPII coat (Connerly et al. 2005). FRAP (fluorescence recovery after photobleaching) shows YFP-Sec16A, expressed at low levels, to be rapidly recycling on and off the membrane (Watson et al. 2006). There are two immediately obvious explanations for this; either Sec16 becomes incorporated into the COPII vesicles, or it is being exchanged between the membrane bound pool on the ER and the cytoplasmic pool (Iinuma et al. 2007; Watson et al. 2006).
Database searching using the central conserved domain of Sec16 also identified a second, shorter, mammalian homologue of Sec16 (Bhattacharyya and Glick 2007). The longer and shorter isoforms of Sec16 have been referred to as Sec16L and Sec16S respectively; here, we use the nomenclature of all other COPII subunit isoforms and refer to the longer isoform as Sec16A and second, shorter one as Sec16B. It is hoped that this nomenclature is also then applicable to species in which there are two isoforms of intermediate length such as Arabidopsis. Sec16B is encoded by a different gene to Sec16A and results in a smaller 117 kDa protein; Sec16B is required for ER export and a GFP-tagged form has been shown to localize to ERES (Bhattacharyya and Glick 2007). There are also two striking differences in their sequence homologies; Sec16B does not contain the small conserved C-terminal domain that is present in both yeast Sec16 and mammalian Sec16A, and is also truncated at the N-terminus by >1,000 amino acids (Bhattacharyya and Glick 2007). Therefore, it has been proposed that Sec16A is analogous to yeast Sec16, and that higher eukaryotes have evolved Sec16B as an additional component.
The two isoforms are not functionally redundant (Bhattacharyya and Glick 2007) suggesting potentially complex functions in COPII-dependent export from the ER. Evidence for Sec16A and Sec16B working in tandem with one another is accumulating. When GFP-tagged and transfected into cells both constructs colocalize completely (Bhattacharyya and Glick 2007). Both Sec16A and Sec16B genes are known to be expressed in multiple tissue types, and when knocked down singly, or together, they both inhibit ER export demonstrating the same phenotypic disruption of ERES (Bhattacharyya and Glick 2007). The two proteins probably also function together in a complex, since co-expression of FLAG-tagged Sec16A or FLAG-tagged Sec16B with GFP-Sec16A or GFP-Sec16B enables co-immunoprecipitation of the two isoforms (Bhattacharyya and Glick 2007). However, depletion of endogenous Sec16B by siRNA appears to have no effect on the phenotypic distribution of Sec16A (Bhattacharyya and Glick 2007; Iinuma et al. 2007). Although Sec16 is thought to be required for ER export, the effect of Sec16 depletion on the transport of the classical transmembrane secretory cargo tsO45-G from the ER is fairly minimal; tsO45-G can still exit the ER but at a slower rate. It is possible that Sec16 is required for efficient export of certain cargo, but overall remains nonessential (Watson et al. 2006). It is important to note that the remaining endogenous levels of Sec16 can still associate to apparently fully functional ERES (Watson et al. 2006), thus it is possible that in fact the steady-state number of ERES in a HeLa cell is in vast excess to what is required and a much smaller number (around 15%) is in fact sufficient for efficient ER-to-Golgi transport. This could have implications for systems in which a rapid up-regulation of ER export is required such as plasma cell differentiation.
The mechanism by which either isoform of Sec16 localized to ERES in metazoans remains unclear. The minimal ERES targeting regions that have been defined show only weak homology (Bhattacharyya and Glick 2007). It is also unclear whether they truly localize to ERES independently of one another. It remains possible that these regions have something to do with hetero-oligomer formation (Bhattacharyya and Glick 2007) and that in fact the ERES targeting information lies elsewhere.
Other potential COPII assembly factors have also been identified including the nucleoside diphosphate kinase Nm23H2 (Kapetanovich et al. 2005), which appears to facilitate COPII vesicle formation independent of its kinase activity. In addition, the Rab-interacting protein Yip1p is also implicated in COPII vesicle formation (Heidtman et al. 2003). Yip1p interacts with the Ypt1p/Rab1 GTPase (Calero and Collins 2002; Yang et al. 1998) and genetically with several COPII subunits (Heidtman et al. 2003). It forms a heterodimeric complex with Yif1p (Matern et al. 2000) and this in turn interacts with Yos1p (Heidtman et al. 2005). Genetic defects in each of these components show reduced COPII vesicle formation. The mammalian orthologue of Yip1p, Yip1A localizes to ERES (Tang et al. 2001). More recent data implicate another S. cerevisiae protein, Smy2p, in the formation of COPII vesicles (Higashio et al. 2007). The precise mode of action of these proteins and roles either directly in vesicle formation, or particularly in the case of Yip1p-Yif1p, later in vesicle tethering and fusion (Barrowman et al. 2003), remains to be fully defined.
Quality control and ER export
A vital aspect to efficient export of proteins from the ER is to ensure that only those proteins that are correctly folded and assembled are competent for export. This is a relatively under-researched area at present that is sparking renewed interest, particularly since the majority of “trafficking-related diseases” (Aridor and Hannan 2000, 2002) are in fact attributable to folding and assembly defects. In order to exit the ER proteins must be properly folded and assembled into their multimeric protein complexes (Ellgaard and Helenius 2003). Misfolded or aggregated proteins are recognized by chaperones as part of a quality control mechanism for proteins leaving the ER (Ellgaard and Helenius 2003). Chaperones cover up ER export signals, or anchor proteins in the ER, as export of incorrectly formed proteins may have detrimental effects if they are transported onwards. One proposed mechanism for overcoming ER retention motifs comes from initial work on T cell and immunoglobulin E receptors (Letourneur et al. 1995; Mallabiabarrena et al. 1992). Masking of the ER retention motif is thought to occur during the association of protein subunits as they fold into their quaternary multimeric complexes; unassembled and incorrectly assembled proteins are prevented from exiting the ER (Letourneur et al. 1995; Mallabiabarrena et al. 1992). Studies in neuronal cells support this theory, showing that the retention of plasma membrane destined multimeric channels and receptors within the ER, occurs via a dibasic motif. Upon protein folding this motif is masked and the resulting complexes can undergo ER export (Ma et al. 2002).
More recently, a second model for quality control of exported proteins has been suggested involving mediation by the chaperone complex 14-3-3 (O’Kelly et al. 2002; Yuan et al. 2003). This mechanism for overcoming basic signal mediated protein retention was discovered as a result of direct binding assays showing a specific interaction between both 14-3-3 and the COPI subunits β-COP (O’Kelly et al. 2002; Yuan et al. 2003). These foundations suggest newly synthesized proteins are continually probed as they are exported from the ER, along microtubules, towards ERGIC or the cis-Golgi. Any misfolding that occurs along route can be detected by 14-3-3 proteins, which adjudicate the subsequent binding of COPI to the exposed dibasic motif so that unassembled cargo can be recycled back to the ER (Nufer and Hauri 2003).
A requirement for multiple cargo export signals helps achieve efficient ER export
Oligomerization of cargo not only helps in overcoming ER retention signals, but also plays a role in mediating efficient ER export (Hurtley and Helenius 1989). Studying the role that oligomerization of cargo proteins play on ER export has proven difficult as preventing proteins from oligomerizing often results in protein misfolding and ER retention by chaperones. The type I membrane protein ERGIC-53 provides and excellent model cargo protein for the study of ER export and has so far formed the basis for much of this research (Appenzeller-Herzog and Hauri 2006; Appenzeller et al. 1999; Ben-Tekaya et al. 2005; Kappeler et al. 1997; Schindler et al. 1993). ERGIC-53 is a mannose-binding lectin, which serves as a receptor packaging selected secretory cargo into COPII vesicles (Appenzeller et al. 1999; Moussalli et al. 1999). ERGIC-53 is an important component of the cargo packaging machinery since humans with an inherited mutation in this gene were unable to package blood clotting factors, Factor V and Factor VIII, resulting in excessive bleeding (Nichols et al. 1998).
Cargo capture via the COPII coat can be mediated by dihydrophobic (e.g. –FF-) or diacidic (e.g. –DxE-) motifs in the cytoplasmic domain (reviewed in Barlowe 2003). For example, export of ERGIC-53 is dependent upon an FF motif in the C-terminus of the cytoplasmic domain (Itin et al. 1995). Indeed other work has shown that C-terminal valine residue, appropriately spaced from the membrane, is alone sufficient to direct efficient export (Nufer et al. 2002). Current thinking favours a model whereby cooperation between features within both the cytoplasmic and transmembrane domains, as well as the correct optimal oligomeric presentation of cargo, are required for efficient export of Type I membrane proteins from the ER (Nufer et al. 2003; Otte and Barlowe 2004; Sato and Nakano 2003). When the FF motif of ERGIC-53 is appended to reporter proteins, it alone is not sufficient for ER export (Kappeler et al. 1997). Subsequently, when the FF motif from the cytoplasmic domain of ERGIC-53 is removed, ERGIC-53 can still export the ER but does so at a slower rate than wild type ERGIC-53 (Itin et al. 1995; Kappeler et al. 1997). This started the search for other signals required for a cooperative export mechanism. Homology searches identified residues that could potentially prove important for export (Nufer et al. 2003). Three possible export determinants were probed further; phenylalanine F509 at position-2 from the C-terminus, glutamine Q501 (working as part of an RSSQE motif) in the cytoplasmic domain, and a non-contiguous motif in the transmembrane domain that is required for the correct oligomerization of ERGIC-53 (Nufer et al. 2002, 2003). In the cytoplasmic domain F484 mediates COPII binding, whereas Q488 accelerates transport (Nufer et al. 2003). The mechanisms for this are still uncertain, current views suggest the less highly conserved Q488 assists F484 by mediating its optimal presentation to the COPII subunit Sec24 (Nufer et al. 2003). This mechanism is likely to be a result of hydrophobicity and structure of the cytoplasmic domain.
Immediately following its synthesis, ERGIC-53 forms homodimers and homohexamers via disulphide bond formation between two highly conserved cysteine residues, C466 and C475, close to the membrane (Nufer et al. 2003). If either, or both, cysteines are absent ERGIC-53 is unable to hexamerize, although ERGIC-53 homodimers remain stable and have similar stability to the wild-type protein. Stabilization of oligomers by disulphide-bond formation is therefore not a requirement for export but instead appears to be a non-obligatory but preferable event resulting in efficient ER export. The results of chemical crosslinking experiments between constructs unable to form disulphide-bonds helped confirm this theory (Nufer et al. 2003). The ability to oligomerize efficiently is contributed to three residues in the TMD, F484, Q488, Y498, whose absence results in the formation of aggregates. Q488 is strictly conserved, whereas both F484 and Y498 aromatics are less highly conserved (Nufer et al. 2003). Helical wheels show all three residues to be present on the same side of the alpha helix; this allows the polar properties of Q488 to drive oligomerization via association of these hydrophobic stretches of the TMD amphipathic helixes in the membrane (Nufer et al. 2003). In summary, dimerization is mediated by disulphide bond formation and further hexamerization occurs via TMD associations between amphipathic helices. Both mechanisms alone are sufficient for ER export, however, when dimerization occurs prior to hexamerization the disulphide bonds formed help stabilize helices and mediate helix packing events via the correct presentation of Q488 (Eilers et al. 2000; Nufer et al. 2003). This cooperation between mechanisms results in the most efficient ER export.
The length of the transmembrane domain aids the sorting of various transmembrane proteins within the exo-endocytic system; this includes protein retention in the Golgi, segregation of membrane proteins into intracellular organelles in Toxoplasma gondii, and tail-anchored protein sorting from the ER to the plasma membrane to name a few (Bulbarelli et al. 2002; Karsten et al. 2004; Munro 1995). For ERGIC-53, the length of the transmembrane domain did not seem to affect membrane association, but instead showed an effect on transport efficiency (Nufer et al. 2003). The optimal transmembrane length for efficient ER export is 21 amino acids, when past this optimal number export from the ER directly slows (Nufer et al. 2003). This suggests that a transmembrane domain of 21 amino acids allows the clustering of ERGIC-53 into ERES domains. The precise means by which TMD length dictates incorporation into COPII clusters remains to be determined. Upon switching the transmembrane domain for a 18 leucine residues, ERGIC-53 became retained in the ER (Nufer et al. 2003). Cooperation between recognition signals and presentation of the optimal oligomeric form achieves an ER export mechanism that is more controlled and selective. This applies in multiple systems (e.g. see Otte and Barlowe 2004; Sato and Nakano 2003) and includes proper recognition of fusion competent SNARE pairings (Allan et al. 2000; Sato and Nakano 2005).
