
Abstract
Background
Mitochondrial disorders are clinically complex and have highly variable phenotypes among all inherited disorders. Mutations in mitochon drial DNA (mtDNA) and nuclear genome or both have been reported in mitochondrial diseases suggesting common pathophysiological pathways. Considering the clinical heterogeneity of mitochondrial encephalopathy, lactic acidosis and stroke-like episodes (MELAS) phenotype including focal neurological deficits, it is important to look beyond mitochondrial gene mutation.
Methods
The clinical, histopathological, biochemical analysis for OXPHOS enzyme activity, and electron microscopic, and neuroimaging analysis was performed to diagnose 11 patients with MELAS syndrome with a multisystem presentation. In addition, whole exome sequencing (WES) and whole mitochondrial genome sequencing were performed to identify nuclear and mitochondrial mutations.
Results
Analysis of whole mtDNA sequence identified classical pathogenic mutation m.3243A > G in seven out of 11 patients. Exome sequencing identified pathogenic mutation in several nuclear genes associated with mitochondrial encephalopathy, sensorineural hearing loss, diabetes, epilepsy, seizure and cardiomyopathy (POLG, DGUOK, SUCLG2, TRNT1, LOXHD1, KCNQ1, KCNQ2, NEUROD1, MYH7) that may contribute to classical mitochondrial disease phenotype alone or in combination with m.3243A > G mutation.
Conclusion
Individuals with MELAS exhibit clinical phenotypes with varying degree of severity affecting multiple systems including auditory, visual, cardiovascular, endocrine, and nervous system. This is the first report to show that nuclear genetic factors influence the clinical outcomes/manifestations of MELAS subjects alone or in combination with m.3243A > G mutation.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Defective mitochondrial function contributes to childhood and adult neurometabolic disorders affecting multiple organs with a projected global prevalence of 1 in 5000 [1]. Patients with mitochondrial disease manifests with a heterogeneous clinical phenotype, varying age of onset and different grades of severity [2]. Previous studies have identified more than 1500 nuclear genome encoded proteins participating in various mitochondrial function [3]. Due to this dual nature of genetic control, mutations in either nuclear or mitochondrial genomes or both may contribute to mitochondrial dysfunction [4, 5]. Mitochondrial diseases are classified based on mutations in either the mitochondrial or the nuclear genome affecting intraorganellar gene expression and function [6].
Nuclear gene defects identified in mitochondrial diseases are primarily responsible for mtDNA replication, transcription and translation, oxidative phosphorylation (OXPHOS) and biogenesis of mtDNA [7]. The other nuclear genes contribute to various mitochondrial disorders involved in nucleoside transport, salvage or synthesis; and maintenance of balanced mitochondrial deoxyribonucleoside triphosphates (dNTP) pool [8]. MELAS is a clinically complex early onset multi-organ mitochondrial disorder with diverse phenotype and varying degree of severity [9, 10]. These patients clinically present with encephalomyopathy, lactic acidosis, and stroke-like episodes and show evidence of ragged red fibers (RRF) on muscle biopsy. In addition, there are a wide range of other clinical symptoms which include diabetes mellitus, cardiomyopathy, migraine, epilepsy, cerebellar ataxia, impaired vision, and hearing loss in varying combination [11]. Majority of these patients harbor mutation in the MT-TL1 gene coding for the tRNALeu (UUR). The m.3243A > G mutation has been reported in 80% of MELAS patients affecting tRNALeu (UUR) aminoacylation and mitochondrial translation of OXPHOS protein subunits [12,13,14]. However, recent study suggests that patients with m.3243A > G mutation exhibiting the vast clinical heterogeneity underpinning the necessity to interrogate nuclear genome for better understanding of complex mitochondrial disorders [15]. This study aimed to sequence the whole mitochondrial genome and the whole exome of clinically characterized MELAS patients and identify mutations in both nuclear and mitochondrial genome and their contribution to its heterogeneous phenotype.
Methods
Patients
A total of eleven patients with clinically suspected MELAS syndrome referred to Department of Neurology, National Institute of Mental Health and Neurosciences, Bangalore, India; and Department of Neurology, Nizam’s Institute of Medical Sciences, Hyderabad, India, were included in this study. All patients had comprehensive clinical evaluation, neurological and ophthalmological examination, biochemical tests, and magnetic resonance imaging (MRI). Routine histopathological examination of skeletal muscle was done in 10 patients. Institutional Ethical Committee (IEC) clearance was obtained from the participating institute (NIMHANS/IEC (BS & NS DIV.) 8th meeting/2017). Patients were enrolled after obtaining the written informed consent.
Pathological studies
Fresh skeletal muscle biopsy of the patients was used for histopathology, electron microscopy (EM) and enzyme histochemistry. Histological [Hematoxylin and Eosin (H&E), modified Gomori trichrome (MGT) staining) and histochemical [cytochrome c oxidase (COX), succinate dehydrogenase (SDH) and combined COX/SDH] analysis was performed using standard protocol [16]. A small fragment of the muscle fixed in 3% glutaraldehyde was subjected to mitochondrial ultra-structural analysis using transmission electron microscopy (JEOL200X, Japan) by established protocols described elsewhere [17, 18].
Biochemical analysis
Tissue homogenates from frozen muscle biopsies were extracted as described earlier [19] and used for the enzyme assay of the four mitochondrial respiratory chain complexes (I, II, III and IV) using spectrophotometry in six patients [19, 20].
