
Abstract
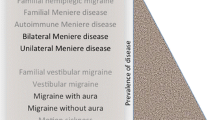
The two most common vestibular disorders are motion sickness and vestibular migraine, affecting 30 and 1–2 % of the population respectively. Both are related to migraine and show a familial trend. Bilateral vestibular hypofunction is a rare condition, and some of patients also present cerebellar ataxia and neuropathy. We present recent advances in the genetics of vestibular disorders with familial aggregation. The clinical heterogeneity observed in different relatives of the same families suggests a variable penetrance and the interaction of several genes in each family. Some Mendelian sensorineural hearing loss also exhibits vestibular dysfunction, including DFNA9, DFNA11, DFNA15 and DFNA28. However, the most relevant finding during the past years is the familial clustering observed in Meniere’s disease. By using whole exome sequencing and combining bioinformatics tools, novel variants in DTNA and FAM136A genes have been identified in familial Meniere’s disease, and this genomic strategy will facilitate the discovery of the genetic basis of familial vestibular disorders.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Vestibular disorders (VDs) are considered a group of diseases leading to transient or permanent loss of vestibular function. Regardless of their clinical heterogeneity, several families with vestibular symptoms affecting different relatives have been described. However, the contribution of genetic variations, either common or rare variants, to develop a specific vestibular disease is largely unknown. Some of the reasons for this missing heritability in VDs are the limitations in clinical phenotyping, the overlapping symptoms among different VDs and the clinical heterogeneity observed in families with variable penetrance (Table 1). According to their clinical presentation, we can distinguish three major syndromes: an episodic vestibular syndrome, a progressive vestibular syndrome and a third group of diseases, which include sensorineural hearing loss (SNHL) with a variable vestibular dysfunction [1, 2].
In this review, we will update the evidences to support the genetic contribution in some of the vestibular disorders with familial aggregation.
Episodic vestibular syndromes
The two most common vestibular disorders are motion sickness and vestibular migraine, affecting 30 and 1–2 % of the population, respectively. Both are complex, multifactorial disorders with a strong genetic contribution. Table 2 summarizes diseases with vestibular involvement and potential genes involved.
Motion sickness
Motion sickness refers to autonomic signs and symptoms occurring during movement, and maybe elicited by self-motion or motion of the environment. It is the most common vestibular condition affecting 30 % of the population. Its symptoms include dizziness, nausea, vomiting, pallor and headache [3]. The condition has been associated with postoperative vomiting, altitude sickness, morning sickness and migraine. A genome-wide association study performed in 80494 individuals has identified 35 common single-nucleotide variants at genome-wide significant level. The top ten genes involved in these regions include PVRL3, GPD2, ACO1, AUTS2, GPR26, UBE2E2, CBLN4, BLOC1S5, LINGO2 and CPNE4. These genes involve a large variety of functions such as ocular and brain development, insulin resistance, otolith biogenesis or iron homeostasis [4].
Vestibular migraine
Definite vestibular migraine (VM) is defined by the occurrence of episodic vestibular symptoms and a history of migraine, demonstrating a temporal association between vestibular and migraine symptoms in at least 50 % of the attacks [5]. VM shows a transient vestibular dysfunction, and it presents overlapping symptoms with Meniere’s disease (MD) during the attacks [6]. Moreover, the association with migraine, episodic vertigo and MD, frequently observed clustered in families, including identical twins, supports the heritability of VM [7].
Epidemiological studies report that around 56 % of MD patients also refer migrainous headaches [8]; in both disorders, a familial predisposition has been postulated. Migraine, per se, may provoke episodic vertigo, and VM has a prevalence of about 1 % of the population [5].
So, genetic variations in ion channels and membrane transporters which regulate fluid homeostasis could explain the relationship between migraine and MD [9, 10].
Different mutations in three genes have been linked to a rare form of migraine called familial hemiplegic migraine (FHM): (a) CACNA1A, provoking a gain of function in a L-type Ca++ channel, (b) ATP1A2, leading to a loss of function of the Na+, K+, -ATPase transporter and (c) SCN1A causing a gain of function of a voltage-gated Na+ channel [11–13]. Based on the observation of families presenting both FHM and common migraine, a shared pathophysiology has been postulated. The cortical spreading depression, for example, has been associated with acute changes in brain extracellular ionic concentration [14]; moreover, an increased capillary endothelial cell Na+, K+, -ATPase activity, leading to an increased Na+ concentration, has been linked to migraine pathophysiology [15]. In a recent paper, common variations in different ion transport genes were examined; none were significant, but an epistatic interaction between KCNB2 gene (encoding a potassium channel) and CACNB2 (encoding a calcium channel) was observed [16]. Conversely, no evidence of an association with genes encoding ion channels linked to FHM has been found in VM [17].
Ionic homeostasis in the inner ear plays an important role for the maintenance of the endocochlear potential. In Jervell and Lange-Nielsen syndrome, a mutation of KCNQ1 and KCNE1 channels leads to a severe SNHL and a collapse of the cochlear scala media [18].
More recently, a variant of ATP1A2 has been associated in a Korean family with a new form of progressive hearing loss with migraine [19].
Progressive vestibular syndromes
Bilateral vestibular hypofunction
Bilateral vestibular hypofunction starts in young adults with episodes of vertigo, triggered by exercise and stress, causing chronic disequilibrium, postural instability and disabling oscillopsia. Bilateral vestibular hypofunction without hearing loss, also referred as bilateral vestibulopathy, was described in 20 families with autosomal dominant inheritance without migraine. Linkage analyses in these families segregated a region in chromosome 6q [20]. No genes have been identified in bilateral vestibular hypofunction.
