
Abstract
Clinical equipoise is widely accepted as the basis of ethics in clinical research and requires investigators to be uncertain of the relative therapeutic merits of trial comparators. When clinical equipoise is in question, innovative trial designs are needed to reduce ethical tension while satisfying regulators’ requirements. We report a novel response-conditional crossover study design used in a Phase 3, randomized, double-blind, placebo-controlled clinical trial of intravenous 10% caprylate-chromatography purified immunoglobulin for chronic inflammatory demyelinating polyradiculoneuropathy. During the initial 24-week period, patients crossed over to the alternative treatment at the first sign of deterioration or if they failed to improve or were unable to maintain improvement at any time after 6 weeks. This trial design addressed concerns about lack of equipoise raised by physicians interested in trial participation and proved acceptable to regulatory authorities. The trial design may be applicable to other studies where clinical equipoise is in question.
Similar content being viewed by others


Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Confirmatory Phase 3 clinical trials typically involve the demonstration of efficacy and safety of a new intervention based on comparisons between a treated group and a control group, which may be placebo or another active treatment. Guidelines on clinical trial design generally state that new interventions should be compared with the best available therapy, and that a placebo comparator should only be used where no effective treatment exists [1]. This creates an ethical dilemma and clinical trial design challenge when regulatory authorities require large-scale, randomised, Phase 3 trials that are placebo controlled (in the absence of an approved treatment option).
As an example, intravenous immunoglobulin (IVIg) has been considered first-line therapy in chronic inflammatory demyelinating polyradiculoneuropathy (CIDP) for several years, based on evidence from small trials that IVIg is beneficial [2]. In the absence of an approved treatment for CIDP, gaining regulatory approval for the use of IVIg in CIDP required the conduct of large-scale, placebo-controlled Phase 3 trials.
A randomized, double-blind, controlled design (RCT) is the gold standard for confirmatory trials. Strengths of the RCT design include reduction or elimination of patient selection bias, minimization of imbalance of covariates such as prognostic factors, and enhanced reliability of statistical analysis due to the introduction of randomization [3]. Yet the RCT design also presents methodological and ethical challenges, including dilemmas in choosing the most suitable comparator, in ensuring an honest null hypothesis, and in designing studies that are not only acceptable ethically to physicians and patients but also meet the rigorous standards for Phase 3 trials required by regulatory authorities to support licensure. The latter challenge is the focus of this article and revolves around the concept of clinical equipoise.
Clinical equipoise
The ethics of clinical research require a state of clinical equipoise to exist between the comparator treatments. The term “clinical equipoise” was introduced in 1987 by Freedman [4], who defined it as a state of genuine uncertainty on the part of the clinical investigator about the relative therapeutic merits of the treatment arms in a trial. An investigator who has good reason to believe that one treatment is superior is obliged ethically to offer that treatment to patients.
While the results of RCTs invariably show that treatments differ in their effects, clinical equipoise at the start of a trial and throughout its duration protects patients from knowingly being exposed to inferior treatments. Progress in clinical medicine relies heavily on the willingness of patients to take part in clinical trials and evidence shows that they are only willing to participate in RCTs if there is an acknowledgment of expert uncertainty and if clinical equipoise exists [5]. Without clinical equipoise, investigators may be unwilling to risk their patients being randomized to a treatment arm (or placebo) that data indicate may be inferior, while patients are unlikely to enroll for the same reason.
The ethical debate surrounding clinical equipoise and RCTs
The principle of clinical equipoise as the basis of clinical research ethics has come under increasing criticism in recent years [6–10]. Miller and colleagues argue that clinical research and therapeutic practice are distinct activities with different goals, and are governed by different ethical principles [8, 9, 11, 12]. In their view, physicians in clinical practice have a moral obligation to provide patients with optimal care, whereas investigators in clinical trials have a primary duty to increase scientific knowledge, which may be at the expense of their secondary duty—to prevent harm to experimental subjects. This view has been regarded as unsatisfactory by many because it requires an implausible moral dissociation whereby trial investigators must ignore the professional obligations that they have as physicians [13].