Kinetic assembly of the COPII coat
Stabilization of the pre-budding complex via a combination of GTPase, GEF and GAP activities
Several pieces of evidence point to a direct role for the controlled rate of GTP hydrolysis by Sar1 as an important factor in the incorporation of cargo into nascent buds. First, inhibition of GTP hydrolysis by Sar1 using a mutation that restricts Sar1 to the GTP-bound state (Sar1H79G) causes transmembrane and soluble cargo to become accumulated in pre-budding complexes that often accumulate in the juxtanuclear region of mammalian cells; in contrast, GPI-anchored cargo remains restricted to the bulk of the ER (Stephens and Pepperkok 2004). Elegant reconstitution experiments have suggested that GTP hydrolysis by Sar1, and its regulation by cargo proteins, acts to ensure that improper cargoes are excluded from nascent buds (Sato and Nakano 2004). Data remain controversial as to whether all COPII vesicles are the same or differ in their composition or mechanism of production. Biochemical data have shown that transmembrane and GPI-anchored cargo can be found in distinct populations of COPII-derived vesicles in yeast (Muniz et al. 2001). Similarly, imaging of cells expressing GFP-tagged procollagen shows that this particular cargo appears in vesicular-tubular transport carriers that appear to exclude other cargo proteins (Stephens and Pepperkok 2002). It remains possible that kinetic differences in the rates of vesicle formation account for these distinctions. Furthermore, it is likely that procollagen presents a particular biophysical problem in terms of incorporating a 300 nm rod-like structure in to a transport container. However, mechanistic data implicate tethering and fusion machineries in the generation and subsequent consumption of these vesicle populations (Morsomme and Riezman 2002; Morsomme et al. 2003).
Initial work on COPII reconstitution showed that it was possible to generate COPII vesicles from minimal components on lipid membranes of appropriate (generally negatively charged) membranes (Antonny et al. 2001; Matsuoka et al. 1998). Recently an assay was developed that enabled analysis of one full cycle of COPII turnover on a membrane incorporating transport-competent cargo. Exploiting fluorescence resonance energy transfer (FRET) between cargo and coat, Sato and Nakano (2005) reconstituted cargo molecules in the form of SNARE proteins in to proteoliposomes along with purified, fluorescently labelled Sec23/24 complexes (Sato and Nakano 2005) and Sar1. This system allows a direct comparison between the time-course of cargo disassembly, as monitored by FRET, alongside the time-course for Sar1-GTP turnover, as monitored by tryptophan fluorescence (Sato and Nakano 2005). The kinetics of Sar1 binding to COPII inner coat subunits were measured using the two fluorophores; CFP-labelled SNARE proteins as donors, and YFP-labelled subunits Sec23/24 as acceptors. Upon addition of GMPPNP, a poorly hydrolysable analogue of GTP resulting in stabilization of COPII subunits on the synthetic liposomes, energy transfer was observed between subunits Sec23/24 and various SNARE proteins (Sato and Nakano 2005). In other systems that lack inclusion of cargo proteins, after GTP hydrolysis, COPII subunits disassociate from the membrane due to lack of stability (Antonny et al. 2003; Matsuoka et al. 1998). In Sato and Nakano’s system Sec23/24 remained membrane bound upon interacting with v-SNARE Bet1p. For stabilization of COPII inner proteins by t-SNARE Sec22p (Sato and Nakano 2005), it was necessary for Sec22 to be present along with its partner proteins as part of the t-SNARE complex presumably to efficiently engage with Sec23/24 (Miller et al. 2003; Mossessova et al. 2003).
These important experiments allowed for analysis of the requirement for Sar1-GTP in the maintenance of cargo-coat interactions. This showed that Sec23/24 remained associated with the membrane (through interaction with cargo) for much longer time frames than the rate of GTP hydrolysis by Sar1. Further to this, the continual reactivation of Sar1 was facilitated by the presence of Sec12pΔlum, the cytosolic domain of the Sar1 GEF, Sec12. In this case, the interaction between Sec23/24 and SNARE proteins is prolonged further still suggesting that multiple rounds of Sar1-GTP cycling facilitate sustained interaction of Sec23/24 with cargo (Sato and Nakano 2005). To test this idea directly, the authors added unlabelled Sec23/24p subunits and found that in the presence of incomplete t-SNARE complexes, exchange of these subunits with their YFP versions on the membrane occurred indicative of continued Sec23/24 turnover. When acting as part of the fully formed t-SNARE complex the exchange of fluorescent subunits for non-fluorescent subunits is not observed, consistent with a sustained interaction of Sec23/24 with the t-SNARE complex. Interactions between membrane recruited Sec23/24 and anionic phospholipids alone cannot bring about stabilization (Matsuoka et al. 1998); this Sar1-independent, SNARE-based, COPII subunit stabilization allows time for the maximum possible accuracy of cargo recognition and sorting. SNAREs have not only been shown to stabilize Sec23/24 on the membrane, but have recently been proposed to play a role in modulating the GAP activity of Sec23 (Sato and Nakano 2004). It is suggested that through binding to SNAREs, Sec23 becomes optimally orientated in order for Sar1 to catalyze GTP hydrolysis (Sato and Nakano 2004).
COPII subunit dynamics in living cells
Initial work using live cell imaging to define COPII dynamics in cells showed that COPII-coated ERES are relatively immobile, undergoing only short range movements within a confined area (Hammond and Glick 2000; Stephens et al. 2000). Occasional long-range movements are observed but these are not directed towards any particular cellular target and could result from repositioning of the underlying ER. More recent work suggests that opposing motor proteins might act in positioning ERES within cells (V. Gupta and D.J.S., unpublished observations) but it remains unclear how the position of ERES relates to their function.
Early measurements using fluorescence recovery after photobleaching (FRAP) of GFP-tagged ERES markers showed relatively slow turnover times of the order of around 40 s (Stephens et al. 2000). The ability to bleach individual ERES, and measure their subsequent recovery rates, has improved sampling frequencies and led to a re-evaluation of this original data. If one bleaches a large area of cytoplasm, then one observes a slow turnover time for COPII subunits (Stephens et al. 2000) owing to bleaching of both the ERES and surrounding cytosol. In contrast, photobleaching of a much smaller area incorporating a single ERES reveals a half time for recovery of fluorescence of around 4 s (Forster et al. 2006). This suggests that there is a local concentration of COPII subunits within the cytosol in the immediate vicinity of ERES. Other work has hinted at such a possibility before (Orci et al. 1991) but it remains unclear as to how this local concentration is achieved.
FRAP analysis of the turnover activities of COPII components Sar1, Sec23, and Sec13 has allowed further analysis of how the balance between GTPase and GAP activity controls the membrane association of the COPII coat (Forster et al. 2006). Advances in imaging technology have enabled us to determine turnover rates of each individual component at a single ERES. Each turnover rate is now of the order of <5 s. It should be noted of course that these measurements are global population measurements for COPII subunits within a single ERES. Experiments in which the amount of cargo within the ER was modulated were also entirely consistent with in vitro data described above in that they showed that membrane association of COPII subunits is modulated (i.e. sustained) by the presence of transport competent cargo in the ER (Forster et al. 2006). This mechanism of cargo regulating coat subunit kinetics is also employed by the COPI coat assembling of VTC and Golgi membranes (Presley et al. 2002). A significant finding in terms of the mechanism of COPII assembly was that the coat components could continue to cycle on and off the membrane in the presence of a GTP-restricted form of Sar1 (Sar1H79G) (Ward et al. 2001), albeit with reduced recovery. These data showed dynamic association of COPII components with membranes despite perturbation of the GTPase cycle of Sar1. Cargo (YFP-p58, the rodent ERGIC-53 orthologue) could also dynamically associate with these structures (Ward et al. 2001), consistent with the idea that these are in many ways reminiscent of pre-budding complexes.
Role of Sec12 in maintaining COPII coat assembly
Sec12 is a type II transmembrane protein that acts primarily as a GEF for Sar1 (Barlowe and Schekman 1993; Weissman et al. 2001). It is required to generate the active pool of Sar1 in cells to trigger COPII recruitment and in vitro is necessary for the formation of COPII on synthetic liposomes in the presence of GTP. In the absence of Sec12, COPII does not assemble unless a non-hydrolysable form of GTP is included in the assay (Futai et al. 2004). The experiments of Sato and Nakano (2005) and Futai et al. (2004) clearly demonstrate a role for Sec12 in the continual supply of Sar1-GTP during cargo capture by COPII. This is presumably necessary to provide a sufficient membrane-associated pool of Sar1 and Sec23/24 for cargo concentration to occur. Further complexity occurs upon recruitment of Sec13/31 to the membrane. This outer layer of COPII further stimulates the GAP activity of Sec23/24 by an order of magnitude (Antonny et al. 2001; Bi et al. 2007). The ratio of inner coat protein subunits Sec23/24 to the exchange factor Sec12 is 14:1 (Futai et al. 2004). This suggests the tenfold increase in GAP activity, occurring on Sec13/31 binding, is required to overcome the approximate tenfold ratio of Sec12 GEF activity to Sec23/24 GAP activity to drive COPII uncoating once fission is complete (Futai et al. 2004). Sec12 binds preferentially to nucleotide-free Sar1 and is predicted to form a β-propeller consisting of 7 WD repeats (Chardin and Callebaut 2002). Upon mutation of a highly conserved asparagine, N40, present in the asparagine-rich loop of the first blade, the catalytic activity of Sec12ΔCp is reduced to only ∼2% of wild-type protein activity (Futai et al. 2004). This mutation, along with three others, shows critical defects in Sar1 binding and Sec12ΔCp activity, which results in diminished coat stability (Futai et al. 2004). This provides further evidence for nucleotide exchange by Sec12p being responsible for stabilising COPII coat assembly. In S. cerevisiae, a putative Sec12 homologue, Sed4p, is present. Sed4p is an integral membrane protein localized on the ER, which shares 45% identity with the cytoplasmic domain of Sec12p (Gimeno et al. 1995). SED4 exhibits genetic interactions with other COPII components SEC12, SEC16, and SAR1 (Gimeno et al. 1995; Saito et al. 1999). The role of Sed4 in COPII vesicle formation is unclear and no metazoan orthologue has been identified.
In summary, after Sar1-GDP activation by membrane-bound Sec12, Sar1-GTP inserts into the membrane and recruits Sec23/24. The presence of cargo dramatically increases the time Sec23/24 spends associated with the membrane after GTP hydrolysis and subsequent membrane release. Therefore, cargo is required to retain Sec23/24 on the membrane via a “holding” mechanism providing time for sustained, efficient cargo sorting (Forster et al. 2006; Futai et al. 2004; Sato and Nakano 2004).
An N-terminal amphipathic helix in Sar1 initiates membrane curvature
Upon activation of Sar1 by the membrane-bound GEF Sec12, Sar1 undergoes a conformational change resulting in the exposure of an N-terminal amphipathic helix, which inserts into the lipid bilayer anchoring Sar1-GTP to the ER (Huang et al. 2001). This membrane recruitment mechanism is reversed by Sar1 GTP hydrolysis inducing a conformational change in Sar1, which results in retraction of the helix from its hydrophobic pocket in the membrane (Huang et al. 2001). Subsequently, Sar1-GDP is released into the cytosol. The effect of this membrane insertion can be witnessed on synthetic liposomes observed by thin-section EM (Bielli et al. 2005; Lee et al. 2005). Incubating Sar1 and liposomes alone showed no distinguishable effects, however, upon addition of GMPPNP these large spherical liposomes deformed into long, narrow tubules (Lee et al. 2005). These tubules have an average diameter of ∼26 nm, much smaller than the average COPII vesicle. This dramatic effect on the membrane was confirmed to be a direct result of Sar1 N-terminal helix insertion as Sar1 mutants were produced that cannot undergo membrane deformation (Lee et al. 2005). These mutants have bulky hydrophobic residues, which line the membrane contacting side of the amphipathic helix, mutation to alanine removes the hydrophobic force which provides the drive required for membrane insertion of amphipathic helices.
Coupling coat protein assembly and cargo selection to membrane curvature
The bilayer couple hypothesis suggests that membrane deformation results from asymmetric insertion of amphipathic compounds into the lipid membrane; the asymmetry between inner and outer lipid bilayers induces spontaneous membrane curvature (Chernomordik and Kozlov 2003; Farsad and De Camilli 2003). However, a second mechanism proposes that after membrane insertion of the Sar1-GTP N-terminal helix it is the subsequent binding of Sec23/24, via electrostatic interactions with the lipid bilayer, that “captures” these deformations (Bi et al. 2002; Lee et al. 2005). In this model, recruitment and binding of outer COPII subunits Sec13/31 acts to propagate membrane curvature from the initial membrane deformation until a fully coated vesicle is formed (Lee et al. 2005).
Like other members of the Ras superfamily of G proteins Sar1 requires GTP hydrolysis to disrupt switch regions in the protein which cause disruption to the hydrophobic pockets in which the amphipathic helix usually resides (Huang et al. 2001); this disruption results in Sar1-GDP membrane release. Upon deletion of the amphipathic helix, Sar1 can still bind and turnover GTP in the cytosol, as well as retaining its ability to recruit and bind Sec23/24 (Lee et al. 2005). As expected, in this case synthetic liposomes remained large and spherical, as membrane deformation could not be induced. Upon restoring the ability of Sar1 to attach to the membrane, the kinetic turnover rates of the membrane-bound truncated form of Sar1 could be determined using tryptophan fluorescence. The GTPase activity of Sar1 was found to be independent of its membrane insertion properties in conditions with and without Sec12, Sec23/24, and Sec13/31. It was possible for the cytosolic domain of Sec12 to stimulate nucleotide exchange on the truncated form of Sar1. Altogether results show that membrane insertion by the N-terminal helix of Sar1 is purely a localization mechanism and does not appear to induce any conformational changes in the protein but does serve to deform the underlying lipid bilayer (Lee et al. 2005).