Canvas
Cerebellar ataxia, neuropathy and bilateral vestibular areflexia syndrome (CANVAS) is a neurodegenerative ganglionopathy. It is a rare late-onset slowly progressive ataxia, characterized by the combination of imbalance due to cerebellar gait and limb ataxia, bilateral vestibular impairment, and nonlength-dependent sensory neuropathy [21]. It has been reported a loss of neurons from the dorsal root and cranial nerve ganglia, but there is a phenotypic heterogeneity in CANVAS patients [22]. Although most of cases are sporadic, few families with several cases suggest genetic heterogeneity. No causal gene has been involved with CANVAS.
Sensorineural hearing loss with variable vestibular dysfunction
Dizziness and episodic vertigo can be combined with moderate or severe hearing loss as a result of a cochleovestibular disorder. So, genes involved in the development of the otic capsule and temporal bone development are likely to cause cochleovestibular disorders. Hereditary deafness may be conductive, sensorineural or a combination of both: syndromic (associated with malformations of the external ear or other organs or with medical problems involving other organ systems) or nonsyndromic (without association to visible abnormalities of the external ear or any related medical problems); and prelingual (before language development) or postlingual (after language development) [23]. Hearing loss heredity can be classified as autosomal dominant (A), autosomal recessive (B), X-linked (X), or based on changes in the mitochondrial DNA. Cerebellar and vestibular disorders show not only overlapping clinical symptoms, but also shared genetic risk factors. Some types of autosomal dominant nonsyndromic deafness genes (DFNA) are known to be associated with vestibular symptoms (DFNA9, DFNA11 and DFNA15).
Dfna9
DFNA9 is a nonsyndromic autosomal dominant SNHL with vestibular dysfunction caused by heterozygous mutations in the COCH (coagulation factor C homology) gene, encoding the secreted protein cochlin. The protein contains an N-terminal signal peptide (SP), an LCCL (limulus factor C, cochlin and late gestation lung protein Lgl1) domain, two von Willebrand factor A-like (vWFA) domains and two short intervening domains (ivd) [24]. It is an adult-onset form of progressive high-frequency SNHL associated with variable vestibular dysfunction that consisted of gait imbalance with instability in the dark and oscillopsia. Vestibular testing showed bilateral vestibular hypofunction [25].
Robertson et al. [26] identified a partial human cDNA for a novel cochlear transcript, hCoch-5B2, later called COCH. High levels of hCoch-5B expression were only seen in human fetal cochlea and vestibule, within a large panel of human fetal and adult tissues. They mapped hCoch-5B2 to human 14q11.2–q13 linked to DFNA9. Additionally, they reported three missense mutations in human COCH, in three unrelated kindreds with DFNA9. All three residues mutated in DFNA9 were conserved in mouse and chicken Coch and were found in a region containing four conserved cysteines with homology to the LCCL domain. Human temporal bones show histopathological findings of an acidophilic ground substance in DFNA9 patients [27].
So far, 21 mutations have been described in the COCH gene. Bae et al. [24] performed a comprehensive analysis of clinical information and molecular findings from DFNA9 patients to identify genotype–phenotype correlations. Five mutations were limited to intracellular cochlin: two vWFA domain mutants that formed high molecular weight aggregates in cell lysates, and three LCCL domain mutants, which were detected as intracellular dimeric cochlins, resulting in earlier age of onset of hearing defects. Further, those with vWFA domain mutations exhibited predominantly hearing loss, while LCCL domain mutations showed accompanying vestibular dysfunction. So, the failure of mutant cochlin transport, which impairs cochlin secretion, induces the formation and retention of dimers and large multimeric intracellular aggregates, which correlates with an earlier onset and progression of hearing loss in DFNA9.
Furthermore, Tsukada et al. [28] reported two novel mutations in p.I372T and p.C542R. The patients with the novel mutations in p.I372T and p.C542R within the vWFA2 domain showed early-onset progressive hearing loss, and those with the p.G88E mutation showed late-onset hearing loss and acute hearing deterioration over a short period. Vestibular symptoms were reported in the patients with p.G88E and p.C542R variants. Vestibular testing was carried out for the family with the p.G88E mutation. The patient had a severe vestibular dysfunction, while his son had no vestibular symptoms and a normal bilateral response in cVEMP, thought he showed a unilateral semicircular canal dysfunction with mild hearing loss.
Dfna11
DFNA11 is a nonsyndromic form of progressive SNHL with postlingual onset caused by MYO7A gene (myosin VIIA, 11q13.5), a further gene known to be associated with vestibular dysfunction. MYO7A is an unconventional myosin involved in the structural organization of hair bundles at the apex of sensory hair cells [29]. DFNA11 is characterized, clinically, by low- and middle-frequency hearing loss and variable vestibular dysfunction [30]. Patients have a late-onset hearing loss, and it progresses to severe impairment. Several mutations in the MYO7A gene have been identified as a cause of DFNA11 [31], and whole genome-sequencing approaches seem to validate this association [32]. Tamagawa et al. [33] described a family with 19 members, eight of which had bilateral progressive hearing impairment. Genetic studies demonstrated that all eight subjects presented a deletion in MYO7A gene, confirming a complete penetrance in the family. The subjects presented bilateral SNHL at all frequencies, five of them showed spontaneous nystagmus while the other three showed bilateral caloric hyporeflexia.