Whole mitochondria genome sequencing
DNA was isolated from both peripheral blood and skeletal muscle tissue using the method described earlier [21]. Whole mitochondrial genome was amplified by using 24 sets of primers to generate overlapping fragments [22]. PCR amplicons were electrophoresed using 2% agarose gel, purified with Exo-SAP and sequenced using BigDye Terminator ready reaction kit (Applied Biosystems, Foster City, USA). Post PCR purification of amplified products were performed by ethanol precipitation and dissolved in Hi-Di formamide. PCR amplicons were sequenced using ABI 3730 automated DNA analyzer (Applied Biosystems). The sequences were aligned with revised Cambridge reference sequence (rCRS) (NC_012920) [23, 24] using Sequence Analysis and AutoAssembler tools. All the mismatched nucleotide sequences were obtained and compared with 300 healthy controls, and further compared with Mitomap database (http://www.mitomap.org). Haplogroup analysis was performed using phylotree [(www.phylotree.org; mtDNA tree Build 17 (18 Feb 2016)]. Further, m.3243A > G heteroplasmic levels of the patients and their available maternal relatives were assessed using the PCR-RFLP method as described earlier [25].
Whole exome sequencing
Whole exome sequencing (WES) was performed using Ion AmpliSeq™ Exome RDY Kit 4 × 2 kit (ThermoFisher Scientific, Waltham, MA, USA) according to manufacturer’s protocol. Library preparation of ultra-high multiplex PCR enriched whole exome amplicons was carried out using Ion AmpliSeq™ Library Kit Plus (Thermo Fisher Scientific) as per the manufacturer’s instructions. Each sample was barcoded using Ion Express Barcode Adapter 1–16 kit (Thermo Fisher Scientific). Quantification of each library was performed on Agilent Bioanalyzer 2100 using Agilent high sensitivity (HS) DNA kit (Agilent Technologies, Santa Clara, CA, USA). WES was performed using Ion Proton PI v3 chip in Ion Proton next generation sequencing platform (Thermo Fisher Scientific) as per the manufacturer’s instruction.
In silico analysis
Sequence read alignment to human reference genome sequence (Hg19) and variant calling were carried out using Torrent Variant Caller v5.12 (TVC) in Torrent Suite software (Thermo Fisher Scientific). After removal of low-quality reads, variants were detected using Ion TVC using optimized parameters (AmpliSeq Designer, Thermo Fisher Scientific) for low-frequency variant detection with minimal false-positive calls. Variant filter used the technical parameters which include 1) variant quality score, 2) variant coverage, and 3) variant allele frequency. Coverage analysis was done using Coverage Analysis Plugin software v5.12. Visual inspection of read alignment and variants called were performed using Integrative Genomic Viewer (IGV v2.8.2) software [26] to investigate for strand biases, homopolymer length and sequencing errors. Variant caller format (VCF) generated by variant caller plug-in was further analyzed using Ion Reporter software v5.12 (Thermo Fisher Scientific) for variant annotation and filtering.
Silent and known germline variants were removed based on population frequency using dbSNP, NHLBI Exome Sequencing Project (https://esp.gs.washington.edu/EVS/) and Exome Aggregation Consortium (http://exac.broadinstitute.org/) (variants with more than 1% minor allele frequency, MAF). Variant prioritization was performed to select variants that alter protein function; these included nonsenses, splice site, coding indel, or missense variants.
Variant analysis
VCF files for each exome datasets from MELAS patients were uploaded to Ingenuity variant analysis (IVA, Qiagen Bioinformatics, USA) program [27]. Each exome dataset was rigorously filtered to identify pathogenic variants based on quality, MAF, deleteriousness, inheritance patterns, conservation and its association with clinical phenotype of each MELAS patients. Additionally, we analyzed heterozygous SNVs identified in MELAS patients as per the American College of Medical Genetics and Genomics (ACMG) guidelines and the prediction algorithms used by Varsome, the allele frequencies in the Genome Aggregation Database (GnomAD), Exome Aggregation Consortium-ExAC (http://exac.broadinstitute.org/) and multiple in silico tools for prediction of pathogenicity which includes DANN, DEOGEN2, EIGEN, FATHMM-MKL, M-CAP, MVP, MutationAssessor, MutationTaster, PrimateAI, REVEL and SIFT [28]. In addition, we have compared the mutations/ prioritized variants with our internal database consisting of 51 whole exome data of other mitochondrial disorder patients.
Copy number variation analysis
Copy number variation (CNV) analysis of whole exome sequencing data of MELAS patients was performed using EXCAVATOR2 v1.1.2 [29]. EXCAVATOR2 employs off target reads aligning outside targeted regions for improved detection of CNVs when compared with other exome-based CNV callers such as XHMM and CoNIFER. Each MELAS exome and control BAM files were analyzed through the EXCAVATOR2 pipeline using the standard settings.