The outer layer: Sec13/31 structure and function
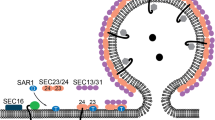
The outer layer of the COPII coat comprises two subunits, Sec13 and Sec31, which assemble into heterotetramers containing two copies of each subunit (Gurkan et al. 2006). These heterotetramers oligomerize forming a cage, which encompasses the prebudding vesicle. Empty COPII cages assemble in vitro from purified recombinant proteins making it possible to solve their structure from cryo-EM and single particle analysis (Stagg et al. 2006). The Sec13/31 cage can assemble into cuboctahedrons, polyhedrons with eight triangular faces and six square faces. This represents the minimal assembly of the outer layer. A cuboctahedron has 12 identical vertices, with two triangles and two squares meeting at each; and 24 identical edges, each separating a triangle from a square. Each vertices consists of four asymmetric edges, and it is the assembly of these vertices that forms the geometric characteristic that directs the assembly of the COPII cage (Stagg et al. 2006). There are two basic structural features behind COPII cage formation, WD40 motifs and α-solenoid motifs (Fath et al. 2007; Stagg et al. 2006). Together they provide the COPII cage with structural stability as well as functional flexibility. The 35 kDa Sec13 subunit is almost entirely composed of a WD40 domain, providing six of the seven β-propellers of the seven-bladed β-propeller motif (Fath et al. 2007). The seventh β-propeller is provided by the N-terminus of Sec31, with the remaining protein comprising two regions of α-solenoid separated by a region of low complexity. These motifs are required to contribute towards protein–protein interactions at the cage vertices (Stagg et al. 2006).
Formation of the cage begins with the association of Sec13 with Sec31 to form one complete WD40 domain (Stagg et al. 2006). The second stage involves the assembly of two Sec13/31 dimers to form a (Sec13-Sec31)–(Sec31-Sec13) heterotetramer with the C-terminal α-solenoid motifs of the Sec31 subunits interacting to form the centre of one asymmetric edge (Stagg et al. 2006). Lastly, the association of multiple heterotetramers, via contacts between four asymmetric edges forming one vertex, finalizes the basic configuration of the COPII cage (Stagg et al. 2006). In total the COPII cage consist of a minimum of 24 Sec13/31 heterotetramers. Ultimately, the number of heterotetramers present determines the vesicle size. This flexibility in size is made possible by making small changes in local contacts that form the vertices; and stems from the ability of both WD40 and α-solenoid motifs to overlap to varying degrees (Stagg et al. 2007). It is also probably that by being comprised of asymmetric associations between edges this allows the generation of greater combinations of asymmetrical vesicle cages (Stagg et al. 2007). The assembly mechanism and a comparison to other coat assembly processes are very nicely described in more detail in (Gurkan et al. 2006).
The initial Sec13/31 crystal structure of the Sec13/31 complex (Fath et al. 2007) is incomplete, missing a large part of the central domain, likely to be the part that contacts the other COPII components. The minimal fragment of Sec31 that can stimulate Sar1-GTPase activity has recently been identified and turns out to lie within this proline rich region. This proline rich domain in fact directly contacts Sar1 and Sec23 (Bi et al. 2007) and the crystal structure of the Sar1-Sec23-Sec31 ternary complex has been solved (Bi et al. 2007). Sec31 directly contributes to the active site on Sar1 positioning it for high efficiency GTPase activity. This important crystallographic work provides a framework for the entire assembly of the COPII coat and provides key insight in to the likely positioning of the inner (Sec23/24) layer relative to the outer (Sec13/31) layer. In addition to the obvious interest for COPII biochemistry, this has key implications in clinical cases where COPII function is defective (see Boyadjiev et al. 2006; Fromme et al. 2007; Lang et al. 2006 and described in detail below). The overall assembly process for the COPII coat is summarized in Fig. 3. Key aspects of this model are discussed in more below. Figure 4 shows the completed minimal cage structure. The positioning of the inner Sec23/24 layer relative to the outer Sec13/31 layer shows the potential accessibility of both the inner layer of the coat and the underlying membrane could be in the final structure. This has important implications for downstream vesicle function such as tethering and signalling.
The role of the COPII coat during and after budding
Until recently, completion of COPII vesicle formation was believed to be rapidly followed by uncoating. In the simplest models of ER-to-Golgi transport in mammalian cells, COPII vesicles uncoat and fuse to form larger carriers that are subsequently transported along microtubules to the Golgi (Mizuno and Singer 1994; Palmer et al. 2005b; Presley et al. 1997; Thyberg and Moskalewski 1985; Watson et al. 2005). The role of homotypic fusion and the role of microtubules in transport are likely to be less significant in yeast owing to the considerably shorter distances along which transport carriers must travel to reach their destination. However, there is now very good evidence for homotypic fusion of COPII vesicles shortly after their formation in mammalian systems.
In contrast to these simple models, recent data suggest that in fact, the COPII coat, and specifically the Sec23/24 layer might in fact act in the recruitment of machinery molecules necessary for subsequent processes. Two key pieces of evidence point to this. First, Sec23 has been shown to recruit the dynactin complex (Watson et al. 2005), which works with dynein to drive motility towards the Golgi (Presley et al. 1997). Second, tethering complexes such as TRAPPI have been shown to associate with the Sec23/24 layer (Cai et al. 2007) suggesting that in fact tethering of vesicles together occurs before full uncoating. This leads one to speculate on the nature of uncoating and perhaps indicates that the Sec13/31 layer is lost at an early point with the inner, Sec23/24 layer being more stably associated. This is emphasized by the recent discovery that Sec23/24 is likely to be positioned beneath the Sec13/31 layer of the COPII coat. In a cuboctahedral structure (Fig. 4) these inner components (including Sec23/24) could therefore be accessible from the cytosolic side.
These findings have prompted the theory that tethers are not simply structural links between donor and acceptor membranes, the role they play in preceding membrane fusion events is anticipated to be more complex. Broadly speaking there are two forms of tethers; long coiled-coil proteins, and more globular multi-subunit complexes. From single particle EM analysis the TRAPP complexes, TRAPPI and TRAPPII, are the best structurally elucidated of all eight tethering complexes (Sacher et al. 2001). The TRAPPI complex acts to specifically promote ER-to-Golgi trafficking and is a large 300 kDa complex with the profile of a flattened dumbbell (Kim et al. 2006; Sacher et al. 2001). Each lobe consists of one of two heterotrimers; either trs20-trs31-bet3, or bet3-trs33-bet5, with a seventh subunit, trs23, linking the two lobes (Kim et al. 2006). TRAPPII is required for intra-Golgi or endosomal trafficking to the late Golgi and includes all of same subunits as TRAPPI, but with an additional three proteins; trs130, trs120, and trs65 (Cai et al. 2005; Sacher et al. 2001).
When COPII vesicles exit the ER in yeast cells they individually target and fuse to the cis-Golgi apparatus. In mammalian cells after COPII budding occurs, the majority of COPII vesicles undergo homotypic fusion events resulting in the formation of larger structures known as ERGIC/VTCs (Xu and Hay 2004). This homotypic fusion step is required for soluble cargo concentration into fewer larger transport carriers, making trafficking between the ER and Golgi a less congested, and more energy efficient process. Tethering events involved in these homotypic fusion events appear at be mediated by the TRAPPI complex (Yu et al. 2006). The TRAPPI subunit mBet3 is the most highly conserved of all the TRAPP complex subunits and has been detected by immuno-EM to be present on both COPII vesicles and budding profiles (Sacher et al. 1998; Yu et al. 2006). Protein binding assays preformed in both yeast and mammalian cells has shown a direct interaction between mBet3 and the COPII inner subunits Sec23 (Cai et al. 2007). Based on the structure of the TRAPPI complex (Kim et al. 2006), a mechanism for TRAPPI-mediated vesicle fusion can be hypothesized. The TRAPPI complex may act as a structural bridge between COPII vesicles with each TRAPPI lobe heterotrimer becoming associated with a different COPII vesicle through an interaction between Bet3 (located at each end of the TRAPPI molecule) and Sec23 (Kim et al. 2006; Sacher et al. 2001). Although TRAPPII also contains one Bet3 subunit per lobe the additional three component subunits could act to obscure the Sec23 binding site (Cai et al. 2007). Thus, one can conclude that for these tethering events to occur COPII vesicles must retain their inner coat subunits until recognized by their specific tethering complexes. Further evidence in support of this idea comes from work showing that the Sec23/24 layer can also interact directly with the GRASP homologue in S. cerevisiae (Behnia et al. 2007). Together these data strongly support the idea that recruitment of tethers precedes full uncoating. The discovery of Rab exchange factors binding coat protein subunits implies a potential regulatory role for Rabs in vesicle uncoating (Cai et al. 2007). It is not yet known at what stage of uncoating vesicle tethering occurs. COPII vesicles may require the loss of Sec13/31 for the Bet3 binding site on Sec23 to become exposed for tethering; likewise Sec13/31 may be required on the budded vesicle for attracting accessory factors associated with this or other processes.
Regulatory role for Rabs in COPII vesicle formation and fusion
The majority of membrane trafficking events within the cell are regulated by Rab GTPases (Gillingham and Munro 2007). Rabs cycle between their cytoplasmic and membrane-bound forms through the actions of GDI (guanine nucleotide inhibitor) and GDF (GDI displacement factor) proteins acting on the membrane-inserting prenyl group. So far over 60 Rab proteins have been identified in mammalian cells (Gillingham and Munro 2007). Different subclasses of Rab GTPases can be defined by their compartment specific properties, and recruitment of downstream signalling effectors. Through interacting with these effectors Rabs regulate multiple trafficking events such as; formation, tethering, docking and fusion of various transport vesicles. Anterograde trafficking from ER to Golgi requires at least two Rab isoforms, Rab1a and Rab1b (Tisdale et al. 1992). These GTPases have been seen to localize at the ER–Golgi interface and within the Golgi apparatus. They are associated with two main roles; they interact with tethering proteins, such as p115, GM130 and golgin-84, as well as playing a role in COPI recruitment (Alvarez et al. 2003; Gillingham and Munro 2007). Each of these features directly impacts on COPII function. Many tethering proteins are known to be either Rab effectors or Rab exchange factors; examples include EEA1 (Christoforidis et al. 1999) and TRAPPI (Cai et al. 2005; Sacher et al. 2001) in endosomal membrane fusion and, more relevant here, TRAPPI (Sacher et al. 2001). Rab1 (known in yeast as Ypt1p), is required to precede SNARE complex formation for yeast homotypic vacuole fusion (Cao et al. 1998). Rab1 is also known to be required for COPII tethering in vitro, with the TRAPPI complex acting as a Rab1 exchange factor (Sacher et al. 2001).
Recent data reinforce the importance of regulation of Rab1 function at the ER/Golgi interface. TBC1D20 has been defined as a GTPase activating protein for Rab1 and Rab2 (Haas et al. 2007). It is localized throughout the ER network in a manner that is dependent, at least in part, on interaction with reticulon proteins. Cells overexpressing TBC1D20 have dramatically altered Golgi morphology (Haas et al. 2007) while retaining unperturbed ERES morphology. It is not itself enriched in COPII-coated structures and could act by ensuring that Rab1 does not become activated at inappropriate locations prior to COPII assembly, requiring Sec23-mediated recruitment of TRAPPI to occur.
Further roles for COPII in the biogenesis of VTCs: recruitment of COPI
Further recent discoveries provide major advances in our understanding of the mechanistic coupling between COPII and COPI. It has been known for many years that these two coat complexes act sequentially during ER-to-Golgi transport (Aridor et al. 1995; Scales et al. 1997; Stephens et al. 2000). When mammalian cells are labelled for COPII components, ERGIC-53, and COPI components both their juxtanuclear and peripheral puncta are seen juxtaposed with one another, and partial, but incomplete, colocalization is observed (Martinez-Menarguez et al. 1999; Stephens et al. 2000) and see Fig. 1c. In many ways these VTC/ERGIC elements can be considered to be functionally similar to the earliest membranes of the Golgi itself—they contain both anterograde and retrograde cargo and recruit COPI thereby being the first physical location for COPI-mediated retrieval. The events that keep these components in close contact are not well understood but it seems logical to suggest there would be a mechanism in place to organize the activities of both coat protein complexes. Such a mechanism for COPII-COPI interplay, based on published data, is shown in Fig. 2. This model indicates that the sequential assembly and disassembly of the two complexes is linked through recruitment and stabilization of these accessory factors.