Dfna15
DFNA15 is characterized by early-onset progressive high-frequency hearing loss, but also by variable vestibular phenotype. It is caused by several missense mutations in POU4F3 gene (POU class 4 homeobox 3, 5q32). This gene encodes a member of the POU-domain family of transcription factors. POU-domain proteins have been observed to play important roles in control of cell identity in several systems [34]. Two affected individuals presented vestibular symptoms, according to their medical history. Vestibular examination (electronystagmography with rotatory chair and caloric tests) in 18 carriers and one phenocopy carrier in a Dutch family with DFNA15 with a L289F mutation in POU4F3 gene showed a great variability in the vestibular function from normal response to complete areflexia, suggesting that additional genes should be involved in the vestibular phenotype [35].
Dfna28
DFNA28 is a progressive dominant SNHL associated with a frameshift mutation of grainyhead-like 2 (GRHL2), but its etiology and mechanism remain unknown. The protein encoded by this gene is a transcription factor that can act as a homodimer or heterodimer with either GRHL1 or GRHL3. In 2002, Peters et al. [36] first associated the DFNA28 locus with mild-to-moderate postlingual progressive bilateral SNHL involving an affected five-generation North American family. Affected members had a heterozygous c.1609_1610insC mutation in exon 13.
Han et al. [37] developed a zebrafish grhl2b (T086) mutant model in which grhl2b expression was interrupted by an insertion of a Tol2 transposon element. The mutants exhibit enlarged otocysts, smaller or absence of otoliths, malformed semicircular canals, insensitiveness to sound stimulation and, interestingly, imbalanced swimming motion. Since grainyhead-like family members can regulate epithelial adhesion, the expression of some genes encoding junction proteins in mutants was examined. They showed that the expression of claudin b (cldnb) and the epithelial cell adhesion molecule (epcam) was abolished or dramatically reduced, and apical junctional complexes were abnormal in otic epithelial cells of mutant embryos. Co-injection of cldnb and epcam mRNA could largely restore the mutant phenotype. Injection of human wild-type GRHL2 mRNA could rescue the inner ear phenotype, but not the mutant GRHL2 mRNA derived from DFNA28 patients, into grhl2b (T086) mutant embryos.
GRHL2 has been confirmed as a deafness gene at the DFNA28 locus. Together with GRHL1, these two homologous proteins have similar sequences and functions. A grhl1 down-regulated zebrafish model exhibited inner ear developmental malformations, including missing otoliths, disordered and abnormal number of hair cells in the inner ear and lateral line, and sound insensitivity [38]. Remarkably, the mutant zebrafish swam in circles, being hair cell apoptosis evident. Using electron microscopy, desmosomes in the otic sensory epithelium were found to be damaged. These defects were partially rescued by treatment with either GRHL1 or its target gene, DSG1 [38].
Dfnb102/103
DFNB102/103 has been described in a Turkish family with recessive hearing loss and vestibular dysfunction. CLIC5 (chloride intracellular channel 5, 6p12.3) gene encodes a member of the chloride intracellular channel (CLIC) family of chloride ion channels. The encoded protein associates with actin-based cytoskeletal structures and may play a role in multiple processes, including hair cell stereocilia formation, myoblast proliferation and glomerular podocyte and endothelial cell maintenance [39].
The mutation was identified in a consanguineous family diagnosed with autosomal recessive SNHL. A homozygous region of 47.4 Mb on chromosome 6p21.1–q15 was shared by the two affected siblings. This region contains 247 genes including the known deafness gene MYO6, but no pathogenic variants were found. Subsequent candidate gene evaluation revealed CLIC5 as an excellent candidate gene. The orthologous mouse gene is mutated in the jitterbug mutant that exhibits progressive hearing impairment and vestibular dysfunction. Mutation analysis of CLIC5 revealed a homozygous nonsense mutation c.96T > A [p.(Cys32Ter)] that segregated with hearing loss. Hearing impairment in the present family had an early childhood onset and progressed, from mild to severe, before the second decade. Impaired hearing was accompanied by vestibular areflexia and in one of the subjects with mild renal dysfunction [40].
EVAS
Enlarged vestibular aqueduct syndrome (EVAS) is the most common form of inner ear malformation caused by a dilated vestibular aqueduct. It has been associated with a mutation in the SLC26A4 gene, which encodes pendrin, an anion exchanger expressed in the endolymphatic sac, in different epithelial cells of the cochlea and vestibular labyrinth, as well as in the kidney and thyroid gland. Mutations in this gene cause DFNB4 and Pendred’s syndrome, both of which are related to EVAS [41]. Some studies, performed in Chinese population, analyzed SLC26A4 gene using direct sequencing observing a total of 23 pathogenic mutations, 13 of which were unique [42], while other studies found a correlation between SLC26A4 mutations with wider aqueducts at the midpoint and more severe hearing loss [43]. Nevertheless, recent studies [44, 45] did not show any correlation between SLC26A4 gene mutations and the nature, degree, and progression of hearing loss [46]. SLC26A4 gene therapy targeted to the endolymphatic sac restored hearing and balance in SLC26A4 mutant mice [47].
Ménière’s disease
Ménière’s disease (MD) is a clinical syndrome that affects the inner ear, and it is characterized by episodes of spontaneous vertigo, usually associated with unilateral fluctuating SNHL, tinnitus and aural fullness [48]. The prevalence of MD is about 0.5–1/1000 individuals, being most of patients with MD considered sporadic, although around 9–10 % are familial cases in European descendant population [10, 49, 50]. Both familial and sporadic cases are clinically indistinguishable [2, 10]. The disorder is associated with an accumulation of endolymph in the scala media of the cochlea and the saccule (endolymphatic hydrops), and it may involve both ears in 10–40 % of patients [51]. Several triggering factors such as allergens, virus or autoantigens have been proposed as contributors to the pathophysiology of the endolymphatic hydrops in susceptible individuals, but no genetic locus has been linked with sporadic MD.