Results
Clinical presentation of MELAS patients
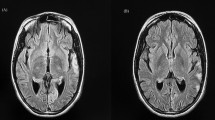
Clinical presentation of 11 patients having MELAS (6 males and 5 females) is summarized in Table 1. The mean age at presentation was 20 \(\pm\) 11.3 years (age range 7–45 years) and mean age at onset was 14.2 \(\pm\) 10.6 years (age range 6–41 years). Majority of the patients presented with seizures, status epilepticus, hearing impairment and had elevated lactate. Magnetic resonance imaging (MRI) of brain showed stroke-like lesions predominantly in parieto-occipital cortex (Fig. 1) in eight patients (Table 1). Histopathology analysis of muscle biopsy showed ragged red fibers (RRF) and ragged blue fibers (RBF) (Fig. 2a–c) in five patients including one patient who was negative for m.3243A > G. However, there were no COX-deficient fibers in any of these cases. Electron microscopy analysis done in five patients, whose muscle biopsy did not reveal any diagnostic pathology on light microscope, showed sub-sarcolemmal aggregation of mitochondria of varying size and altered cristae pattern (Fig. 2d). In addition, increased pinocytic vesicles were noted in the endothelial cells of blood vessels (Fig. 2e–f). Respiratory chain complex assay was performed in six patients and revealed isolated complex I (P10, P11) and IV deficiency (P2, P9) in two patients each and multiple complex deficiency in two patients (P4, P5) (Table 1).

CT and MRI brain of MELAS patients with nuclear gene mutations. Axial sections of CT and MRI of brain show shifting cortical-based signal changes that do not conform any arterial territory in patient P8 (A–F). CT brain shows an ill-defined focal hypodensity in the left parietal region in 2008 (a), Brain MRI shows focal wedge-shaped hyperintensity in the left temporal region resembling an ‘infarct’ in 2010 (b, c), focal hyperintensity in the right frontal region in 2012, while the lesion seen in the left temporal region in 2010 has resolved completely (d, e) and recurrence of focal hyperintensity in the left temporal region in 2014 (f). Axial sections of MRI of brain show shifting hyperintensities involving the cortex that do not conform an arterial territory in patient P9 (g–j). Brain MRI shows focal hyperintensity in the left parieto-occipital region in September 2012 (g), focal hyperintensity in the right temporal region in November 2012 (h), focal hyperintensity in left medial temporal region in January 2013 (i) and hyperintensity in bilateral frontal region in August 2016 (j)

Light and electron microscopic features of muscle biopsy from a patient negative for m.3243A > G mutation showing evidence of mitochondrial insult. a ragged red fibers (MGT); b Ragged blue fibers (SDH); c No COX-deficient fiber (COX- SDH); d Aggregates of mitochondria with altered cristae in the sub-sarcolemmal region; e, f Increased pinocytic vesicles in the endothelial cells of blood vessels
Mutations in mitochondrial genome
Whole mitochondrial genome sequencing analysis identified pathogenic mutation m.3243A > G of MT-TL1 gene in seven patients, and none in the remaining four patients (Table 1). In addition, one patient (P2) with m.3243A > G was also carrying m.4317A > G in MT-TI gene. Another patient (P11) negative to m.3243A > G was carrying m.13135G > A in MT-ND5 gene that is reported as possible hypertrophic cardiomyopathy susceptible factor. Phylogenetic analysis showed that the patients were from different haplogroup background (Table 1).
Mutations in nuclear genome
Whole exome sequencing analysis of eleven patients generated an average of 34 million mapped reads with more than 92% reads on target in all MELAS patients in Ion Proton NGS platform (Supplementary Table 1). The mean per base depth of coverage for the exome consensus coding sequence was 97-fold, with 93% of bases covered more than 20-fold (Supplementary Table 1). Patients with the MELAS phenotype present with wide spectrum of clinical features ranging from seizures, stroke-like episodes, ataxia, sensorineural hearing loss, diabetes and epilepsy. Phenotype-driven analysis and prioritization of causal mutations in each MELAS patient was performed using Ingenuity variant analysis software. Phenotype-driven analysis identified deleterious protein coding mutations in nuclear genes with known mitochondrial function previously reported in mitochondrial disorders (POLG, DGUOK, SUCLG2, TRNT1) (Table 2).
POLG (DNA polymerase subunit gamma) is solely responsible for replication of mitochondrial DNA [30]. POLG mutation has been reported in both autosomal dominant and autosomal recessive mitochondrial disorders [31]. In our study, we identified two patients (P6 and P7) with homozygous missense POLG mutation (p.Trp748Ser), previously reported in patients with progressive neurological disorder (Table 2) [31, 32]. Additionally, we identified heterozygous POLG mutation (p.Arg1187Trp) in three patients (P1, P3 and P5), which was previously reported in two cases with mitochondrial respiratory chain diseases [33, 34].
DGUOK encodes enzyme deoxyguanosine kinase, which is essential for maintaining building blocks of mitochondrial DNA. Mutation in DGUOK is well documented in patients with mitochondrial DNA depletion syndrome leading to neurological conditions with liver dysfunction [35]. We identified homozygous frameshift deletion in DGUOK (p.Phe156LeufsTer45) in two patients (P10 and P11; Table 2). The heterozygous mutation in SUCLG2, c.235G > T (p.Glu79*) has been identified in three MELAS patients (P1, P3 and P5). In addition, homozygous frameshift insertion in TRNT1 (p.Glu318Argfs*11) was observed in one patient (P1) (Table 2).