Schematic model for COPII–COPI coupling. The model shows the assembly and putative coupling of COPI and COPII function in mammalian cells. Starting from the top, the model shows the early stages of assembly of the COPII complex, dependent on Sar1 and Sec16 function. The blue box indicates events at the ERES and the protein components are labelled as detailed in the legend. This model is based on published work and includes known interactions. What is unclear is precisely how many of these interactions occur together i.e. just how large the complexes/subcomplexes can get. For example, we have no real idea whether there is a point at which Sec16 and COPI can be present on the same membrane/vesicle. Sec16 is drawn as part of the budded vesicle as the true fate of Sec16 during the COPII assembly/budding process remains unclear. At the stage of nascent VTC/tethering complex assembly (light blue) we have suggested that Sec13/31 dissociates. This is based on observations linking Sec23/24 to multiple important components that operate post budding such as TRAPPI and dynactin. The nascent VTC stage (light yellow) precedes complete loss of the COPII components, after which one is left with a COPI-coated VTC
The basis for functional coupling of COPII to COPI revolves around the TRAPPI tether and the Arf1-GEF, GBF1. TRAPPI is a guanine nucleotide exchange factor (GEF) for Rab1 (Jones et al. 2000). Gbf1 has a key role in membrane dynamics at the ER–Golgi interface through its direct action of the activation of Arf1 (Garcia-Mata et al. 2003; Niu et al. 2005; Szul et al. 2005, 2007; Zhao et al. 2002, 2006). It is also likely the direct target of brefeldin A at this point in the secretory pathway (which stabilizes an abortive complex of Arf1/Gbf1/GDP). GBF1 has recently been identified as a new Rab1b effector mediating COPI recruitment to the ER-Golgi interface (Monetta et al. 2007) thus linking Rab1b activation (by TRAPPI) to Gbf1 recruitment. Rab1b has been shown to mediate the membrane association properties of GBF1. When expressed in its GTP-restricted form, Rab1b causes an increase in GBF1 and COPI association to peripheral structures located at ERES (Alvarez et al. 2003; Monetta et al. 2007). Subsequently, when Rab1b is depleted from cells Arf1 membrane association to ERES and the Golgi is affected (Monetta et al. 2007). Together these observations suggest a model whereby Rab1b becomes recruited from the cytoplasm to membranes at the ER-Golgi interface (Alvarez et al. 2003). Gbf1 also interacts with the tethering protein p115 (Garcia-Mata and Sztul 2003). GTP-bound Rab1b binds the N-terminal domain of GBF1 (Monetta et al. 2007), with the C-terminal domain of GBF1 able to bind the Golgi membrane tethering protein p115 (Allan et al. 2000). p115 is known to be required at an early stage of vesicle tethering (Alvarez et al. 1999) and is directly involved in recruitment of other key molecules such as GM130 (Moyer et al. 2001). Coiled-coil tethering factor p115 interacts with specific COPII associated SNAREs that are required for homotypic tethering of COPII vesicles, and subsequent VTC formation (Allan et al. 2000). As well as p115 bringing COPII vesicles into close association for SNARE pairings, p115 initiates SNARE complex formation via its N-terminal coiled-coil region being weakly homologous to a SNARE motif (Shorter et al. 2002). Thus a complex network of protein:protein interactions directs the tethering and fusion of COPII-derived vesicles. These processes functionally link the COPII and COPI complexes. Many questions remain unanswered here including the means of regulation of this system and just how many of these multiprotein assemblies are present together at any one time. Is there a sequential aspect to the process whereby for example following recruitment of Gbf1 by TRAPPI, subsequent activation of Rab1b by Gbf1 requires dissociation of TRAPPI?
There is considerable evidence that COPI recruitment is indeed required for anterograde trafficking (e.g. Orci et al. 1997; Shima et al. 1999). It remains unclear whether this is in fact a direct role, for example in the sustained recruitment and activation of dynein, or whether it is in fact solely involved in mediated retrograde recycling of proteins back to the ER but that its physical presence on these membranes is necessary for their function. One could envisage a series of control checks in such a system by which COPI recruitment is required for subsequent recruitment/activation steps but in which COPI does not itself play a direct role.
While this model has its basis in published work there is much that we are unclear about. Does Sec16 incorporate in to vesicles budding from the mammalian ER? Just how long do COPII components remain associated with nascent structures that are beginning to recruit COPI? One must remember that biochemical experiments on permeabilized cells or in vitro assembly assays might not fully reflect endogenous processes, notably in terms of thermodynamics; similarly, imaging experiments are resolution limited or hindered by fixation and processing artefacts. Photobleaching experiments currently provide only an ensemble average of multiple COPII proteins at ERES and not selectively those engaged in vesicle formation versus those associated with the ER membrane. Clearly, a combination of these approaches is essential to a full understanding of these events.
How do COPII vesicles prevent from fusing with the ER? The answer to this appears to be complex but a specific mutant of Tip20p in yeast provides major clues. Tip20p is a component of the SNARE complex mediating fusion of retrograde COPI-derived vesicles with the ER. A specific mutant, Tip20-8, results in cells in which COPII vesicles can fuse back with the ER (Kamena and Spang 2004). This strongly suggests that active mechanisms exist that prevent this “back-fusion” and ensure directional trafficking.
Regulation of COPII coat formation via signalling mechanisms
A key function of the ER in most eukaryotic cells is the sequestration and storage of calcium from the cytosol. In response to extracellular signals, calcium from the ER can undergo rapid release and reuptake in order to trigger intracellular signalling cascades (Berridge et al. 2000). To date, the downstream effects of highly fluctuating calcium concentrations on the ER membrane surface have been speculative as little research has been done into the effects of calcium fluctuation on early secretory pathway trafficking. A recent observation regarding the intracellular localization of penta-EF-hand Ca2+-binding protein ALG-2, apoptosis linked gene 2, has prompted research into the potential role of calcium signalling in COPII vesicle formation. When ALG-2 was expressed in HeLa cells, it localizes to ERES (Shibata et al. 2007; Yamasaki et al. 2006). To confirm this, ALG-2 and ERES marker p125 (Tani et al. 1999) were co-expressed and their partial colocalization was observed (Yamasaki et al. 2006). When intracellular calcium levels were increased, ALG-2 was distributed at the juxtanuclear area colocalizing with COPII outer subunit Sec31A (la Cour et al. 2007). Upon lowering intracellular calcium concentrations ALG-2 is seen to disperse from the juxtanuclear area, which also shows a loss of Sec31A staining as Sec31A dissociated from the membrane and remains cytosolic (la Cour et al. 2007). Experiments undertaken to knockdown and mislocalize ALG-2 also exhibit a decrease in the number of Sec31A puncta within the cells (la Cour et al. 2007; Shibata et al. 2007). Although ALG-2 clearly affects intracellular distribution of Sec31A, there appears to be no effect on function as the amount of Sec13 binding to Sec31 in ALG-2 knockdown cells remains unchanged (Shibata et al. 2007).
Interestingly, this effect of ALG-2 upon Sec31 is isoform specific and only appears to affect Sec31A; ALG-2 shows no phenotypic effect on Sec31B (la Cour et al. 2007). This provides the first key functional distinction between these isoforms (Stankewich et al. 2006). Reasons for this are unknown. Current results implicate a model in which calcium binds to ALG-2 when intracellular calcium levels reach a certain threshold value (la Cour et al. 2007). This interaction induces a conformational change in ALG-2, which allows ALG-2 binding to a proline-rich region of Sec31 (Yamasaki et al. 2006). The conformational change in ALG-2 may confer its ability to discriminate between Sec31 subunit isoforms. A further conformational change in Sec31A could result in a tighter association between the outer coat of COPII and the inner coat, stabilizing Sec31A at ERES.
Protein phosphorylation regulates many events in traffic between the ER and Golgi. Roles have been proposed for protein phosphorylation in the regulation of anterograde transport (Aridor and Balch 2000; Muniz et al. 1996), export of specific cargo (Scott et al. 2003), and in retrograde transport back to the ER (Cabrera et al. 2003). Many of these effects were initially ascribed to protein kinase A (PKA) (Muniz et al. 1996) based on use of the inhibitor H-89; more recent evidence suggests that H-89 may be acting through inhibition of a different kinase (Aridor and Balch 2000; Jamora et al. 1999; Lee and Linstedt 2000). PCTAIRE protein kinases interact directly with COPII and regulate secretory cargo transport (Palmer et al. 2005a). PCTAIREs are cyclin-dependent kinase (cdk) family members with a single substitution (Ser→Cys) in the classical cyclin binding motif. PCTAIREs do not appear to be regulated during the cell cycle, or to associate with any cyclin (Graeser et al. 2002). All cells and tissues examined express at least one PCTAIRE isoform. These data (Palmer et al. 2005a) show that mutations in the ATP binding site that abolish kinase activity (K194R) (Graeser et al. 2002) result in fragmentation of the Golgi and an inhibition of anterograde cargo transport (transport of tsO45-G-GFP); expression of activated mutants causes an equivalent increase in the rate of cargo transport.
COPII-dependent budding from the ER has long been known to be ATP- as well as GTP-dependent (Balch et al. 1986) and can be inhibited by the kinase inhibitor H-89 (Aridor and Balch 2000); it is conceivable that PCTAIRE activity represents this ATP requirement. PCTAIRE kinases bind 14-3-3 proteins (Sladeczek et al. 1997), which have themselves been implicated in the control of ER export. 14-3-3 proteins are believed to act in export by masking specific retention signals in proteins, facilitating their forward transport (O’Kelly et al. 2002). The association of 14-3-3 with some cargoes (e.g. the KCNK3 K+ channel) in this scheme is regulated by phosphorylation (O’Kelly et al. 2002); it is possible that PCTAIRE mediates cargo phosphorylation and acts to transfer 14-3-3 to facilitate cargo export. A recent large-scale affinity column proteomic analysis has identified the Sec23/24 complex as interacting with 14-3-3 proteins (Rubio et al. 2004). Thus 14-3-3 proteins could integrate PCTAIRE and COPII function. PCTAIRE kinases are highly expressed in terminally differentiated and transformed cells and appear to regulate neurite outgrowth (Graeser et al. 2002).
So far the mechanisms of ERES formation, COPII vesicle formation, and efficient ER export have been discussed in terms of cooperative protein–protein interactions. Additional protein–lipid, and lipid–lipid interactions provide regulated modification of ER membrane phospholipid composition which are required to support the cooperative interactions required for selective transport. These include phospholipase D (PLD, Pathre et al. 2003). Phospholipase D catalyses the hydrolysis of the phosphodiester bond of glycerophospholipids to generate phosphatidic acid (PA) and a free head group. Transport from the ER to the Golgi is PLD dependent (Bi et al. 1997); the role of PLD in COPII vesicle formation is complex. It does not directly enhance COPII recruitment to membranes but appears to act in facilitating the recruitment of Sec23/24 following Sar1 recruitment (Pathre et al. 2003). The authors invoke a model of localized Sar1-stimulated phosphatidic acid production in COPII vesicle formation. This would certainly provide a spatial cue for COPII assembly but is difficult to reconcile with the localization of Sec12 to the entire ER membrane thereby presumably leading to Sar1 activation across the entire ER. Once more, the mechanism of restriction of COPII dependent budding to ERES remains the elusive component. The activation of Sar1p only supports coat recruitment when a high proportion of acidic phospholipids are present (Matsuoka et al. 1998). PA could be required to stabilize activated Sar1 and Sec23/24 as both inner COPII subunits have basic residues exposed on their surface (Bi et al. 2002) where possible PA interactions occur. It is likely that these protein lipid interactions are transient, indicating a role in the initial recruitment; sustained stability could then be provided through subunit protein–protein interactions. PA could also play an allosteric role, modulating Sec23/24 for cargo recognition. The Sec23-interacting protein p125 (Tani et al. 1999) has been identified that appears to play a role in the organization of ERES (Shimoi et al. 2005). The protein shows homology to phosphatidic acid preferring phospholipase A1. Recruitment to ERES appears to be mediated by a combination of the PA-binding domain and interaction with Sec23. The cholesterol content of the membrane also has a direct effect on the export of secretory cargo from the ER (Runz et al. 2006). Specifically, reduction in the sterol content of cells caused reduced mobility of a model cargo protein within the ER membrane that correlated with a reduced incorporation in to ERES. The rate of turnover of Sec23 on the membrane was also reduced in sterol-depleted cells. The interplay between COPII and sterol biochemistry is illustrated beautifully but our understanding of the pathways that sense the sterol requirements of cells. In mammalian cells, sterol synthesis is regulated by sterols themselves. Sterols regulatory element binding proteins (SREBPs) require transport to the Golgi and subsequent proteolytic cleavage in order to translocate to the nucleus and act on the relevant genes for cholesterol synthesis. High levels of sterols block the ER-to-Golgi transport of SREBP cleavage-activating protein (SCAP), a sterol-sensing protein that escorts SREBPs (Espenshade et al. 2002). The mechanism underlying this appears to be the selective inhibition of SCAP incorporation in to COPII vesicles. Thus, SREBP/SCAP complexes remain in the ER and SREBP remains transcriptionally inactive. Incorporation of SCAP into COPII vesicles is mediated through binding of a sorting motif-MELADL in SCAP to Sec23/24 (Sun et al. 2005). A further component of the system called Insig renders this motif inaccessible when bound (Sun et al. 2007). The reduction in sterols in the ER leads to dissociation of Insig and transport of SCAP/SREBP to the Golgi.
Further data supporting a role for lipid composition in regulating protein assembly at ERES comes from studies of phosphatidylinositiol-4-kinases (PI4k) (Blumental-Perry et al. 2006). The authors show that the FAPP1-PH domain, through binding to PI4P inhibits ER export. Further to this, they showed apparent recruitment of a GFP-FAPP1-PH-domain to ERES in permeabilized cells. Interestingly, addition of PI4P appears to overcome the requirement for ATP in COPII recruitment in vitro (Blumental-Perry et al. 2006). Much of the data within this paper relies on the use of the FAPPI-PH domain as a PI4P biosensor. It has been well documented that this in fact requires the dual binding of both PI4P and Arf1 for its recruitment to Golgi membranes (Godi et al. 2004; Shin and Nakayama 2004); given the proximity of Arf1 activation to COPII assembly, and the fact that COPII and COPI show such close interplay in terms of function (for examples see Scales et al. 1997; Stephens et al. 2000), one could conclude that the observed effects could reflect PI4P-dependent processes adjacent to rather than at ERES. Furthermore, use of the PH domain of FAPP1 likely sequesters PI4P and potentially activates Arf1-GTP, which could cause significant perturbation of ERES/VTC function.