Several comorbidities are associated with MD, including migraine, obesity, psoriasis and systemic autoimmune disorders [52]. Although its exact etiology and pathogenesis are still unknown, and 1/3 of MD cases may have an anomalous response of the innate or adaptive system, the mechanisms involved remain unknown [53]. This hypothesis is supported by some evidence, including the response to steroids therapy, the finding of elevated levels of autoantibodies or circulating immune complexes (CIC) in the serum of some patients with MD against inner ear antigens. Remarkably, several allelic variants in immune innate response genes MICA, TLR10 and NFKB1 influence the hearing outcome in sporadic MD [54–56].
The hypothesis of a channelopathy has supported aquaporins (AQP) as molecular targets in MD. These proteins are water channels, the function of which is to transport water and solutes along the osmotic gradient, and four of them (AQP 1–4) are expressed in the human inner ear [57]. Water permeability of AQP is regulated by vasopressin [58], which has been reported to be increased in MD subjects before a vertigo attack. Vertigo attacks may arise from interplay of increased vasopressin release and allelic variants in AQP genes [59]. To date, genotyping studies on AQP have produced controversial results in AQP2 and AQP4 genes [58], and a homozygous c.105G- > C synonymous variant in AQP3 was found in 11 of 34 patients with MD [60].
Moreover, the association with allelic variants of two voltage-gated potassium channel genes, KCNE1 and KCNE3 in Japanese population, produced contrasting results, and it could not be replicated [61, 62].
Finally, in a case–control study, the predisposition to develop MD was linked to a common variant in the ADD1 gene, encoding a cytoskeletal protein, which interacts with the Na+, K+, -ATPase transporter, leading to its faster activity [63]; however, these findings were not replicated in an independent cohort [64].
Familial Ménière’s disease
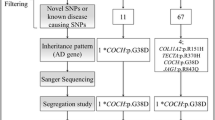
Familial MD (FMD) should be considered if at least two family members (first or second degree) fulfill all the criteria of definite or probable MD [10]. Most families described have an autosomal dominant pattern of inheritance, but, as FMD shows clinical heterogeneity, mitochondrial and recessive inheritance patterns are also observed in some families [10]. Linkage studies in FMD have found candidate loci at 5q14–15 in a German family [65] and 12p12.3 in a large Swedish family [66], but the genes were not identified. By exome sequencing, Requena et al. [67] have identified mutations in DTNA and FAM136A genes in an autosomal dominant Spanish family with MD segregating the phenotype in three women in consecutive generations. DTNA encodes alpha-dystrobrevin, a dystrophin-associated protein which interacts with transmembrane proteins and actin in the basolateral membranes of epithelial cells, and it is associated with tight junction reorganization [68]. The identified mutation at position chr18: 32462094G > T in the DTNA gene is a rare splice-site acceptor skipping in exon 21. FAM136A is a mitochondrial protein of unknown function, and the mutation chr2:70527974C > T leads to a novel stop codon which shortens the protein product. Both FAM136A and DTNA proteins are expressed in the neurosensorial epithelium of the crista ampullaris of the rat. Further studies are needed to investigate DTNA and FAM136A genes in sporadic MD.
Conclusion
The improvement in phenotyping families with vestibular symptoms combined with exome sequencing will facilitate the identification of rare variants and genes involved in familial vestibular disorders.
References
Gazquez I, Lopez-Escamez JA (2011) Genetics of recurrent vertigo and vestibular disorders. Curr Genomics 12(6):443–450. doi:10.2174/138920211797248600
Requena T, Espinosa-Sanchez JM, Lopez-Escamez JA (2014) Genetics of dizziness: cerebellar and vestibular disorders. Curr Opin Neurol 27(1):98–104. doi:10.1097/wco.0000000000000053
Golding JF, Gresty MA (2015) Pathophysiology and treatment of motion sickness. Curr Opin Neurol 28(1):83–88. doi:10.1097/wco.0000000000000163
Hromatka BS, Tung JY, Kiefer AK, Do CB, Hinds DA, Eriksson N (2015) Genetic variants associated with motion sickness point to roles for inner ear development, neurological processes and glucose homeostasis. Hum Mol Genet 24(9):2700–2708. doi:10.1093/hmg/ddv028