The question arises as to whether clinical equipoise can ever exist in late-stage development trials. Some evidence for the new treatment under test always exists before a confirmatory RCT is conducted, including data from in vitro and animal experiments, data from uncontrolled clinical studies, evidence for the same treatment in other diseases, and evidence for similar treatments in the same disease [14–16]. Clinical equipoise may also be challenged by the accumulation of data during an RCT. To maintain clinical equipoise, investigators are usually prevented from looking at the accumulating data during the study, and an independent data monitoring committee may be tasked with stopping or modifying the trial if the accumulating data indicate that this is necessary. This approach is generally effective, with the notable exception of situations where test drugs become identifiable by virtue of having noticeably different side-effect profiles.
A novel response-conditional crossover trial design to ease concerns about lack of equipoise
When clinical equipoise is in question, as is undoubtedly the case with placebo-controlled trials, drug manufacturers and regulatory bodies share an obligation to utilize clinical trial designs that remove investigators’ ethical objections and protect patients while providing the regulators with appropriate evidence to grant approval. Without this imperative, there is a risk of patients being denied access to an effective treatment for a condition where no equivalent treatment option exists.
Response-adaptive clinical trial designs utilize outcome data that accumulate as the trial progresses to assign more patients to the better treatment arm. An alternative novel approach is to use a response-conditional crossover study design. This study design was adopted for a randomized, double-blind, placebo-controlled, pivotal, Phase 3 trial of 10% caprylate-chromatography purified immune globulin intravenous (IGIV-C; Gamunex®, Talecris Biotherapeutics, Research Triangle Park, NC, USA) for the treatment of CIDP [17]. This response-conditional crossover design differed from a typical crossover design and a response-adaptive design in a number of ways, as outlined in Table 1.
At the start of the IGIV-C CIDP efficacy (ICE) study, there was a lack of clinical equipoise. Three of four small, short-term, placebo-controlled studies had suggested that IVIg was efficacious in patients with CIDP [2]. Subsequently, a meta-analysis concluded that IVIg improved disability in patients with CIDP for at least 2–6 weeks compared with placebo, and had similar efficacy to plasma exchange and oral prednisolone [2]. Furthermore, IVIg was being used in several countries to treat patients with CIDP (even though the labelling for IVIg did not include CIDP as an indication), and IVIg was recommended as a first-line treatment option for CIDP in clinical practice guidelines [18–20]. Because of the lack of equipoise, investigators were unwilling to expose patients to long-term placebo treatment. Therefore, a trial had to be designed in which exposure to placebo was minimized.
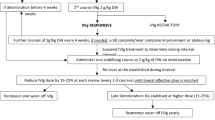
The prospectively designed, randomized, double-blind, placebo-controlled ICE study included an initial treatment period incorporating response-conditional rescue crossover that is the focus of this paper and an extension phase (Fig. 1). After screening, patients were randomised in a 1:1 ratio to receive either IGIV-C or placebo. Patients randomized to IGIV-C received a loading dose of 2 g/kg over 2–4 days followed by a maintenance infusion of 1 g/kg over 1–2 days every 3 weeks for up to 24 weeks. Albumin (0.1%) was used as the placebo.

During this first period, patients either remained on their randomized treatment or switched to the alternative treatment depending on their treatment responses [17]. Nonresponders or “rescued” patients received the alternative treatment for up to 24 weeks and were withdrawn from the study if no improvement was seen after one infusion of the alternative treatment or if improvement was not maintained during the crossover period. Patients who showed maintained improvement and completed 24 weeks of treatment in either the first or crossover periods were eligible for re-randomization in a blinded 24-week extension phase. During the extension phase, patients were again withdrawn from the trial if improvement was not maintained.
This response-conditional crossover trial design enabled the inclusion of a placebo arm to meet regulatory requirements, while minimizing ethical concerns about placebo treatment (the duration of exposure to treatment was short for any therapy that did not provide sustained improvement in the patient’s condition) [17]. Importantly, the design was valid for the primary endpoint: completion of the first period without crossover (IGIV-C responders 54.2% vs placebo responders 20.7%; p = 0.0002). The results from the crossover period provided verification of these findings (IGIV-C 57.8% vs placebo crossover completers 21.7%; p = 0.005). Overall, the study design reduced patient exposure to the inferior treatment in favour of the agent with superior efficacy (see Table 2).