MELAS patients in our study showed a wide range of clinical presentation including hearing loss, diabetes, epilepsy, seizures and impaired vision. We identified heterozygous missense mutation in NEUROD1 (p.Glu59Gln) in one patient (P2), which was previously reported to be associated with maturity-onset diabetes of the young (MODY) phenotype in Indian population [36, 37]. Compound heterozygous mutation in LOXHD1 c.4690C > T (p.Leu1564Phe) c.2054G > A (p.Arg685His) and c.5813G > A (p.Arg1938His), c.1191G > T (p.Trp397Cys) was observed in two patients (P2, P4) with hearing impairment. Additionally, we identified heterozygous KCNQ1 mutation c.575G > A (p.Arg192His) and heterozygous KCNQ2 mutation c.598C > A (p.Leu200Met) in one MELAS patient (P9) (Table 2).
Influence of nuclear gene mutations on m.3243A > G
We looked at the control of nuclear gene mutations on the clinical expression of m.3243A > G and found patient P2 harbor variants in two genes namely LOXHD1 (p.Leu1564Phe, p.Arg685His) and NEUROD1 (p.Glu59Gln) which is reported to be associated with non-syndromic hearing loss and maturity-onset diabetes, respectively. This is likely that these variations could have contributed to deafness in patient and the mother, and diabetes mellitus in multiple family members (Table 1). Another patient P3 harbored variants in two genes namely POLG (p.Arg1187Trp) and SUCLG2 (p.Glu79*) reported to be associated with mitochondrial respiratory chain diseases and mtDNA depletion, impaired respiratory complex subunits and mitochondrial encephalopathy, respectively. These gene variants may be modifiers for the severe clinical course and premature death.
Copy number variants identified in MELAS syndrome
CNV analysis of MELAS exome datasets identified recurrent copy number amplification in 2q34 (ERBB4), 4q12 (ADGRL3), 5p12 (HCN1), 8q23 (CSMD3), 11p14 (LRRC4C) and 14q11.2 (FOXG1). We observed copy number gain in 5p12 (HCN1) and 14q11.2 (FOXG1) in multiple MELAS individuals (Supplementary Table 2).
Discussion
Mitochondrial disorders have complex clinical phenotypes and thus pose a significant challenge in the clinical diagnosis. These are caused by genetic mutations in mitochondrial DNA, nuclear DNA or both [39, 40]. We have performed a detailed clinical, biochemical, imaging, and molecular analysis of 11 patients with suspected MELAS syndrome. Whole mitochondrial genome sequencing identified a pathogenic m.3243A > G mutation in seven patients. Recent studies have identified nuclear gene mutations in suspected MELAS patients and other classical mitochondrial phenotypes highlighting the need to screen nuclear gene mutations and function for improved diagnosis of mitochondrial disorders [38,39,40,41,42]. To achieve this, we have performed WES of 11 MELAS patients and identified pathogenic mutation in nuclear encoded genes associated with mitochondrial encephalopathy (POLG, DGUOK, SUCLG2, TRNT1), sensorineural hearing loss, seizures, epileptic encephalopathy, and cardiomyopathy (LOXHD1, KCNQ1, KCNQ2 and MYH7). Mutations in POLG and DGUOK have been previously implicated in mitochondrial disorders with common phenotype such as mitochondrial respiratory complex defect, mitochondrial DNA depletion and encephalopathy [42, 43].
Homozygous POLG mutation c.2243G > C (p.Trp748Ser) identified in two patients with m.3243A > G (P6 and P7) was previously reported in ataxia patients of European origin [31]. POLG mutation is also associated with multiple mtDNA deletion syndrome [31]. However, histopathology of muscle in these two patients (P6 and P7) in our study did not show any abnormality as reported previously in European patients with this mutation [32]. Additionally, both P6 and P7 did not have classical MELAS phenotype based on the available clinical data. It is interesting to note that sequencing of mitochondrial genome along with exome sequencing identified these double pathogenic mutations in two patients (P6 and P7). Hence, our observations indicate that pathogenic mutation in both nuclear (POLG) and mitochondrial (MT‐TL1, m.3243A > G) in two patients (P6 and P7) corroborates the role of dual genetic mutations in MELAS phenotype. In contrast to our observation, a study identified POLG1 mutations in MELAS-like phenotype but negative for common mtDNA mutations [42]. Another study reported heterozygous POLG1 variants p.Arg627Gln and p.Gly848Ser in a patient with stroke-like episodes typical of MELAS phenotype negative for m.3243A > G [44].
DGUOK mutation (p.Phe156LeufsTer45) was seen in two patients negative for m.3243A > G (P10 and P11). Interestingly, patient (P10) showed RRF, RBF and aggregates of mitochondria with altered cristae in the sub-sarcolemmal region (Fig. 2a–d). Functional role of DGUOK in mitochondrial DNA maintenance is well established [35]. Mutation in DGUOK has been previously reported in patients with neurological abnormality [43]. Mutation in TRNT1 and SUCLA2 has been implicated in patients with mitochondrial encephalopathy [45, 46].