Lempert T, Olesen J, Furman J, Waterston J, Seemungal B, Carey J, Bisdorff A, Versino M, Evers S, Newman-Toker D (2012) Vestibular migraine: diagnostic criteria. J Vestib Res Equilib Orientat 22(4):167–172. doi:10.3233/ves-2012-0453
Lopez-Escamez JA, Dlugaiczyk J, Jacobs J, Lempert T, Teggi R, von Brevern M, Bisdorff A (2014) Accompanying symptoms overlap during attacks in Meniere’s disease and vestibular migraine. Front Neurol 5:265. doi:10.3389/fneur.2014.00265
Cha YH, Kane MJ, Baloh RW (2008) Familial clustering of migraine, episodic vertigo, and Meniere’s disease. Otol Neurotol Off Publ Am Otol Soc Am Neurotol Soc Eur Acad Otol Neurotol 29(1):93–96. doi:10.1097/mao.0b013e31815c2abb
Radtke A, Lempert T, Gresty MA, Brookes GB, Bronstein AM, Neuhauser H (2002) Migraine and Meniere’s disease: is there a link? Neurology 59(11):1700–1704
Rudkjobing LA, Esserlind AL, Olesen J (2012) Future possibilities in migraine genetics. J Headache Pain 13(7):505–511. doi:10.1007/s10194-012-0481-2
Requena T, Espinosa-Sanchez JM, Cabrera S, Trinidad G, Soto-Varela A, Santos-Perez S, Teggi R, Perez P, Batuecas-Caletrio A, Fraile J, Aran I, Martin E, Benitez J, Perez-Fernandez N, Lopez-Escamez JA (2014) Familial clustering and genetic heterogeneity in Meniere’s disease. Clin Genet 85(3):245–252. doi:10.1111/cge.12150
Dichgans M, Freilinger T, Eckstein G, Babini E, Lorenz-Depiereux B, Biskup S, Ferrari MD, Herzog J, van den Maagdenberg AM, Pusch M, Strom TM (2005) Mutation in the neuronal voltage-gated sodium channel SCN1A in familial hemiplegic migraine. Lancet 366(9483):371–377. doi:10.1016/s0140-6736(05)66786-4
De Fusco M, Marconi R, Silvestri L, Atorino L, Rampoldi L, Morgante L, Ballabio A, Aridon P, Casari G (2003) Haploinsufficiency of ATP1A2 encoding the Na+/K+ pump alpha2 subunit associated with familial hemiplegic migraine type 2. Nat Genet 33(2):192–196. doi:10.1038/ng1081
Ducros A, Denier C, Joutel A, Cecillon M, Lescoat C, Vahedi K, Darcel F, Vicaut E, Bousser MG, Tournier-Lasserve E (2001) The clinical spectrum of familial hemiplegic migraine associated with mutations in a neuronal calcium channel. N Engl J Med 345(1):17–24. doi:10.1056/nejm200107053450103
Kraig RP, Nicholson C (1978) Extracellular ionic variations during spreading depression. Neuroscience 3(11):1045–1059
Harrington MG, Fonteh AN, Arakaki X, Cowan RP, Ecke LE, Foster H, Huhmer AF, Biringer RG (2010) Capillary endothelial Na(+), K(+), ATPase transporter homeostasis and a new theory for migraine pathophysiology. Headache 50(3):459–478. doi:10.1111/j.1526-4610.2009.01551.x
Nyholt DR, LaForge KS, Kallela M, Alakurtti K, Anttila V, Farkkila M, Hamalainen E, Kaprio J, Kaunisto MA, Heath AC, Montgomery GW, Gobel H, Todt U, Ferrari MD, Launer LJ, Frants RR, Terwindt GM, de Vries B, Verschuren WM, Brand J, Freilinger T, Pfaffenrath V, Straube A, Ballinger DG, Zhan Y, Daly MJ, Cox DR, Dichgans M, van den Maagdenberg AM, Kubisch C, Martin NG, Wessman M, Peltonen L, Palotie A (2008) A high-density association screen of 155 ion transport genes for involvement with common migraine. Hum Mol Genet 17(21):3318–3331. doi:10.1093/hmg/ddn227
von Brevern M, Ta N, Shankar A, Wiste A, Siegel A, Radtke A, Sander T, Escayg A (2006) Migrainous vertigo: mutation analysis of the candidate genes CACNA1A, ATP1A2, SCN1A, and CACNB4. Headache 46(7):1136–1141. doi:10.1111/j.1526-4610.2006.00504.x
Friedmann I, Fraser GR, Froggatt P (1966) Pathology of the ear in the cardioauditory syndrome of Jervell and Lange-Nielsen (recessive deafness with electrocardiographic abnormalities). J Laryngol Otol 80(5):451–470
Oh SK, Baek JI, Weigand KM, Venselaar H, Swarts HG, Park SH, Hashim Raza M, da Jung J, Choi SY, Lee SH, Friedrich T, Vriend G, Koenderink JB, Kim UK, Lee KY (2015) A missense variant of the ATP1A2 gene is associated with a novel phenotype of progressive sensorineural hearing loss associated with migraine. Eur J Hum Genet EJHG 23(5):639–645. doi:10.1038/ejhg.2014.154
Jen JC, Wang H, Lee H, Sabatti C, Trent R, Hannigan I, Brantberg K, Halmagyi GM, Nelson SF, Baloh RW (2004) Suggestive linkage to chromosome 6q in families with bilateral vestibulopathy. Neurology 63(12):2376–2379
Szmulewicz DJ, Waterston JA, MacDougall HG, Mossman S, Chancellor AM, McLean CA, Merchant S, Patrikios P, Halmagyi GM, Storey E (2011) Cerebellar ataxia, neuropathy, vestibular areflexia syndrome (CANVAS): a review of the clinical features and video-oculographic diagnosis. Ann N Y Acad Sci 1233:139–147. doi:10.1111/j.1749-6632.2011.06158.x