The response-conditional crossover trial design has some limitations. It requires investigators to be vigilant to ensure that crossover is applied correctly to avoid patients remaining on therapy to which they are not responding. In addition, due to differences in duration of drug exposure, the safety data need to be adjusted to provide incidences of adverse events per infusion.
A greater limitation is the reduction in the data that can be collected during the latter stages of the study, due to enhanced patient crossover or drop-out (withdrawal). In the case of the ICE study, secondary efficacy endpoints included changes in grip strength and nerve conductions from baseline to the end of the first period (up to week 24). The results for these secondary endpoints were supportive of the results for the primary endpoint, but as patients could be crossed-over to the alternative therapy at various time points during the first period, the number of patients providing data to week 24 was reduced. Furthermore, the number of patients in the extension phase was reduced because patients who were crossed over to the alternative treatment were discontinued if they failed to improve and maintain improvement based on the adjusted INCAT disability score.
Despite the limitations discussed above, the response-conditional crossover trial design appears to provide a rigorous evaluation of the drug under test. The ICE study results [17] confirmed the efficacy of IGIV-C in treating CIDP observed previously [2] and resulted in successful licensure in the US, Canada, Europe, and elsewhere.
Conclusions
The development of innovative trial designs may help to ease concern about the lack of clinical equipoise in clinical trials while providing the clinical trial data required to support regulatory approvals. In the case of the ICE study, a novel response-conditional crossover design addressed concerns about lack of equipoise raised by physicians interested in trial participation. The design minimized patient exposure to the inferior treatment and proved acceptable to both investigators and regulatory authorities. We conclude that trial sponsors should collaborate with investigators, experts in the field, and regulatory authorities at the protocol design stage to ensure that any proposed study design minimizes ethical concerns regarding lack of equipoise.
References
World Medical Association (2008) World Medical Association Declaration of Helsinki: ethical principles for medical research involving human subjects. World Medical Association. http://www.wma.net/en/30publications/10policies/b3/. Accessed 11 April 2011
Van Schaik IN, Winer JB, De Haan R, Vermeulen M (2002) Intravenous immunoglobulin for chronic inflammatory demyelinating polyradiculoneuropathy. Cochrane Database Syst Rev CD001797
Royall RM, Bartlett RH, Cornell RG, Byar DP, Dupont WD, Levine RJ, Lindley F, Simes RJ, Zelen M (1991) Ethics and statistics in randomized clinical trials. Stat Sci 6:52–88
Freedman B (1987) Equipoise and the ethics of clinical research. N Engl J Med 317:141–145
Mills N, Donovan JL, Smith M, Jacoby A, Neal DE, Hamdy FC (2003) Perceptions of equipoise are crucial to trial participation: a qualitative study of men in the ProtecT study. Control Clin Trials 24:272–282
Gifford F (2007) So-called “clinical equipoise” and the argument from design. J Med Philos 32:135–150
Gifford F (2007) Pulling the plug on clinical equipoise: a critique of Miller and Weijer. Kennedy Inst Ethics J 17:203–226
Joffe S, Miller FG (2008) Bench to bedside: mapping the moral terrain of clinical research. Hastings Cent Rep 38:30–42
Miller FG, Brody H (2003) A critique of clinical equipoise. Therapeutic misconception in the ethics of clinical trials. Hastings Cent Rep 33:19–28
Miller FG, Brody H (2007) Clinical equipoise and the incoherence of research ethics. J Med Philos 32:151–165
Brody H, Miller FG (2003) The clinician-investigator: unavoidable but manageable tension. Kennedy Inst Ethics J 13:329–346
Miller FG, Joffe S (2011) Equipoise and the dilemma of randomized clinical trials. N Engl J Med 364:476–480
Lemmens T, Miller PB (2002) Avoiding a Jekyll-and-Hyde approach to the ethics of clinical research and practice. Am J Bioeth 2:14–17