SUCLG2 encodes a GTP-specific beta subunit of succinate-CoA ligase (SUCL), an important enzyme in TCA cycle, and primarily expressed in liver [47]. Homozygous knockout model of SUCLG2 in mice showed recessive lethality [48]. Patients with SUCL deficiency showed mtDNA depletion, impaired respiratory complex subunits and mitochondrial encephalomyopathy [47,48,49]. Mutation in SUCLG2 has not been reported so far in any patients with mitochondrial disorder. In this study, we report for the first time, a heterozygous mutation in SUCLG2 gene, which introduces a nonsense codon in the ATP-grasp_2 domain. The glutamate residue in the 79 position in the SUCLG2 protein is responsible for substrate specificity. However, a detailed functional analysis is needed to establish the role of SUCLG2 nonsense mutation (p.Glu79*) in the mitochondrial dysfunction identified in our study. TRNT1 (CCA-adding transfer RNA nucleotidyl transferase) is the key enzyme which plays an important role in cytosolic and mitochondrial tRNA modification and protein translation [50]. We identified a homozygous frameshift insertion in TRNT1 (p.Glu318Argfs*11) in one patient (P1). A previous study reported wide range of phenotypes with TRNT1 mutations both in childhood and adult onset retinitis pigmentosa, encephalopathy, hepatosplenomegaly and pancreatic insufficiency [46].
In our study, patients with sensorineural hearing loss, seizures, epileptic encephalopathy and cardiomyopathy showed mutations in LOXHD1, KCNQ1, KCNQ2 and MYH7 genes. These may contribute to the severity of diverse clinical phenotype of MELAS syndrome alone or in combination with mitochondrial mutations. LOXHD1 mutation has been previously reported in patients with non-syndromic hearing loss (NSHL) [51, 52]. Mutations in both KCNQ1 and KCNQ2 identified in our study were previously reported as pathogenic in the ClinVar database. Autosomal dominant mutation in KCNQ1 causes congenital long-QT syndrome and cardiac arrhythmia [53]. KCNQ2, a brain-specific voltage-gated potassium-channel subunit, has been implicated in patients with epileptic encephalopathy and neonatal‐onset seizures [54]. In this study, the patient (P9) carrying KCNQ2 mutation was positive for NMDAR (N-methyl-D-aspartate receptor) antibodies. A previous case report has shown the association of anti-NMDAR antibodies in MELAS syndrome with m.3243A > G mutation [55]. Autosomal dominant MYH7 mutation has been reported in patients with hypertrophic cardiomyopathy [56]. We observed one patient (P8) with MYH7 mutation which was identified as likely pathogenic mutation according to the ACMG guidelines and ClinVar database (Table 2). Most of the patients showed early onset in our cohort. The patients with m.3243A > G mutations harboring nuclear gene mutations showed varying clinical features (Supplementary Table 3). Hence, nuclear gene mutations may act as modifiers in the disease phenotype. Another interesting observation is that the patients with m.3243A > G negative showed mutations in DGUOK, KCNQ1, KCNQ2 and MYH7. Further, functional validation will elucidate the role of these genes in MELAS phenotype.
MELAS syndrome patients with predominant neurodegenerative symptoms may gradually develop wide spectrum of endocrine disorders including diabetes. We have identified NEUROD1 missense mutation (p.Glu59Gln) in one MELAS patient with diabetes. NEUROD1 (p.Glu59Gln) mutation identified in our study has been previously reported in maturity-onset diabetes in an Indian cohort [36, 37]. Copy number variation analysis of exome sequencing data identified recurrent copy number gains in chromosomes 5p12 and 14q11.2 which harbors HCN1 and FOXG1 which has been previously reported in epilepsy, a clinical phenotype commonly observed in MELAS patients [57, 58]. Hence, these CNVs could contribute to the diverse phenotypes of MELAS. Studies have shown advances in the diagnostic pathways for mitochondrial diseases and ‘genetic first approach’ is preferred [59,60,61]. In this study, we identified nuclear mutations in MELAS patients who are negative for m.3243A > G mutation. Biopsy may not be required in patient with characteristic clinical syndrome feature harboring m.3243A > G mutation; however, patients with atypical feature and who are negative for m.3243A > G mutation require MRI, muscle biopsy and RCC assay are useful and complementary to aid for genetic diagnosis.
Conclusion
Mitochondrial diseases are challenging to diagnose due to their wide spectrum of clinical features and genetic heterogeneity. Previous studies have highlighted the contribution of nuclear gene mutations in mitochondrial disorders. This study identified both nuclear and mtDNA mutations in clinically established MELAS patients. Our study demonstrated that the contribution of nuclear genetic background may influence the clinical heterogeneity in m.3243A > G‐related classical mitochondrial disease. This has far-reaching implications which opens the possibility of identifying new mechanisms in well-characterized larger patient cohort and pedigree analysis. A detailed evaluation of these novel interactions between nuclear genetic factors and m.3243A > G may facilitate development of more accurate tools for improved diagnosis, prognosis and better genetic counseling of patients with mitochondrial disorders. Limited number of MELAS cases evaluated in this study is the shortcoming; however, it opens up promising avenues for a larger patient cohort.
Data availability
The data that support the findings of this study are available from the corresponding author upon request.