Szmulewicz DJ, McLean CA, Rodriguez ML, Chancellor AM, Mossman S, Lamont D, Roberts L, Storey E, Halmagyi GM (2014) Dorsal root ganglionopathy is responsible for the sensory impairment in CANVAS. Neurology 82(16):1410–1415. doi:10.1212/wnl.0000000000000352
Smith RJH, Shearer AE, Hildebrand MS, Van Camp G (1993) Deafness and Hereditary Hearing Loss Overview. In: Pagon RA, Adam MP, Ardinger HH et al (eds) GeneReviews(R). University of Washington, Seattle, Seattle
Bae SH, Robertson NG, Cho HJ, Morton CC, da Jung J, Baek JI, Choi SY, Lee J, Lee KY, Kim UK (2014) Identification of pathogenic mechanisms of COCH mutations, abolished cochlin secretion, and intracellular aggregate formation: genotype-phenotype correlations in DFNA9 deafness and vestibular disorder. Hum Mutat 35(12):1506–1513. doi:10.1002/humu.22701
Robertson NG, Cremers CW, Huygen PL, Ikezono T, Krastins B, Kremer H, Kuo SF, Liberman MC, Merchant SN, Miller CE, Nadol JB Jr, Sarracino DA, Verhagen WI, Morton CC (2006) Cochlin immunostaining of inner ear pathologic deposits and proteomic analysis in DFNA9 deafness and vestibular dysfunction. Hum Mol Genet 15(7):1071–1085. doi:10.1093/hmg/ddl022
Robertson NG, Skvorak AB, Yin Y, Weremowicz S, Johnson KR, Kovatch KA, Battey JF, Bieber FR, Morton CC (1997) Mapping and characterization of a novel cochlear gene in human and in mouse: a positional candidate gene for a deafness disorder, DFNA9. Genomics 46(3):345–354. doi:10.1006/geno.1997.5067
Robertson NG, Lu L, Heller S, Merchant SN, Eavey RD, McKenna M, Nadol JB Jr, Miyamoto RT, Linthicum FH Jr, Lubianca Neto JF, Hudspeth AJ, Seidman CE, Morton CC, Seidman JG (1998) Mutations in a novel cochlear gene cause DFNA9, a human nonsyndromic deafness with vestibular dysfunction. Nat Genet 20(3):299–303. doi:10.1038/3118
Tsukada K, Ichinose A, Miyagawa M, Mori K, Hattori M, Nishio SY, Naito Y, Kitajiri SI, Usami SI (2015) Detailed hearing and vestibular profiles in the patients with COCH mutations. Ann Otol Rhinol Laryngol. doi:10.1177/0003489415573074
Ernest S, Rosa FM (2014) A genomic region encompassing a newly identified exon provides enhancing activity sufficient for normal myo7aa expression in zebrafish sensory hair cells. Dev Neurobiol. doi:10.1002/dneu.22263
Sun Y, Chen J, Sun H, Cheng J, Li J, Lu Y, Lu Y, Jin Z, Zhu Y, Ouyang X, Yan D, Dai P, Han D, Yang W, Wang R, Liu X, Yuan H (2011) Novel missense mutations in MYO7A underlying postlingual high- or low-frequency non-syndromic hearing impairment in two large families from China. J Hum Genet 56(1):64–70. doi:10.1038/jhg.2010.147
Sang Q, Yan X, Wang H, Feng R, Fei X, Ma D, Xing Q, Li Q, Zhao X, Jin L, He L, Li H, Wang L (2013) Identification and functional study of a new missense mutation in the motor head domain of myosin VIIA in a family with autosomal dominant hearing impairment (DFNA11). PLoS one 8(1):e55178. doi:10.1371/journal.pone.0055178
Riahi Z, Bonnet C, Zainine R, Lahbib S, Bouyacoub Y, Bechraoui R, Marrakchi J, Hardelin JP, Louha M, Largueche L, Ben Yahia S, Kheirallah M, Elmatri L, Besbes G, Abdelhak S, Petit C (2015) Whole exome sequencing identifies mutations in Usher syndrome genes in profoundly deaf Tunisian patients. PLoS one 10(3):e0120584. doi:10.1371/journal.pone.0120584
Tamagawa Y, Ishikawa K, Ishikawa K, Ishida T, Kitamura K, Makino S, Tsuru T, Ichimura K (2002) Phenotype of DFNA11: a nonsyndromic hearing loss caused by a myosin VIIA mutation. Laryngosc 112(2):292–297. doi:10.1097/00005537-200202000-00017
Baek JI, Oh SK, Kim DB, Choi SY, Kim UK, Lee KY, Lee SH (2012) Targeted massive parallel sequencing: the effective detection of novel causative mutations associated with hearing loss in small families. Orphanet J Rare Dis 7:60. doi:10.1186/1750-1172-7-60
van Drunen FJ, Pauw RJ, Collin RW, Kremer H, Huygen PL, Cremers CW (2009) Vestibular impairment in a Dutch DFNA15 family with an L289F mutation in POU4F3. Audiol Neuro Otol 14(5):303–307. doi:10.1159/000212109
Peters LM, Anderson DW, Griffith AJ, Grundfast KM, San Agustin TB, Madeo AC, Friedman TB, Morell RJ (2002) Mutation of a transcription factor, TFCP2L3, causes progressive autosomal dominant hearing loss, DFNA28. Hum Mol Genet 11(23):2877–2885
Han Y, Mu Y, Li X, Xu P, Tong J, Liu Z, Ma T, Zeng G, Yang S, Du J, Meng A (2011) Grhl2 deficiency impairs otic development and hearing ability in a zebrafish model of the progressive dominant hearing loss DFNA28. Hum Mol Genet 20(16):3213–3226. doi:10.1093/hmg/ddr234