Defects in secretory pathway functions can lead to disease
Our increased knowledge of the fundamental mechanism underlying ER-to-Golgi transport has enabled us to gain insight in to the molecular basis of a number of disease states. As mentioned above, many diseases that are attributed to “trafficking defects” are in fact primary disorders of protein folding and assembly. The trafficking element here really relates to the important question of quality control in terms of export of properly assembled cargo. However, an increasing number of disease states are now directly attributable to defects in trafficking machinery. A good example of this is the resulting defects in secretion of blood clotting factors and delivery of lysosomal enzymes in patients with mutations in ERGIC-53, a key cargo receptor for ER export. This is described in previous sections and covered in depth elsewhere (e.g. Zhang et al. 2003). Here, we will deal with mutations in the core trafficking machinery i.e. COPII.
Cranio-lenticulosutural dysplasia and integration of COPII coat assembly
Cranio-lenticulo sutural dysplasia (CLSD) is an autosomal recessive syndrome that results from a missense mutation in Sec23A (F382L). This disorder affects cranio-facial development and skeletogenesis and appears to be due to a defect in the deposition of extracellular matrix (Boyadjiev et al. 2006). A chemical mutagenesis screen for phenotypes affecting craniofacial development in zebrafish generated a series of mutants with similar phenotypes (Neuhauss et al. 1996). One such mutant, crusher, is caused by a nonsense mutation L402X in Sec23A and results in a truncated form of Sec23A that is not functionally active (Lang et al. 2006). The truncated domain is known to interact with Sar1; however, as this truncation affects full Sec23A activity it is difficult to specify the outcome of the mutation (Lang et al. 2006). Cartilage of the craniofacial region primarily consists of type II collagen and is derived from cranial neural crest cells that differentiate into chondrocytes. These collagen-secreting cells with Sec23A mutations show a large build up of procollagen which is retained inside distended ER structures (Boyadjiev et al. 2006). The Sec23A mutation is not lethal as presumably the majority of secretory cargoes can still become exported from the ER. One interpretation is that this results from the ongoing, normal, function of the second Sec23 isoform Sec23B. This could be compounded by a preferential requirement for Sec23A in the biogenesis of collagen-containing transport carriers. Such isoform selectivity has been shown for other cargoes (Wendeler et al. 2007). Recent data support the first of these conclusions (Bi et al. 2007; Fromme et al. 2007). Cells from CLSD patients show a redistribution of Sec13/31 to the cytoplasm (Boyadjiev et al. 2006); this suggests that a failure to recruit and stabilize Sec13/31 at ERES prevents procollagen secretion. Now Orci, Schekman and colleagues (Fromme et al. 2007) show that in vitro assays for COPII-dependent budding, Sec23A containing the F382L mutation is less efficient in coupling to the Sec13/31 layer and consequently inhibits COPII-dependent budding. They further show that Sec23B is only expressed at very low levels in calvarial osteoblasts (responsible for ossification of the cranial suture which is significantly defective in CLSD and fibroblasts) which could explain several other aspects of the clinical presentation of CLSD (Boyadjiev et al. 2003). Thus, the combination of mutation in Sec23A and deficiency of Sec23B is proposed to result in inefficient coupling of Sec13/31 and the ensuing phenotypes. Further complexity was revealed in these experiments in that Sar1 isoform selectivity in budding assays was revealed for the first time (Fromme et al. 2007). Intriguingly, Sar1A, but not Sar1B, was found to partially rescue the defect in budding observed when paired with Sec23A-F382L. Sar1A is in fact expressed at high level in calvarial osteoblasts but evidently not a sufficient level to overcome the defect arising from F382L mutation in Sec23A (Fromme et al. 2007). This suggests that Sar1A is not a limiting factor in COPII-dependent budding and the authors suggest that in fact this is caused by the low expression level of Sec23B. An alternative explanation would be that impaired recruitment of Sec13/31 has a greater effect on the ER export of certain cargoes such as collagen and other extracellular matrix components. This could be caused by a specific requirement to generate a transport carrier of such unusual dimensions [to accommodate the large, rod-like structure of assembled collagen protofibrils (Canty and Kadler 2005)]. Some evidence exists for this possibility; coupling of Sar1A with Sec23A-F382L leads to robust packaging of ERGIC-53 (Fromme et al. 2007). siRNA silencing of Sec13 expression in mammalian cells does not significantly reduce ER-to-Golgi transport of tsO45-G (A.K. Townley and D.J.S., unpublished observations).
A key outcome of the analysis of CLSD patient cells is the clear accumulation of budding profiles on distended ER (Fromme et al. 2007). These images show clearly that COPII vesicle fission is inhibited in these cells. The nature of COPII vesicle scission has remained elusive, notably in the absence of any constricting force generator. These new data suggest that the defect in coupling of Sec23/24 to Sec13/31 causing inefficient recruitment of the outer layer leads to a failure of vesicle scission. This is elegantly reinforced by crystallographic data showing that Sec31 directly contacts Sec23 in very close proximity to the F382 residue (Bi et al. 2007). Furthermore, Sec31 also contacts Sar1 directly (Bi et al. 2007) and significantly, this interface lies on the same surface as the F382 residue in Sec23A, i.e. in direct apposition with Sec31. Mechanistically, Sec31 contributes to the GTP binding site of Sar1 and facilitates rapid GTP hydrolysis by optimising the geometry of the active site. In a complete vesicle, this would also position Sec23/24 directly underneath Sec13/31 in the vesicle coat structure (Bi et al. 2007; Fath et al. 2007; Stagg et al. 2006). Thus, Sec13/31 itself provides the driving force for COPII vesicle scission. This role for Sec13/31 is shown in Fig. 3 in which recruitment of Sec13/31 occurs after membrane curvature and cargo accumulation but precedes scission of the vesicle (Fig. 4).

Sequential assembly of the COPII coat structure. Following activation of Sar1-GDP to Sar1-GTP by the membrane-bound GEF Sec12, Sec23/24 is recruited to the ER membrane. Sec23/24 provides the major cargo binding function of the coat. Sec12 could be geometrically excluded from ERES because of membrane curvature. The final outer layer coat components Sec13/13 are recruited just prior to scission events, forming a vesicle. The minimal cage structure is shown; vesicle size would be dictated by cargo incorporation and potentially by other factors. These are schematic illustrations generated using Autodesk Maya 8.0 and are not intended as accurate rendering of the precise atomic coordinates from crystallographic data

Sec16 spatially restricts assembly of the COPII coat. a Outer coat components Sec13/31 form cuboctahedrons in their minimal state. The proline-rich region of Sec13 contacts Sec23/24 and Sar1 GTP stabilizing contacts between the inner and outer coat complexes. This also has the capacity to position Sec23/24 underneath the edges of the cage making the membrane surface (and associated proteins) more accessible. These are schematic illustrations generated using Autodesk Maya 8.0 and are not intended as accurate rendering of the precise atomic coordinates from crystallographic data
Chylomicron retention diseases
Mutation in Sar1B, one of the two human isoforms of Sar1, causes clinically important defects in lipoprotein metabolism (Jones et al. 2003). A number of diseases that can be classified together as chylomicron retention diseases (CMRDs) have now been confirmed to be caused by a specific mutation in the SARA2 gene encoding Sar1B. The mutations either cause truncation or significant changes in the immediate vicinity of the GTP binding site of the protein. It is likely that these patients thrive because they retain functional Sar1A and yet display a specific defect in the assembly and secretion of a subset of apolipoprotein particles, chylomicrons. It remains unclear whether there is a direct defect in the packaging of lipoprotein particles here or if some other related pathway is perturbed. The reader is referred to other literature on this subject for a more detailed description of these disorders and the underlying mutations (Jones et al. 2003; Shoulders et al. 2004). Sar1A and Sar1B show 90% amino acid sequence identity and do not show any significant differences in their intracellular localization (DJS, unpublished observations). Clearly Sar1B has at least one function that is non-redundant with Sar1A; this difference could lie in coupling to a specific packaging receptor for lipoprotein particles, or involve the mobilization of lipid rather than protein. As described above recent data show that Sar1 can directly couple to the Sec13/31 outer layer of the COPII coat (Bi et al. 2007; Fromme et al. 2007). The subtle differences that have been uncovered between Sar1A and Sar1B in these studies might also provide an explanation for the molecular basis of the defect in CMRD. One possibility is that the weaker affinity of Sar1B for Sec13/31 facilitates the formation of a flexible cage required for the packaging of chylomicron lipoproteins. This is probably over-simplistic since other atypically large cargoes should also show defective packaging and therefore one might expect further clinical symptoms in patients in which Sar1B is mutated. A clear definition of the molecular basis for CMRD remains to be determined.
Hijacking of COPII pathways by viruses and microbial pathogens
Many pathogenic bacteria and viruses are known to modulate host cell trafficking machineries. We know most about modulation of endocytic systems during viral or bacterial entry, notably through hijacking of clathrin-dependent endocytic pathways (see Veiga et al. 2007 for an elegant recent illustration of this). Increasingly, examples are coming to light of viruses that exploit the COPII pathway during their life cycle, for example the biogenesis of the poliovirus particles (Rust et al. 2001) and the formation of vacuoles used by Legionella for its replication (Robinson and Roy 2006). Bacterial systems also co-opt host pathways to their own advantage. A good recent example of this comes from the NleA protein from Escherichia coli. This protein is transferred from the bacteria to host cells through a type III secretion system where it localizes primarily to a juxtanuclear compartment (Gruenheid et al. 2004). Recent use of biochemical reconstitution of COPII-dependent budding has shown that NleA in fact binds to Sec24 modulates COPII vesicle formation from the ER (Kim et al. 2007). NleA appears to stabilize the binding of Sec23/24 to liposomes in the presence of GTP, inducing the formation of irregular aggregates of COPII proteins. These data suggest that inhibition of COPII function is a contributing factor to bacterial virulence. Many of these factors also affect other closely linked pathways such as the COPI system [for recent examples see (Belov et al. 2007; Wessels et al. 2006)] and highlight the importance of a full molecular understanding of host membrane trafficking pathways.
Conclusions
Clearly, our knowledge of membrane dynamics at the ER/Golgi interface is far from complete. The combination of technology available to us today, including yeast genetics, biochemical reconstitution, live cell imaging and whole organism physiology, should ensure that we rapidly reach a greater understanding of the mechanistic basis for selective cargo export and intracellular organization in health and disease. Increasingly, our understanding will tend more towards physiologically relevant systems and the differences that underlie intracellular patterning and function in different species and cell types during and after differentiation.