References
Skladal D, Halliday J, Thorburn DR (2003) Minimum birth prevalence of mitochondrial respiratory chain disorders in children. Brain 126:1905–1912. https://doi.org/10.1093/brain/awg170
Ylikallio E, Suomalainen A (2012) Mechanisms of mitochondrial diseases. Ann Med 44:41–59. https://doi.org/10.3109/07853890.2011.598547
Calvo SE, Clauser KR, Mootha VK (2016) MitoCarta2.0: an updated inventory of mammalian mitochondrial proteins. Nucleic Acids Res 44:D1251-1257. https://doi.org/10.1093/nar/gkv1003
Kirby DM, McFarland R, Ohtake A et al (2004) Mutations of the mitochondrial ND1 gene as a cause of MELAS. J Med Genet 41:784–789. https://doi.org/10.1136/jmg.2004.020537
Wong L-JC, Yim D, Bai R-K et al (2006) A novel mutation in the mitochondrial tRNA(Ser(AGY)) gene associated with mitochondrial myopathy, encephalopathy, and complex I deficiency. J Med Genet 43:e46. https://doi.org/10.1136/jmg.2005.040626
Gorman GS, Schaefer AM, Ng Y et al (2015) Prevalence of nuclear and mitochondrial DNA mutations related to adult mitochondrial disease. Ann Neurol 77:753–759. https://doi.org/10.1002/ana.24362
Boczonadi V, Horvath R (2014) Mitochondria: impaired mitochondrial translation in human disease. Int J Biochem Cell Biol 48:77–84. https://doi.org/10.1016/j.biocel.2013.12.011
Lamperti C, Zeviani M (2009) Encephalomyopathies caused by abnormal nuclear-mitochondrial intergenomic cross-talk. Acta Myol 28:2–11
Goto Y, Tojo M, Tohyama J et al (1992) A novel point mutation in the mitochondrial tRNA(Leu)(UUR) gene in a family with mitochondrial myopathy. Ann Neurol 31:672–675. https://doi.org/10.1002/ana.410310617
Sato W, Hayasaka K, Shoji Y et al (1994) A mitochondrial tRNA(Leu)(UUR) mutation at 3,256 associated with mitochondrial myopathy, encephalopathy, lactic acidosis, and stroke-like episodes (MELAS). Biochem Mol Biol Int 33:1055–1061
Nesbitt V, Pitceathly RDS, Turnbull DM et al (2013) The UK MRC Mitochondrial Disease Patient Cohort Study: clinical phenotypes associated with the m.3243A\textgreaterG mutation–implications for diagnosis and management. J Neurol Neurosurg Psychiatry 84:936–938. https://doi.org/10.1136/jnnp-2012-303528
Chomyn A, Enriquez JA, Micol V et al (2000) The mitochondrial myopathy, encephalopathy, lactic acidosis, and stroke-like episode syndrome-associated human mitochondrial tRNALeu(UUR) mutation causes aminoacylation deficiency and concomitant reduced association of mRNA with ribosomes. J Biol Chem 275:19198–19209. https://doi.org/10.1074/jbc.M908734199
Flierl A, Reichmann H, Seibel P (1997) Pathophysiology of the MELAS 3243 transition mutation. J Biol Chem 272:27189–27196. https://doi.org/10.1074/jbc.272.43.27189
Ikeda T, Osaka H, Shimbo H et al (2018) Mitochondrial DNA 3243A>T mutation in a patient with MELAS syndrome. Hum Genome Var 5:25. https://doi.org/10.1038/s41439-018-0026-6
Pickett SJ, Grady JP, Ng YS et al (2018) Phenotypic heterogeneity in m.3243A\textgreaterG mitochondrial disease: the role of nuclear factors. Ann Clin Transl Neurol 5:333–345. https://doi.org/10.1002/acn3.532
Sunitha B, Gayathri N, Kumar M et al (2016) Muscle biopsies from human muscle diseases with myopathic pathology reveal common alterations in mitochondrial function. J Neurochem 138:174–191. https://doi.org/10.1111/jnc.13626
Dubowitz V, Sewry CA, Oldfors A (2013) Muscle biopsy: a practical approach, 4th edn. Saunders, Oxford
Govindaraj P, Khan NA, Gopalakrishna P et al (2011) Mitochondrial dysfunction and genetic heterogeneity in chronic periodontitis. Mitochondrion 11:504–512. https://doi.org/10.1016/j.mito.2011.01.009
Kirby DM, Thorburn DR, Turnbull DM, Taylor RW (2007) Biochemical assays of respiratory chain complex activity. Methods Cell Biol 80:93–119. https://doi.org/10.1016/S0091-679X(06)80004-X
Sonam K, Bindu PS, Srinivas Bharath M et al (2017) Mitochondrial oxidative phosphorylation disorders in children: phenotypic, genotypic and biochemical correlations in 85 patients from South India. Mitochondrion 32:42–49. https://doi.org/10.1016/j.mito.2016.11.002
Thangaraj K, Joshi MB, Reddy AG et al (2002) CAG repeat expansion in the androgen receptor gene is not associated with male infertility in Indian populations. J Androl 23:815–818
Rieder MJ, Taylor SL, Tobe VO, Nickerson DA (1998) Automating the identification of DNA variations using quality-based fluorescence re-sequencing: analysis of the human mitochondrial genome. Nucleic Acids Res 26:967–973. https://doi.org/10.1093/nar/26.4.967
Anderson S, Bankier AT, Barrell BG et al (1981) Sequence and organization of the human mitochondrial genome. Nature 290:457–465. https://doi.org/10.1038/290457a0
Andrews RM, Kubacka I, Chinnery PF et al (1999) Reanalysis and revision of the Cambridge reference sequence for human mitochondrial DNA. Nat Genet 23:147. https://doi.org/10.1038/13779