Liu F, Yang F, Wen D, Xia W, Hao L, Hu J, Zong J, Shen X, Ma J, Jiang N, Sun S, Zhang J, Wang H, Wang X, Ma Z, Ma D (2015) Grhl1 deficiency affects inner ear development in zebrafish. Int J Dev Biol. doi:10.1387/ijdb.140230FL
Gagnon LH, Longo-Guess CM, Berryman M, Shin JB, Saylor KW, Yu H, Gillespie PG, Johnson KR (2006) The chloride intracellular channel protein CLIC5 is expressed at high levels in hair cell stereocilia and is essential for normal inner ear function. J Neurosci Off J Soc Neurosci 26(40):10188–10198. doi:10.1523/jneurosci.2166-06.2006
Seco CZ, Oonk AM, Dominguez-Ruiz M, Draaisma JM, Gandia M, Oostrik J, Neveling K, Kunst HP, Hoefsloot LH, del Castillo I, Pennings RJ, Kremer H, Admiraal RJ, Schraders M (2015) Progressive hearing loss and vestibular dysfunction caused by a homozygous nonsense mutation in CLIC5. Eur J Hum Genet EJHG 23(2):189–194. doi:10.1038/ejhg.2014.83
Duman D, Tekin M (2012) Autosomal recessive nonsyndromic deafness genes: a review. Front Biosci (Landmark Ed) 17:2213–2236
Hu H, Wu L, Feng Y, Pan Q, Long Z, Li J, Dai H, Xia K, Liang D, Niikawa N, Xia J (2007) Molecular analysis of hearing loss associated with enlarged vestibular aqueduct in the mainland Chinese: a unique SLC26A4 mutation spectrum. J Hum Genet 52(6):492–497. doi:10.1007/s10038-007-0139-0
Madden C, Halsted M, Meinzen-Derr J, Bardo D, Boston M, Arjmand E, Nishimura C, Yang T, Benton C, Das V, Smith R, Choo D, Greinwald J (2007) The influence of mutations in the SLC26A4 gene on the temporal bone in a population with enlarged vestibular aqueduct. Arch Otolaryngol Head Neck Surg 133(2):162–168. doi:10.1001/archotol.133.2.162
Reyes S, Wang G, Ouyang X, Han B, Du LL, Yuan HJ, Yan D, Dai P, Liu XZ (2009) Mutation analysis of SLC26A4 in mainland Chinese patients with enlarged vestibular aqueduct. Otolaryngol Head Neck Surg Off J Am Acad Otolaryngol Head Neck Surg 141(4):502–508. doi:10.1016/j.otohns.2009.07.004
Jonard L, Niasme-Grare M, Bonnet C, Feldmann D, Rouillon I, Loundon N, Calais C, Catros H, David A, Dollfus H, Drouin-Garraud V, Duriez F, Eliot MM, Fellmann F, Francannet C, Gilbert-Dussardier B, Gohler C, Goizet C, Journel H, Mom T, Thuillier-Obstoy MF, Couderc R, Garabedian EN, Denoyelle F, Marlin S (2010) Screening of SLC26A4, FOXI1 and KCNJ10 genes in unilateral hearing impairment with ipsilateral enlarged vestibular aqueduct. Int J Pediatr Otorhinolaryngol 74(9):1049–1053. doi:10.1016/j.ijporl.2010.06.002
Gopen Q, Zhou G, Whittemore K, Kenna M (2011) Enlarged vestibular aqueduct: review of controversial aspects. Laryngosc 121(9):1971–1978. doi:10.1002/lary.22083
Li X, Sanneman JD, Harbidge DG, Zhou F, Ito T, Nelson R, Picard N, Chambrey R, Eladari D, Miesner T, Griffith AJ, Marcus DC, Wangemann P (2013) SLC26A4 targeted to the endolymphatic sac rescues hearing and balance in Slc26a4 mutant mice. PLoS Genet 9(7):e1003641. doi:10.1371/journal.pgen.1003641
Lopez-Escamez JA, Carey J, Chung WH, Goebel JA, Magnusson M, Mandala M, Newman-Toker DE, Strupp M, Suzuki M, Trabalzini F, Bisdorff A (2015) Diagnostic criteria for Meniere’s disease. J Vestib Res Equilib Orientat 25(1):1–7. doi:10.3233/ves-150549
Vrabec JT (2010) Genetic investigations of Meniere’s disease. Otolaryngol Clin North Am 43(5):1121–1132. doi:10.1016/j.otc.2010.05.010
Morrison AW, Bailey ME, Morrison GA (2009) Familial Meniere’s disease: clinical and genetic aspects. J Laryngol Otol 123(1):29–37. doi:10.1017/s0022215108002788
Lopez-Escamez JA, Viciana D, Garrido-Fernandez P (2009) Impact of bilaterality and headache on health-related quality of life in Meniere’s disease. Ann Otol Rhinol Laryngol 118(6):409–416
Tyrrell JS, Whinney DJ, Ukoumunne OC, Fleming LE, Osborne NJ (2014) Prevalence, associated factors, and comorbid conditions for Meniere’s disease. Ear Hear 35(4):e162–e169. doi:10.1097/aud.0000000000000041
Greco A, Gallo A, Fusconi M, Marinelli C, Macri GF, de Vincentiis M (2012) Meniere’s disease might be an autoimmune condition? Autoimmun Rev 11(10):731–738. doi:10.1016/j.autrev.2012.01.004
Gazquez I, Moreno A, Aran I, Soto-Varela A, Santos S, Perez-Garrigues H, Lopez-Nevot A, Requena T, Lopez-Nevot MA, Lopez-Escamez JA (2012) MICA-STR A.4 is associated with slower hearing loss progression in patients with Meniere’s disease. Otol Neurotol Off Publ Am Otol Soc Am Neurotol Soc Eur Acad Otol Neurotol 33(2):223–229. doi:10.1097/MAO.0b013e31824296c8