References
Allan BB, Moyer BD, Balch WE (2000) Rab1 recruitment of p115 into a cis-SNARE complex: programming budding COPII vesicles for fusion. Science 289:444–448
Alvarez C, Fujita H, Hubbard A, Sztul E (1999) ER to Golgi transport: requirement for p115 at a pre-Golgi VTC stage. J Cell Biol 147:1205–1222
Alvarez C, Garcia-Mata R, Brandon E, Sztul E (2003) COPI Recruitment is modulated by a Rab1b-dependent mechanism. Mol Biol Cell 14:2116–2127
Antonny B, Madden D, Hamamoto S, Orci L, Schekman R (2001) Dynamics of the COPII coat with GTP and stable analogues. Nat Cell Biol 3:531–537
Antonny B, Gounon P, Schekman R, Orci L (2003) Self-assembly of minimal COPII cages. EMBO Rep 4:419–424
Appenzeller-Herzog C, Hauri HP (2006) The ER-Golgi intermediate compartment (ERGIC): in search of its identity and function. J Cell Sci 119:2173–2183
Appenzeller C, Andersson H, Kappeler F, Hauri HP (1999) The lectin ERGIC-53 is a cargo transport receptor for glycoproteins. Nat Cell Biol 1:330–334
Aridor M, Bannykh SI, Rowe T, Balch WE (1995) Sequential coupling between COPII and COPI vesicle coats in endoplasmic reticulum to Golgi transport. J Cell Biol 131:875–893
Aridor M, Weissman J, Bannykh S, Nuoffer C, Balch WE (1998) Cargo selection by the COPII budding machinery during export from the ER. J Cell Biol 141:61–70
Aridor M, Balch WE (2000) Kinase signaling initiates coat complex II (COPII) recruitment and export from the mammalian endoplasmic reticulum. J Biol Chem 275:35673–35676
Aridor M, Hannan LA (2000) Traffic jam: a compendium of human diseases that affect intracellular transport processes. Traffic 1:836–851
Aridor M, Hannan LA (2002) Traffic jams II: an update of diseases of intracellular transport. Traffic 3:781–790
Balch WE, Elliott MM, Keller DS (1986) ATP-coupled transport of vesicular stomatitis virus G protein between the endoplasmic reticulum and the Golgi. J Biol Chem 261:14681–14689
Bannykh SI, Rowe T, Balch WE (1996) The organization of endoplasmic reticulum export complexes. J Cell Biol 135:19–35
Bannykh SI, Balch WE (1997) Membrane dynamics at the endoplasmic reticulum–Golgi interface. J Cell Biol 138:1–4
Barlowe C, Schekman R (1993) SEC12 encodes a guanine-nucleotide-exchange factor essential for transport vesicle budding from the ER. Nature 365:347–349
Barlowe C, Orci L, Yeung T, Hosobuchi M, Hamamoto S, Salama N, Rexach MF, Ravazzola M, Amherdt M, Schekman R (1994) COPII: a membrane coat formed by Sec proteins that drive vesicle budding from the endoplasmic reticulum. Cell 77:895–907
Barlowe C (2003) Signals for COPII-dependent export from the ER: what’s the ticket out? Trends Cell Biol 13:295–300
Barrowman J, Wang W, Zhang Y, Ferro-Novick S (2003) The Yip1p.Yif1p complex is required for the fusion competence of endoplasmic reticulum-derived vesicles. J Biol Chem 278:19878–19884
Behnia R, Barr FA, Flanagan JJ, Barlowe C, Munro S (2007) The yeast orthologue of GRASP65 forms a complex with a coiled-coil protein that contributes to ER to Golgi traffic. J Cell Biol 176:255–261
Belov GA, Altan-Bonnet N, Kovtunovych G, Jackson CL, Lippincott-Schwartz J, Ehrenfeld E (2007) Hijacking components of the cellular secretory pathway for replication of poliovirus RNA. J Virol 81:558–567
Ben-Tekaya H, Miura K, Pepperkok R, Hauri HP (2005) Live imaging of bidirectional traffic from the ERGIC. J Cell Sci 118:357–367
Berridge MJ, Lipp P, Bootman MD (2000) The versatility and universality of calcium signalling. Nat Rev Mol Cell Biol 1:11–21
Bevis BJ, Hammond AT, Reinke CA, Glick BS (2002) De novo formation of transitional ER sites and Golgi structures in Pichia pastoris. Nat Cell Biol 4:750–756
Bhattacharyya D, Glick BS (2007) Two mammalian Sec16 homologues have nonredundant functions in endoplasmic reticulum (ER) export and transitional ER organization. Mol Biol Cell 18:839–849
Bi K, Roth MG, Ktistakis NT (1997) Phosphatidic acid formation by phospholipase D is required for transport from the endoplasmic reticulum to the Golgi complex. Curr Biol 7:301–307
Bi X, Corpina RA, Goldberg J (2002) Structure of the Sec23/24-Sar1 pre-budding complex of the COPII vesicle coat. Nature 419:271–277
Bi X, Mancias JD, Goldberg J (2007) Insights into COPII coat nucleation from the structure of Sec23*Sar1 complexed with the active fragment of Sec31. Dev Cell 13:635–645
Bielli A, Haney CJ, Gabreski G, Watkins SC, Bannykh SI, Aridor M (2005) Regulation of Sar1 NH2 terminus by GTP binding and hydrolysis promotes membrane deformation to control COPII vesicle fission. J Cell Biol 171:919–924
Blumental-Perry A, Haney CJ, Weixel KM, Watkins SC, Weisz OA, Aridor M (2006) Phosphatidylinositol 4-phosphate formation at ER exit sites regulates ER export. Dev Cell 11:671–682
Bonifacino JS, Glick BS (2004) The mechanisms of vesicle budding and fusion. Cell 116:153–166
Boyadjiev SA, Justice CM, Eyaid W, McKusick VA, Lachman RS, Chowdry AB, Jabak M, Zwaan J, Wilson AF, Jabs EW (2003) A novel dysmorphic syndrome with open calvarial sutures and sutural cataracts maps to chromosome 14q13-q21. Hum Genet 113:1–9
Boyadjiev SA, Fromme JC, Ben J, Chong SS, Nauta C, Hur DJ, Zhang G, Hamamoto S, Schekman R, Ravazzola M et al (2006) Cranio-lenticulo-sutural dysplasia is caused by a SEC23A mutation leading to abnormal endoplasmic-reticulum-to-Golgi trafficking. Nat Genet 38:1192–1197
Bulbarelli A, Sprocati T, Barberi M, Pedrazzini E, Borgese N (2002) Trafficking of tail-anchored proteins: transport from the endoplasmic reticulum to the plasma membrane and sorting between surface domains in polarised epithelial cells. J Cell Sci 115:1689–1702
Cabrera M, Muniz M, Hidalgo J, Vega L, Martin ME, Velasco A (2003) The retrieval function of the KDEL receptor requires PKA phosphorylation of its C-terminus. Mol Biol Cell 14:4114–4125
Cai H, Zhang Y, Pypaert M, Walker L, Ferro-Novick S (2005) Mutants in trs120 disrupt traffic from the early endosome to the late Golgi. J Cell Biol 171:823–833
Cai H, Yu S, Menon S, Cai Y, Lazarova D, Fu C, Reinisch K, Hay JC, Ferro-Novick S (2007) TRAPPI tethers COPII vesicles by binding the coat subunit Sec23. Nature 445:941–944
Calero M, Collins RN (2002) Saccharomyces cerevisiae Pra1p/Yip3p interacts with Yip1p and Rab proteins. Biochem Biophys Res Commun 290:676–681
Canty EG, Kadler KE (2005) Procollagen trafficking, processing and fibrillogenesis. J Cell Sci 118:1341–1353
Cao X, Ballew N, Barlowe C (1998) Initial docking of ER-derived vesicles requires Uso1p and Ypt1p but is independent of SNARE proteins. Embo J 17:2156–2165
Chardin P, Callebaut I (2002) The yeast Sar exchange factor Sec12, and its higher organism orthologs, fold as beta-propellers. FEBS Lett 525:171–173
Chernomordik LV, Kozlov MM (2003) Protein–lipid interplay in fusion and fission of biological membranes. Annu Rev Biochem 72:175–207
Christoforidis S, McBride HM, Burgoyne RD, Zerial M (1999) The Rab5 effector EEA1 is a core component of endosome docking. Nature 397:621–625
Connerly PL, Esaki M, Montegna EA, Strongin DE, Levi S, Soderholm J, Glick BS (2005) Sec16 is a determinant of transitional ER organization. Curr Biol 15:1439–1447
Dudognon P, Maeder-Garavaglia C, Carpentier JL, Paccaud JP (2004) Regulation of a COPII component by cytosolic O-glycosylation during mitosis. FEBS Lett 561:44–50
Eilers M, Shekar SC, Shieh T, Smith SO, Fleming PJ (2000) Internal packing of helical membrane proteins. Proc Natl Acad Sci USA 97:5796–5801
Ellgaard L, Helenius A (2003) Quality control in the endoplasmic reticulum. Nat Rev Mol Cell Biol 4:181–191
Espenshade P, Gimeno RE, Holzmacher E, Teung P, Kaiser CA (1995) Yeast SEC16 gene encodes a multidomain vesicle coat protein that interacts with Sec23p. J Cell Biol 131:311–324
Espenshade PJ, Li WP, Yabe D (2002) Sterols block binding of COPII proteins to SCAP, thereby controlling SCAP sorting in ER. Proc Natl Acad Sci USA 99:11694–11699
Farsad K, De Camilli P (2003) Mechanisms of membrane deformation. Curr Opin Cell Biol 15:372–381
Fath S, Mancias JD, Bi X, Goldberg J (2007) Structure and organization of coat proteins in the COPII cage. Cell 129:1325–1336
Forster R, Weiss M, Zimmermann T, Reynaud EG, Verissimo F, Stephens DJ, Pepperkok R (2006) Secretory cargo regulates the turnover of COPII subunits at single ER exit sites. Curr Biol 16:173–179
Fromme JC, Ravazzola M, Hamamoto S, Al-Balwi M, Eyaid W, Boyadjiev SA, Cosson P, Schekman R, Orci L (2007) The genetic basis of a craniofacial disease provides insight into COPII coat assembly. Dev Cell 13:623–634
Futai E, Hamamoto S, Orci L, Schekman R (2004) GTP/GDP exchange by Sec12p enables COPII vesicle bud formation on synthetic liposomes. EMBO J 23:4146–4155
Garcia-Mata R, Sztul E (2003) The membrane-tethering protein p115 interacts with GBF1, an ARF guanine-nucleotide-exchange factor. EMBO Rep 4:320–325
Garcia-Mata R, Szul T, Alvarez C, Sztul E (2003) ADP-ribosylation factor/COPI-dependent events at the endoplasmic reticulum–Golgi interface are regulated by the guanine nucleotide exchange factor GBF1. Mol Biol Cell 14:2250–2261
Gillingham AK, Munro S (2007) The small g proteins of the arf family and their regulators. Annu Rev Cell Dev Biol 23:579–611
Gimeno RE, Espenshade P, Kaiser CA (1995) SED4 encodes a yeast endoplasmic reticulum protein that binds Sec16p and participates in vesicle formation. J Cell Biol 131:325–338
Gimeno RE, Espenshade P, Kaiser CA (1996) COPII coat subunit interactions: Sec24p and Sec23p bind to adjacent regions of Sec16p. Mol Biol Cell 7:1815–1823
Godi A, Di Campli A, Konstantakopoulos A, Di Tullio G, Alessi DR, Kular GS, Daniele T, Marra P, Lucocq JM, De Matteis MA (2004) FAPPs control Golgi-to-cell-surface membrane traffic by binding to ARF and PtdIns(4)P. Nat Cell Biol 6:393–404
Gould SJ, McCollum D, Spong AP, Heyman JA, Subramani S (1992) Development of the yeast Pichia pastoris as a model organism for a genetic and molecular analysis of peroxisome assembly. Yeast 8:613–628
Graeser R, Gannon J, Poon RY, Dubois T, Aitken A, Hunt T (2002) Regulation of the CDK-related protein kinase PCTAIRE-1 and its possible role in neurite outgrowth in Neuro-2A cells. J Cell Sci 115:3479–3490
Gruenheid S, Sekirov I, Thomas NA, Deng W, O’Donnell P, Goode D, Li Y, Frey EA, Brown NF, Metalnikov P et al (2004) Identification and characterization of NleA, a non-LEE-encoded type III translocated virulence factor of enterohaemorrhagic Escherichia coli O157:H7. Mol Microbiol 51:1233–1249
Gurkan C, Stagg SM, Lapointe P, Balch WE (2006) The COPII cage: unifying principles of vesicle coat assembly. Nat Rev Mol Cell Biol 7:727–738
Haas AK, Yoshimura S, Stephens DJ, Preisinger C, Fuchs E, Barr FA (2007) Analysis of GTPase-activating proteins reveals that Rab1 and Rab43 are key Rabs required for the maintenance of a functional Golgi in human cells. J Cell Sci 120:2997–3010
Hammond AT, Glick BS (2000) Dynamics of transitional endoplasmic reticulum sites in vertebrate cells. Mol Biol Cell 11:3013–3030
Hanton SL, Bortolotti LE, Renna L, Stefano G, Brandizzi F (2005) Crossing the divide-transport between the endoplasmic reticulum and Golgi apparatus in plants. Traffic 6:267–277
Heidtman M, Chen CZ, Collins RN, Barlowe C (2003) A role for Yip1p in COPII vesicle biogenesis. J Cell Biol 163:57–69
Heidtman M, Chen CZ, Collins RN, Barlowe C (2005) Yos1p is a novel subunit of the Yip1p–Yif1p complex and is required for transport between the endoplasmic reticulum and the Golgi complex. Mol Biol Cell 16:1673–1683
Hicke L, Schekman R (1989) Yeast Sec23p acts in the cytoplasm to promote protein transport from the endoplasmic reticulum to the Golgi complex in vivo and in vitro. Embo J 8:1677–1684
Hicke L, Yoshihisa T, Schekman R (1992) Sec23p and a novel 105-kDa protein function as a multimeric complex to promote vesicle budding and protein transport from the endoplasmic reticulum. Mol Biol Cell 3:667–676