Govindaraj P, Khan NA, Rani B et al (2014) Mitochondrial DNA variations associated with hypertrophic cardiomyopathy. Mitochondrion 16:65–72. https://doi.org/10.1016/j.mito.2013.10.006
Thorvaldsdóttir H, Robinson JT, Mesirov JP (2013) Integrative Genomics Viewer (IGV): high-performance genomics data visualization and exploration. Brief Bioinformatics 14:178–192. https://doi.org/10.1093/bib/bbs017
Steinberg KM, Yu B, Koboldt DC et al (2015) Exome sequencing of case-unaffected-parents trios reveals recessive and de novo genetic variants in sporadic ALS. Sci Rep 5:9124. https://doi.org/10.1038/srep09124
Kopanos C, Tsiolkas V, Kouris A et al (2019) VarSome: the human genomic variant search engine. Bioinformatics 35:1978–1980. https://doi.org/10.1093/bioinformatics/bty897
D’Aurizio R, Pippucci T, Tattini L et al (2016) Enhanced copy number variants detection from whole-exome sequencing data using EXCAVATOR2. Nucleic Acids Res 44:e154. https://doi.org/10.1093/nar/gkw695
Nurminen A, Farnum GA, Kaguni LS (2017) Pathogenicity in POLG syndromes: DNA polymerase gamma pathogenicity prediction server and database. BBA Clin 7:147–156. https://doi.org/10.1016/j.bbacli.2017.04.001
Tzoulis C, Bindoff LA (2012) Acute mitochondrial encephalopathy reflects neuronal energy failure irrespective of which genome the genetic defect affects. Brain 135:3627–3634. https://doi.org/10.1093/brain/aws223
Van Goethem G, Luoma P, Rantamäki M et al (2004) POLG mutations in neurodegenerative disorders with ataxia but no muscle involvement. Neurology 63:1251–1257. https://doi.org/10.1212/01.wnl.0000140494.58732.83
Reichenbach J, Schubert R, Horvàth R et al (2006) Fatal neonatal-onset mitochondrial respiratory chain disease with T cell immunodeficiency. Pediatr Res 60:321–326. https://doi.org/10.1203/01.pdr.0000233252.60457.cf
Rouzier C, Chaussenot A, Serre V et al (2014) Quantitative multiplex PCR of short fluorescent fragments for the detection of large intragenic POLG rearrangements in a large French cohort. Eur J Hum Genet 22:542–550. https://doi.org/10.1038/ejhg.2013.171
Freisinger P, Fütterer N, Lankes E et al (2006) Hepatocerebral mitochondrial DNA depletion syndrome caused by deoxyguanosine kinase (DGUOK) mutations. Arch Neurol 63:1129–1134. https://doi.org/10.1001/archneur.63.8.1129
Chapla A, Mruthyunjaya MD, Asha HS et al (2015) Maturity onset diabetes of the young in India—a distinctive mutation pattern identified through targeted next-generation sequencing. Clin Endocrinol (Oxf) 82:533–542. https://doi.org/10.1111/cen.12541
Doddabelavangala Mruthyunjaya M, Chapla A, Hesarghatta Shyamasunder A et al (2017) Comprehensive maturity onset diabetes of the young (MODY) gene screening in pregnant women with diabetes in India. PLoS ONE 12:e0168656. https://doi.org/10.1371/journal.pone.0168656
Kraja AT, Liu C, Fetterman JL et al (2019) Associations of mitochondrial and nuclear mitochondrial variants and genes with seven metabolic traits. Am J Hum Genet 104:112–138. https://doi.org/10.1016/j.ajhg.2018.12.001
Garone C, D’Souza AR, Dallabona C et al (2017) Defective mitochondrial rRNA methyltransferase MRM2 causes MELAS-like clinical syndrome. Hum Mol Genet 26:4257–4266. https://doi.org/10.1093/hmg/ddx314
Peter B, Waddington CL, Oláhová M et al (2018) Defective mitochondrial protease LonP1 can cause classical mitochondrial disease. Hum Mol Genet 27:1743–1753. https://doi.org/10.1093/hmg/ddy080
Taylor RW, Pyle A, Griffin H et al (2014) Use of whole-exome sequencing to determine the genetic basis of multiple mitochondrial respiratory chain complex deficiencies. JAMA 312:68–77. https://doi.org/10.1001/jama.2014.7184
Cheldi A, Ronchi D, Bordoni A et al (2013) POLG1 mutations and stroke like episodes: a distinct clinical entity rather than an atypical MELAS syndrome. BMC Neurol 13:8. https://doi.org/10.1186/1471-2377-13-8
Mandel H, Szargel R, Labay V et al (2001) The deoxyguanosine kinase gene is mutated in individuals with depleted hepatocerebral mitochondrial DNA. Nat Genet 29:337–341. https://doi.org/10.1038/ng746
Deschauer M, Tennant S, Rokicka A et al (2007) MELAS associated with mutations in the POLG1 gene. Neurology 68:1741–1742. https://doi.org/10.1212/01.wnl.0000261929.92478.3e
Ostergaard E, Hansen FJ, Sorensen N et al (2007) Mitochondrial encephalomyopathy with elevated methylmalonic acid is caused by SUCLA2 mutations. Brain 130:853–861. https://doi.org/10.1093/brain/awl383
Wedatilake Y, Niazi R, Fassone E et al (2016) TRNT1 deficiency: clinical, biochemical and molecular genetic features. Orphanet J Rare Dis 11:90. https://doi.org/10.1186/s13023-016-0477-0