Requena T, Gazquez I, Moreno A, Batuecas A, Aran I, Soto-Varela A, Santos-Perez S, Perez N, Perez-Garrigues H, Lopez-Nevot A, Martin E, Sanz R, Perez P, Trinidad G, Alarcon-Riquelme ME, Teggi R, Zagato L, Lopez-Nevot MA, Lopez-Escamez JA (2013) Allelic variants in TLR10 gene may influence bilateral affectation and clinical course of Meniere’s disease. Immunogenetics 65(5):345–355. doi:10.1007/s00251-013-0683-z
Cabrera S, Sanchez E, Requena T, Martinez-Bueno M, Benitez J, Perez N, Trinidad G, Soto-Varela A, Santos-Perez S, Martin-Sanz E, Fraile J, Perez P, Alarcon-Riquelme ME, Batuecas A, Espinosa-Sanchez JM, Aran I, Lopez-Escamez JA (2014) Intronic variants in the NFKB1 gene may influence hearing forecast in patients with unilateral sensorineural hearing loss in Meniere’s disease. PLoS one 9(11):e112171. doi:10.1371/journal.pone.0112171
Beitz E, Kumagami H, Krippeit-Drews P, Ruppersberg JP, Schultz JE (1999) Expression pattern of aquaporin water channels in the inner ear of the rat. The molecular basis for a water regulation system in the endolymphatic sac. Hear Res 132(1–2):76–84
Knepper MA, Inoue T (1997) Regulation of aquaporin-2 water channel trafficking by vasopressin. Curr Opin Cell Biol 9(4):560–564
Takeda T, Takeda S, Kakigi A, Okada T, Nishioka R, Taguchi D, Nishimura M, Nakatani H (2010) Hormonal aspects of Meniere’s disease on the basis of clinical and experimental studies. ORL J Oto Rhino Laryngol Relat Spec 71(Suppl 1):1–9. doi:10.1159/000265113
Candreia C, Schmuziger N, Gurtler N (2010) Molecular analysis of aquaporin genes 1 to 4 in patients with Meniere’s disease. Cell Physiol Biochem Int J Exp Cell Physiol Biochem Pharmacol 26(4–5):787–792. doi:10.1159/000322346
Doi K, Sato T, Kuramasu T, Hibino H, Kitahara T, Horii A, Matsushiro N, Fuse Y, Kubo T (2005) Meniere’s disease is associated with single nucleotide polymorphisms in the human potassium channel genes, KCNE1 and KCNE3. ORL J Oto Rhino Laryngol Relat Spec 67(5):289–293. doi:10.1159/000089410
Campbell CA, Della Santina CC, Meyer NC, Smith NB, Myrie OA, Stone EM, Fukushima K, Califano J, Carey JP, Hansen MR, Gantz BJ, Minor LB, Smith RJ (2010) Polymorphisms in KCNE1 or KCNE3 are not associated with Meniere disease in the Caucasian population. Am J Med Genet Part A 152A(1):67–74. doi:10.1002/ajmg.a.33114
Teggi R, Lanzani C, Zagato L, Delli Carpini S, Manunta P, Bianchi G, Bussi M (2008) Gly460Trp alpha-adducin mutation as a possible mechanism leading to endolymphatic hydrops in Meniere’s syndrome. Otol Neurotol Off Publ Am Otol Soc Am Neurotol Soc Eur Acad Otol Neurotol 29(6):824–828. doi:10.1097/MAO.0b013e318180a4b1
Hietikko E, Kotimaki J, Okuloff A, Sorri M, Mannikko M (2012) A replication study on proposed candidate genes in Meniere’s disease, and a review of the current status of genetic studies. Int J Audiol 51(11):841–845. doi:10.3109/14992027.2012.705900
Arweiler-Harbeck D, Horsthemke B, Jahnke K, Hennies HC (2011) Genetic aspects of familial Meniere’s disease. Otol Neurotol Off Publ Am Otol Soc Am Neurotol Soc Eur Acad Otol Neurotol 32(4):695–700. doi:10.1097/MAO.0b013e318216074a
Klar J, Frykholm C, Friberg U, Dahl N (2006) A Meniere’s disease gene linked to chromosome 12p12.3. Am J Med Genet Part B Neuropsychiatr Genet Off Publ Int Soc Psychiatr Genet 141B(5):463–467. doi:10.1002/ajmg.b.30347
Requena T, Cabrera S, Martin-Sierra C, Price SD, Lysakowski A, Lopez-Escamez JA (2014) Identification of two novel mutations in FAM136A and DTNA genes in autosomal-dominant familial Meniere’s disease. Hum Mol Genet. doi:10.1093/hmg/ddu524
Sjo A, Magnusson KE, Peterson KH (2005) Association of alpha-dystrobrevin with reorganizing tight junctions. J Membr Biol 203(1):21–30. doi:10.1007/s00232-004-0728-1
Acknowledgments
The authors received funding for this study from MENIERE’S SOCIETY, PI13/1242 from INSTITUTO DE SALUD CARLOS III by FEDER Funds from the EU and by the Italian Headache foundation, FICEF (Fondazione Italiana Cefalee).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflicts of interest
On behalf of all authors, the corresponding author states that there is no conflict of interest.
Additional information
This manuscript is part of a supplement sponsored by the German Federal Ministry of Education and Research within the funding initiative for integrated research and treatment centers.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Frejo, L., Giegling, I., Teggi, R. et al. Genetics of vestibular disorders: pathophysiological insights. J Neurol 263 (Suppl 1), 45–53 (2016). https://doi.org/10.1007/s00415-015-7988-9
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00415-015-7988-9