Higashio H, Sato K, Nakano A (2007) Smy2p participates in COPII vesicle formation through the interaction with Sec23p/Sec24p subcomplex. Traffic doi: 10.1111/j.1600–0854.2007.00668.x
Horton AC, Ehlers MD (2004) Secretory trafficking in neuronal dendrites. Nat Cell Biol 6:585–591
Huang M, Weissman JT, Beraud-Dufour S, Luan P, Wang C, Chen W, Aridor M, Wilson IA, Balch WE (2001) Crystal structure of Sar1-GDP at 1.7 A resolution and the role of the NH2 terminus in ER export. J Cell Biol 155:937–948
Hurtley SM, Helenius A (1989) Protein oligomerization in the endoplasmic reticulum. Annu Rev Cell Biol 5:277–307
Iinuma T, Shiga A, Nakamoto K, O’Brien MB, Aridor M, Arimitsu N, Tagaya M, Tani K (2007) Mammalian Sec16/p250 plays a role in membrane traffic from the endoplasmic reticulum. J Biol Chem 282:17632–17639
Itin C, Schindler R, Hauri HP (1995) Targeting of protein ERGIC-53 to the ER/ERGIC/cis-Golgi recycling pathway. J Cell Biol 131:57–67
Jamora C, Yamanouye N, Van Lint J, Laudenslager J, Vandenheede JR, Faulkner DJ, Malhotra V (1999) Gbetagamma-mediated regulation of Golgi organization is through the direct activation of protein kinase D. Cell 98:59–68
Jones B, Jones EL, Bonney SA, Patel HN, Mensenkamp AR, Eichenbaum-Voline S, Rudling M, Myrdal U, Annesi G, Naik S et al (2003) Mutations in a Sar1 GTPase of COPII vesicles are associated with lipid absorption disorders. Nat Genet 34:29–31
Jones S, Newman C, Liu F, Segev N (2000) The TRAPP complex is a nucleotide exchanger for Ypt1 and Ypt31/32. Mol Biol Cell 11:4403–4411
Kaiser CA, Schekman R (1990) Distinct sets of SEC genes govern transport vesicle formation and fusion early in the secretory pathway. Cell 61:723–733
Kamena F, Spang A (2004) Tip20p prohibits back-fusion of COPII vesicles with the endoplasmic reticulum. Science 304:286–289
Kapetanovich L, Baughman C, Lee TH (2005) Nm23H2 facilitates coat protein complex II assembly and endoplasmic reticulum export in mammalian cells. Mol Biol Cell 16:835–848
Kappeler F, Klopfenstein DR, Foguet M, Paccaud JP, Hauri HP (1997) The recycling of ERGIC-53 in the early secretory pathway. ERGIC-53 carries a cytosolic endoplasmic reticulum-exit determinant interacting with COPII. J Biol Chem 272:31801–31808
Karsten V, Hegde RS, Sinai AP, Yang M, Joiner KA (2004) Transmembrane domain modulates sorting of membrane proteins in Toxoplasma gondii. J Biol Chem 279:26052–26057
Kikuno R, Nagase T, Nakayama M, Koga H, Okazaki N, Nakajima D, Ohara O (2004) HUGE: a database for human KIAA proteins, a 2004 update integrating HUGEppi and ROUGE. Nucleic Acids Res 32:D502–D504
Kim J, Thanabalasuriar A, Chaworth-Musters T, Fromme JC, Frey EA, Lario PI, Metalnikov P, Rizg K, Thomas NA, Lee SF et al (2007) The bacterial virulence factor NleA inhibits cellular protein secretion by disrupting mammalian COPII function. Cell Host Microb 2:160–171
Kim YG, Raunser S, Munger C, Wagner J, Song YL, Cygler M, Walz T, Oh BH, Sacher M (2006) The architecture of the multisubunit TRAPP I complex suggests a model for vesicle tethering. Cell 127:817–830
Kirk SJ, Ward TH (2007) COPII under the microscope. Semin Cell Dev Biol
Kuge O, Dascher C, Orci L, Rowe T, Amherdt M, Plutner H, Ravazzola M, Tanigawa G, Rothman JE, Balch WE (1994) Sar1 promotes vesicle budding from the endoplasmic reticulum but not Golgi compartments. J Cell Biol 125:51–65
la Cour JM, Mollerup J, Berchtold MW (2007) ALG-2 oscillates in subcellular localization, unitemporally with calcium oscillations. Biochem Biophys Res Commun 353:1063–1067
Ladinsky MS, Mastronarde DN, McIntosh JR, Howell KE, Staehelin LA (1999) Golgi structure in three dimensions: functional insights from the normal rat kidney cell. J Cell Biol 144:1135–1149
Lang MR, Lapierre LA, Frotscher M, Goldenring JR, Knapik EW (2006) Secretory COPII coat component Sec23a is essential for craniofacial chondrocyte maturation. Nat Genet 38:1198–1203
Lederkremer GZ, Cheng Y, Petre BM, Vogan E, Springer S, Schekman R, Walz T, Kirchhausen T (2001) Structure of the Sec23p/24p and Sec13p/31p complexes of COPII. Proc Natl Acad Sci USA 98:10704–10709
Lee MC, Orci L, Hamamoto S, Futai E, Ravazzola M, Schekman R (2005) Sar1p N-terminal helix initiates membrane curvature and completes the fission of a COPII vesicle. Cell 122:605–617
Lee MC, Miller EA (2007) Molecular mechanisms of COPII vesicle formation. Semin Cell Dev Biol
Lee TH, Linstedt AD (2000) Potential role for protein kinases in regulation of bidirectional endoplasmic reticulum-to-Golgi transport revealed by protein kinase inhibitor H89. Mol Biol Cell 11:2577–2590
Letourneur F, Hennecke S, Demolliere C, Cosson P (1995) Steric masking of a dilysine endoplasmic reticulum retention motif during assembly of the human high affinity receptor for immunoglobulin E. J Cell Biol 129:971–978
Ma D, Zerangue N, Raab-Graham K, Fried SR, Jan YN, Jan LY (2002) Diverse trafficking patterns due to multiple traffic motifs in G protein-activated inwardly rectifying potassium channels from brain and heart. Neuron 33:715–729
Mallabiabarrena A, Fresno M, Alarcon B (1992) An endoplasmic reticulum retention signal in the CD3 epsilon chain of the T-cell receptor. Nature 357:593–596
Mancias JD, Goldberg J (2005) Exiting the endoplasmic reticulum. Traffic 6:278–285
Martinez-Menarguez JA, Geuze HJ, Slot JW, Klumperman J (1999) Vesicular tubular clusters between the ER and Golgi mediate concentration of soluble secretory proteins by exclusion from COPI-coated vesicles. Cell 98:81–90
Matern H, Yang X, Andrulis E, Sternglanz R, Trepte HH, Gallwitz D (2000) A novel Golgi membrane protein is part of a GTPase-binding protein complex involved in vesicle targeting. Embo J 19:4485–4492
Matsuoka K, Orci L, Amherdt M, Bednarek SY, Hamamoto S, Schekman R, Yeung T (1998) COPII-coated vesicle formation reconstituted with purified coat proteins and chemically defined liposomes. Cell 93:263–275
Miller EA, Beilharz TH, Malkus PN, Lee MC, Hamamoto S, Orci L, Schekman R (2003) Multiple cargo binding sites on the COPII subunit Sec24p ensure capture of diverse membrane proteins into transport vesicles. Cell 114:497–509
Mizuno M, Singer SJ (1994) A possible role for stable microtubules in intracellular transport from the endoplasmic reticulum to the Golgi apparatus. J Cell Sci 107:1321–1331
Monetta P, Slavin I, Romero N, Alvarez C (2007) Rab1b interacts with GBF1 and modulates both ARF1 dynamics and COPI association. Mol Biol Cell 18:2400–2410
Morsomme P, Riezman H (2002) The Rab GTPase Ypt1p and tethering factors couple protein sorting at the ER to vesicle targeting to the Golgi apparatus. Dev Cell 2:307–317
Morsomme P, Prescianotto-Baschong C, Riezman H (2003) The ER v-SNAREs are required for GPI-anchored protein sorting from other secretory proteins upon exit from the ER. J Cell Biol 162:403–412
Mossessova E, Bickford LC, Goldberg J (2003) SNARE Selectivity of the COPII Coat. Cell 114:483–495
Moussalli M, Pipe SW, Hauri HP, Nichols WC, Ginsburg D, Kaufman RJ (1999) Mannose-dependent endoplasmic reticulum (ER)-Golgi intermediate compartment-53-mediated ER to Golgi trafficking of coagulation factors V and VIII. J Biol Chem 274:32539–32542
Moyer BD, Allan BB, Balch WE (2001) Rab1 interaction with a GM130 effector complex regulates COPII vesicle cis-Golgi tethering. Traffic 2:268–276
Muniz M, Alonso M, Hidalgo J, Velasco A (1996) A regulatory role for cAMP-dependent protein kinase in protein traffic along the exocytic route. J Biol Chem 271:30935–30941
Muniz M, Morsomme P, Riezman H (2001) Protein sorting upon exit from the endoplasmic reticulum. Cell 104:313–320
Munro S (1995) An investigation of the role of transmembrane domains in Golgi protein retention. Embo J 14:4695–4704
Nakano A, Brada D, Schekman R (1988) A membrane glycoprotein, Sec12p, required for protein transport from the endoplasmic reticulum to the Golgi apparatus in yeast. J Cell Biol 107:851–863
Nakano A, Muramatsu M, Murakami A, Kimura K (1989) A novel GTP-binding protein, Sar1p, is involved in transport from the endoplasmic reticulum to the Golgi apparatus. J Cell Biol 109:2677–2691
Neuhauss SC, Solnica-Krezel L, Schier AF, Zwartkruis F, Stemple DL, Malicki J, Abdelilah S, Stainier DY, Driever W (1996) Mutations affecting craniofacial development in zebrafish. Development 123:357–367
Nichols WC, Seligsohn U, Zivelin A, Terry VH, Hertel CE, Wheatley MA, Moussalli MJ, Hauri HP, Ciavarella N, Kaufman RJ, Ginsburg D (1998) Mutations in the ER-Golgi intermediate compartment protein ERGIC-53 cause combined deficiency of coagulation factors V and VIII. Cell 93:61–70
Niu TK, Pfeifer AC, Lippincott-Schwartz J, Jackson CL (2005) Dynamics of GBF1, a Brefeldin A-sensitive Arf1 exchange factor at the Golgi. Mol Biol Cell 16:1213–1222
Novick P, Field C, Schekman R (1980) Identification of 23 complementation groups required for post-translational events in the yeast secretory pathway. Cell 21:205–215
Novick P, Ferro S, Schekman R (1981) Order of events in the yeast secretory pathway. Cell 25:461–469
Nufer O, Guldbrandsen S, Degen M, Kappeler F, Paccaud JP, Tani K, Hauri HP (2002) Role of cytoplasmic C-terminal amino acids of membrane proteins in ER export. J Cell Sci 115:619–628
Nufer O, Hauri HP (2003) ER export: call 14-3-3. Curr Biol 13:R391–R393
Nufer O, Kappeler F, Guldbrandsen S, Hauri HP (2003) ER export of ERGIC-53 is controlled by cooperation of targeting determinants in all three of its domains. J Cell Sci 116:4429–4440
O’Kelly I, Butler MH, Zilberberg N, Goldstein SA (2002) Forward transport. 14-3-3 binding overcomes retention in endoplasmic reticulum by dibasic signals. Cell 111:577–588
Orci L, Ravazzola M, Meda P, Holcomb C, Moore HP, Hicke L, Schekman R (1991) Mammalian Sec23p homologue is restricted to the endoplasmic reticulum transitional cytoplasm. Proc Natl Acad Sci USA 88:8611–8615
Orci L, Stamnes M, Ravazzola M, Amherdt M, Perrelet A, Söllner TH, Rothman JE (1997) Bidirectional transport by distinct populations of COPI-coated vesicles. Cell 90:335–349
Otte S, Barlowe C (2004) Sorting signals can direct receptor-mediated export of soluble proteins into COPII vesicles. Nature Cell Biology 6:1189–1194
Palade G (1975) Intracellular aspects of the process of protein synthesis. Science 189:347–358
Palmer KJ, Konkel JE, Stephens DJ (2005a) PCTAIRE protein kinases interact directly with the COPII complex and modulate secretory cargo transport. J Cell Sci
Palmer KJ, Watson P, Stephens DJ (2005b) The role of microtubules in transport between the endoplasmic reticulum and Golgi apparatus in mammalian cells. Biochem Soc Symp 1–13
Pathre P, Shome K, Blumental-Perry A, Bielli A, Haney CJ, Alber S, Watkins SC, Romero G, Aridor M (2003) Activation of phospholipase D by the small GTPase Sar1p is required to support COPII assembly and ER export. Embo J 22:4059–4069
Payne WE, Kaiser CA, Bevis BJ, Soderholm J, Fu D, Sears IB, Glick BS (2000) Isolation of Pichia pastoris genes involved in ER-to-Golgi transport. Yeast 16:979–993
Presley JF, Cole NB, Schroer TA, Hirschberg K, Zaal KJ, Lippincott-Schwartz J (1997) ER-to-Golgi transport visualized in living cells. Nature 389:81–85
Presley JF, Ward TH, Pfeifer AC, Siggia ED, Phair RD, Lippincott-Schwartz J (2002) Dissection of COPI and Arf1 dynamics in vivo and role in Golgi membrane transport. Nature 417:187–193
Ralston E, Ploug T, Kalhovde J, Lomo T (2001) Golgi complex, endoplasmic reticulum exit sites, and microtubules in skeletal muscle fibers are organized by patterned activity. J Neurosci 21:875–883
Robinson CG, Roy CR (2006) Attachment and fusion of endoplasmic reticulum with vacuoles containing Legionella pneumophila. Cell Microbiol 8:793–805
Rossanese OW, Soderholm J, Bevis BJ, Sears IB, O’Connor J, Williamson EK, Glick BS (1999) Golgi structure correlates with transitional endoplasmic reticulum organization in Pichia pastoris and Saccharomyces cerevisiae. J Cell Biol 145:69–81