Donti TR, Masand R, Scott DA et al (2016) Expanding the phenotypic spectrum of Succinyl-CoA ligase deficiency through functional validation of a new SUCLG1 variant. Mol Genet Metab 119:68–74. https://doi.org/10.1016/j.ymgme.2016.07.007
Kacso G, Ravasz D, Doczi J et al (2016) Two transgenic mouse models for β-subunit components of succinate-CoA ligase yielding pleiotropic metabolic alterations. Biochem J 473:3463–3485. https://doi.org/10.1042/BCJ20160594
Donti TR, Stromberger C, Ge M et al (2014) Screen for abnormal mitochondrial phenotypes in mouse embryonic stem cells identifies a model for succinyl-CoA ligase deficiency and mtDNA depletion. Dis Model Mech 7:271–280. https://doi.org/10.1242/dmm.013466
Hou Y-M (2010) CCA addition to tRNA: implications for tRNA quality control. IUBMB Life 62:251–260. https://doi.org/10.1002/iub.301
Minami SB, Mutai H, Namba K et al (2016) Clinical characteristics of a Japanese family with hearing loss accompanied by compound heterozygous mutations in LOXHD1. Auris Nasus Larynx 43:609–613. https://doi.org/10.1016/j.anl.2016.02.010
Shen N, Wang T, Li D et al (2019) Whole-exome sequencing identifies a novel missense variant within LOXHD1 causing rare hearing loss in a Chinese family. BMC Med Genet 20:30. https://doi.org/10.1186/s12881-019-0758-2
Wu J, Sakaguchi T, Takenaka K et al (2019) A trafficking-deficient KCNQ1 mutation, T587M, causes a severe phenotype of long QT syndrome by interfering with intracellular hERG transport. J Cardiol 73:343–350. https://doi.org/10.1016/j.jjcc.2018.10.011
Zhang J, Kim EC, Chen C et al (2020) Identifying mutation hotspots reveals pathogenetic mechanisms of KCNQ2 epileptic encephalopathy. Sci Rep 10:4756. https://doi.org/10.1038/s41598-020-61697-6
Finke C, Prüss H, Scheel M et al (2012) Anti-NMDA receptor antibodies in a case of MELAS syndrome. J Neurol 259:582–584. https://doi.org/10.1007/s00415-011-6229-0
Chung H, Kim Y, Cho S-M et al (2020) Differential contributions of sarcomere and mitochondria-related multigene variants to the endophenotype of hypertrophic cardiomyopathy. Mitochondrion 53:48–56. https://doi.org/10.1016/j.mito.2020.04.010
Poduri A (2014) HCN1 Gain-Of-Function Mutations - A New Cause of Epileptic Encephalopathy. Epilepsy Curr 14:348–349. https://doi.org/10.5698/1535-7597-14.6.348
Seltzer LE, Ma M, Ahmed S et al (2014) Epilepsy and outcome in FOXG1-related disorders. Epilepsia 55:1292–1300. https://doi.org/10.1111/epi.12648
Kerr M, Hume S, Omar F et al (2020) MITO-FIND: A study in 390 patients to determine a diagnostic strategy for mitochondrial disease. Mol Genet Metab. https://doi.org/10.1016/j.ymgme.2020.08.009
Liang C, Ahmad K, Sue CM (2014) The broadening spectrum of mitochondrial disease: shifts in the diagnostic paradigm. Biochim Biophys Acta 1840:1360–1367. https://doi.org/10.1016/j.bbagen.2013.10.040
Schon KR, Ratnaike T, van den Ameele J et al (2020) Mitochondrial diseases: a diagnostic revolution. Trends Genet 36:702–717. https://doi.org/10.1016/j.tig.2020.06.009
Acknowledgments
The authors thank the patients and family members who participated in this study. The infrastructure funding from DST-FIST, TIFAC-CORE, VGST Karnataka and Manipal Academy of Higher Education is gratefully acknowledged. PG was supported by Science and Engineering Research Board (SERB), Department of Science and Technology (DST), Government of India (PDF/2016/001625) and DBT-Research Associateship from Department of Biotechnology (DBT), Government of India. BPS was supported by a clinical fellowship from Mito-Foundation Australia. KT was supported by research Grant and JC Bose Fellowship from SERB, DST, Government of India.
Funding
Open access funding provided by Manipal Academy of Higher Education, Manipal.
Author information
Authors and Affiliations
Contributions
Study concept and design: KS, KT; acquisition, analysis and interpretation of data: SC, PG, BPS, MN, SD, JNJP, HRA, AKM, RKJ, SS, NG, ABT; bioinformatics analysis: SC, SM, PJ; manuscript drafting: SC, PG; critical revision for intellectual content: SC, PG, BPS, SPK, NG, ABT, KT, KS.
Corresponding author
Ethics declarations
Conflicts of interest
All authors declare that they have no conflit of interest.
Ethical approval
Institutional Ethical Committee (IEC) clearance was obtained from the participating institute (NIMHANS/IEC (BS & NS DIV.) 8th meeting/2017). Patients were enrolled after obtaining the written informed consent.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Chakrabarty, S., Govindaraj, P., Sankaran, B.P. et al. Contribution of nuclear and mitochondrial gene mutations in mitochondrial encephalopathy, lactic acidosis, and stroke-like episodes (MELAS) syndrome. J Neurol 268, 2192–2207 (2021). https://doi.org/10.1007/s00415-020-10390-9
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00415-020-10390-9