Rubio MP, Geraghty KM, Wong BH, Wood NT, Campbell DG, Morrice N, Mackintosh C (2004) 14-3-3-affinity purification of over 200 human phosphoproteins reveals new links to regulation of cellular metabolism, proliferation and trafficking. Biochem J 379:395–408
Runz H, Miura K, Weiss M, Pepperkok R (2006) Sterols regulate ER-export dynamics of secretory cargo protein ts-O45-G. Embo J 25:2953–2965
Rust RC, Landmann L, Gosert R, Tang BL, Hong W, Hauri HP, Egger D, Bienz K (2001) Cellular COPII proteins are involved in production of the vesicles that form the poliovirus replication complex. J Virol 75:9808–9818
Sacher M, Jiang Y, Barrowman J, Scarpa A, Burston J, Zhang L, Schieltz D, Yates JR 3rd, Abeliovich H, Ferro-Novick S (1998) TRAPP, a highly conserved novel complex on the cis-Golgi that mediates vesicle docking and fusion. Embo J 17:2494–2503
Sacher M, Barrowman J, Wang W, Horecka J, Zhang Y, Pypaert M, Ferro-Novick S (2001) TRAPP I implicated in the specificity of tethering in ER-to-Golgi transport. Mol Cell 7:433–442
Saito-Nakano Y, Nakano A (2000) Sed4p functions as a positive regulator of Sar1p probably through inhibition of the GTPase activation by Sec23p. Genes Cells 5:1039–1048
Saito Y, Yamanushi T, Oka T, Nakano A (1999) Identification of SEC12, SED4, truncated SEC16, and EKS1/HRD3 as multicopy suppressors of ts mutants of Sar1 GTPase. J Biochem (Tokyo) 125:130–137
Salama NR, Schekman RW (1995) The role of coat proteins in the biosynthesis of secretory proteins. Curr Opin Cell Biol 7:536–543
Salama NR, Chuang JS, Schekman RW (1997) Sec31 encodes an essential component of the COPII coat required for transport vesicle budding from the endoplasmic reticulum. Mol Biol Cell 8:205–217
Sato K, Nakano A (2003) Oligomerization of a cargo receptor directs protein sorting into COPII-coated transport vesicles. Mol Biol Cell 14:3055–3063
Sato K, Nakano A (2004) Reconstitution of coat protein complex II (COPII) vesicle formation from cargo-reconstituted proteoliposomes reveals the potential role of GTP hydrolysis by Sar1p in protein sorting. J Biol Chem 279:1330–1335
Sato K, Nakano A (2005) Dissection of COPII subunit-cargo assembly and disassembly kinetics during Sar1p-GTP hydrolysis. Nat Struct Mol Biol 12:167–174
Scales SJ, Pepperkok R, Kreis TE (1997) Visualization of ER-to-Golgi transport in living cells reveals a sequential mode of action for COPII and COPI. Cell 90:1137–1148
Schekman R, Novick P (2004) 23 genes, 23-years later. Cell 116:S13–S15, 11 p following S19
Schindler R, Itin C, Zerial M, Lottspeich F, Hauri HP (1993) ERGIC-53, a membrane protein of the ER–Golgi intermediate compartment, carries an ER retention motif. Eur J Cell Biol 61:1–9
Schweizer A, Fransen JA, Matter K, Kreis TE, Ginsel L, Hauri HP (1990) Identification of an intermediate compartment involved in protein transport from endoplasmic reticulum to Golgi apparatus. Eur J Cell Biol 53:185–196
Scott DB, Blanpied TA, Ehlers MD (2003) Coordinated PKA and PKC phosphorylation suppresses RXR-mediated ER retention and regulates the surface delivery of NMDA receptors. Neuropharmacology 45:755–767
Sesso A, de Faria FP, Iwamura ESM, Correa H (1992) A three-dimensional reconstruction study of the rough ER–Golgi interface in serial thin sections of the pancreatic acinar cell of the rat. J Cell Sci 107:517–528
Shaywitz DA, Espenshade PJ, Gimeno RE, Kaiser CA (1997) COPII subunit interactions in the assembly of the vesicle coat. J Biol Chem 272:25413–25416
Shibata H, Suzuki H, Yoshida H, Maki M (2007) ALG-2 directly binds Sec31A and localizes at endoplasmic reticulum exit sites in a Ca2+-dependent manner. Biochem Biophys Res Commun 353:756–763
Shima DT, Scales SJ, Kreis TE, Pepperkok R (1999) Segregation of COPI-rich and anterograde-cargo-rich domains in endoplasmic-reticulum-to-Golgi transport complexes. Curr Biol 9:821–824
Shimoi W, Ezawa I, Nakamoto K, Uesaki S, Gabreski G, Aridor M, Yamamoto A, Nagahama M, Tagaya M, Tani K (2005) p125 is localized in endoplasmic reticulum exit sites and involved in their organization. J Biol Chem 280:10141–10148
Shin HW, Nakayama K (2004) Dual control of membrane targeting by PtdIns(4)P and ARF. Trends Biochem Sci 29:513–515
Shorter J, Beard MB, Seemann J, Dirac-Svejstrup AB, Warren G (2002) Sequential tethering of Golgins and catalysis of SNAREpin assembly by the vesicle-tethering protein p115. J Cell Biol 157:45–62
Shoulders CC, Stephens DJ, Jones B (2004) The intracellular transport of chylomicrons requires the small GTPase, Sar1b. Curr Opin Lipidol 15:191–197
Sladeczek F, Camonis JH, Burnol AF, Le Bouffant F (1997) The Cdk-like protein PCTAIRE-1 from mouse brain associates with p11 and 14-3-3 proteins. Mol Gen Genet 254:571–577
Stagg SM, Gurkan C, Fowler DM, LaPointe P, Foss TR, Potter CS, Carragher B, Balch WE (2006) Structure of the Sec13/31 COPII coat cage. Nature 439:234–238
Stagg SM, LaPointe P, Balch WE (2007) Structural design of cage and coat scaffolds that direct membrane traffic. Curr Opin Struct Biol 17:221–228
Stankewich MC, Stabach PR, Morrow JS (2006) Human Sec31B: a family of new mammalian orthologues of yeast Sec31p that associate with the COPII coat. J Cell Sci 119:958–969
Stephens DJ, Lin-Marq N, Pagano A, Pepperkok R, Paccaud JP (2000) COPI-coated ER-to-Golgi transport complexes segregate from COPII in close proximity to ER exit sites. J Cell Sci 113:2177–2185
Stephens DJ, Pepperkok R (2002) Imaging of procollagen transport reveals COPI-dependent cargo sorting during ER-to-Golgi transport in mammalian cells. J Cell Sci 115:1149–1160
Stephens DJ (2003) De novo formation, fusion and fission of mammalian COPII-coated endoplasmic reticulum exit sites. EMBO Rep 4:210–217
Stephens DJ, Pepperkok R (2004) Differential effects of a GTP-restricted mutant of Sar1p on segregation of cargo during export from the endoplasmic reticulum. J Cell Sci 117:3635–3644
Stinchcombe JC, Nomoto H, Cutler DF, Hopkins CR (1995) Anterograde and retrograde traffic between the rough endoplasmic reticulum and the Golgi complex. J Cell Biol 131:1387–1401
Sun LP, Li L, Goldstein JL, Brown MS (2005) Insig required for sterol-mediated inhibition of Scap/SREBP binding to COPII proteins in vitro. J Biol Chem 280:26483–26490
Sun LP, Seemann J, Goldstein JL, Brown MS (2007) Sterol-regulated transport of SREBPs from endoplasmic reticulum to Golgi: Insig renders sorting signal in Scap inaccessible to COPII proteins. Proc Natl Acad Sci USA 104:6519–6526
Supek F, Madden DT, Hamamoto S, Orci L, Schekman R (2002) Sec16p potentiates the action of COPII proteins to bud transport vesicles. J Cell Biol 158:1029–1038
Szul T, Garcia-Mata R, Brandon E, Shestopal S, Alvarez C, Sztul E (2005) Dissection of membrane dynamics of the ARF-guanine nucleotide exchange factor GBF1. Traffic 6:374–385
Szul T, Grabski R, Lyons S, Morohashi Y, Shestopal S, Lowe M, Sztul E (2007) Dissecting the role of the ARF guanine nucleotide exchange factor GBF1 in Golgi biogenesis and protein trafficking. J Cell Sci
Tang BL, Ong YS, Huang B, Wei S, Wong ET, Qi R, Horstmann H, Hong W (2001) A membrane protein enriched in endoplasmic reticulum exit sites interacts with COPII. J Biol Chem 276:40008–40017
Tang BL, Wang Y, Ong YS, Hong W (2005) COPII and exit from the endoplasmic reticulum. Biochim Biophys Acta 1744:293–303
Tani K, Mizoguchi T, Iwamatsu A, Hatsuzawa K, Tagaya M (1999) p125 is a novel mammalian Sec23p-interacting protein with structural similarity to phospholipid-modifying proteins. J Biol Chem 274:20505–20512
Thyberg J, Moskalewski S (1985) Microtubules and the organization of the Golgi complex. Exp Cell Res 159:1–16
Tisdale EJ, Bourne JR, Khosravi-Far R, Der CJ, Balch WE (1992) GTP-binding mutants of rab1 and rab2 are potent inhibitors of vesicular transport from the endoplasmic reticulum to the Golgi complex. J Cell Biol 119:749–761
Veiga E, Guttman JA, Bonazzi M, Boucrot E, Toledo-Arana A, Lin AE, Enninga J, Pizarro-Cerda J, Finlay BB, Kirchhausen T, Cossart P (2007) Invasive and adherent bacterial pathogens Co-Opt host clathrin for infection. Cell Host Microb 2:340–351
Ward TH, Polishchuk RS, Caplan S, Hirschberg K, Lippincott-Schwartz J (2001) Maintenance of Golgi structure and function depends on the integrity of ER export. J Cell Biol 155:557–570
Watson P, Forster R, Palmer KJ, Pepperkok R, Stephens DJ (2005) Coupling of ER exit to microtubules through direct interaction of COPII with dynactin. Nat Cell Biol 7:48–55
Watson P, Townley AK, Koka P, Palmer KJ, Stephens DJ (2006) Sec16 defines endoplasmic reticulum exit sites and is required for secretory cargo export in mammalian cells. Traffic 7:1678–1687
Weissman JT, Plutner H, Balch WE (2001) The mammalian guanine nucleotide exchange factor mSec12 is essential for activation of the Sar1 GTPase directing endoplasmic reticulum export. Traffic 2:465–475
Wendeler MW, Paccaud JP, Hauri HP (2007) Role of Sec24 isoforms in selective export of membrane proteins from the endoplasmic reticulum. EMBO Rep 8:258–264
Wessels E, Duijsings D, Niu TK, Neumann S, Oorschot VM, de Lange F, Lanke KH, Klumperman J, Henke A, Jackson CL et al (2006) A viral protein that blocks Arf1-mediated COP-I assembly by inhibiting the guanine nucleotide exchange factor GBF1. Dev Cell 11:191–201
Xu D, Hay JC (2004) Reconstitution of COPII vesicle fusion to generate a pre-Golgi intermediate compartment. J Cell Biol 167:997–1003
Yamasaki A, Tani K, Yamamoto A, Kitamura N, Komada M (2006) The Ca2+-binding protein ALG-2 is recruited to endoplasmic reticulum exit sites by Sec31A and stabilizes the localization of Sec31A. Mol Biol Cell 17:4876–4887
Yang X, Matern HT, Gallwitz D (1998) Specific binding to a novel and essential Golgi membrane protein (Yip1p) functionally links the transport GTPases Ypt1p and Ypt31p. Embo J 17:4954–4963
Yoshihisa T, Barlowe C, Schekman R (1993) Requirement for a GTPase-activating protein in vesicle budding from the endoplasmic reticulum. Science 259:1466–1468
Yu S, Satoh A, Pypaert M, Mullen K, Hay JC, Ferro-Novick S (2006) mBet3p is required for homotypic COPII vesicle tethering in mammalian cells. J Cell Biol 174:359–368
Yuan H, Michelsen K, Schwappach B (2003) 14-3-3 dimers probe the assembly status of multimeric membrane proteins. Curr Biol 13:638–646
Zhang B, Cunningham MA, Nichols WC, Bernat JA, Seligsohn U, Pipe SW, McVey JH, Schulte-Overberg U, de Bosch NB, Ruiz-Saez A et al (2003) Bleeding due to disruption of a cargo-specific ER-to-Golgi transport complex. Nat Genet 34:220–225
Zhao X, Lasell TK, Melancon P (2002) Localization of large ADP-ribosylation factor-guanine nucleotide exchange factors to different Golgi compartments: evidence for distinct functions in protein traffic. Mol Biol Cell 13:119–133
Zhao X, Claude A, Chun J, Shields DJ, Presley JF, Melancon P (2006) GBF1, a cis-Golgi and VTCs-localized ARF-GEF, is implicated in ER-to-Golgi protein traffic. J Cell Sci 119:3743–3753
Acknowledgments
We thank Pete Watson for Fig. 1 and constructive comments throughout. Work in the Stephens lab is supported by Non-Clinical Senior Fellowship from the MRC and research grants from BBSRC and the Royal Society. HH is funded through a BBSRC Doctoral Training Account. We would particularly like to thank Angus Laurie-Pile for his work in generating the 3D rendered images used in Figs. 3 and 4.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License ( https://creativecommons.org/licenses/by-nc/2.0 ), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
Hughes, H., Stephens, D.J. Assembly, organization, and function of the COPII coat. Histochem Cell Biol 129, 129–151 (2008). https://doi.org/10.1007/s00418-007-0363-x
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00418-007-0363-x