Avins AL (1998) Can unequal be more fair? Ethics, subject allocation, and randomised clinical trials. J Med Ethics 24:401–408
Lilford RJ (2003) Ethics of clinical trials from a bayesian and decision analytic perspective: whose equipoise is it anyway? BMJ 326:980–981
Fries JF, Krishnan E (2004) Equipoise, design bias, and randomized controlled trials: the elusive ethics of new drug development. Arthritis Res Ther 6:R250–R255
Hughes RA, Donofrio P, Bril V, Dalakas MC, Deng C, Hanna K, Hartung HP, Latov N, Merkies IS, van Doorn PA ICE, Group Study (2008) Intravenous immune globulin (10% caprylate-chromatography purified) for the treatment of chronic inflammatory demyelinating polyradiculoneuropathy (ICE study): a randomised placebo-controlled trial. Lancet Neurol 7:136–144
Berger AR, Bradley WG, Brannagan TH, Neuropathy Association, Medical Advisory Committee et al (2003) Guidelines for the diagnosis and treatment of chronic inflammatory demyelinating polyneuropathy. J Peripher Nerv Syst 8:282–284
Gorson KC, Ropper AH (2003) Chronic inflammatory demyelinating polyradiculoneuropathy (CIDP): a review of clinical syndromes and treatment approaches in clinical practice. J Clin Neuromuscul Dis 4:174–189
Joint task force of the European Federation of Neurological Societies and the Peripheral Nerve Society (2005) European Federation of Neurological Societies/Peripheral Nerve Society guideline on management of chronic inflammatory demyelinating polyradiculoneuropathy. Report of a joint task force of the European Federation of Neurological Societies and the Peripheral Nerve Society. J Peripher Nerv Syst 10:220–228
Acknowledgments
The ICE study was funded by Talecris Biotherapeutics Center for Science and Education. Technical editorial assistance was provided under the direction of the authors by Susan Robinson, PhD, and Nick Rusbridge, PhD, of PAREXEL and was funded by Talecris Biotherapeutics, Inc.
Conflicts of interest
CD and KH are fulltime employees of Grifols Inc. (formerly Talecris Biotherapeutics) and hold stock and/or stock options in this company. VB, MCD, PD, and H-PH received honoraria for participation on the ICE Study Steering Committee. VB previously received consultancy fees and unrestricted educational grants for studies in myasthenia gravis (paid to her institution) from Talecris Biotherapeutics. MCD has received a grant, consultancy fees and support for travel to meetings for study purposes from Talecris Biotherapeutics, Baxter and Octapharma. He also previously received payment for lectures from these same companies. PD has received a grant (paid to his institution), consultancy fees, fees for participation in review activities, and support for writing, medicines, equipment, and administration from Talecris Biotherapeutics. He previously received consultancy fees from CSL Behring, Octapharma and Squid, along with payment for lectures and for the development of educational presentations from DIME. PAvD and his institution were paid honoraria from Talecris Biotherapeutics for his membership of the ICE Study Steering Committee. H-PH received a research grant from Baxter (paid to his institution), consultancy fees from Talecris Biotherapeutics, BayerHealthcare, and CSL Behring, and travel support from Talecris Biotherapeutics and CSL Behring. He previously received consultancy fees from BayerHealthcare, BiogenIdec, Genzyme, MerckSerono, Novartis, and Teva Sonofi Aventis. H-PH has also received grants from BayerHealthcare, BiogenIdec, MerckSerono and Teva (paid to his institution), as well as payments for lectures from these same companies. ISJM previously received payments for being a Steering Committee member of the ICE trial (Talecris Biotherapeutics) and a CIDP study (CSL Behring) along with consultancy fees, paid to his institution, for the latter study (CSL Behring). ISJM previously received grants for the GBS/CIDP International foundation for the PeriNomS study, Talents program for the PeriNomS study, plus the Peripheral Nerve Society for the PeriNomS study. ISJM’s institution has also received payments from Talecris Biotherapeutics for presentations given by ISJM during 2009-2010.
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License (https://creativecommons.org/licenses/by-nc/2.0), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
Deng, C., Hanna, K., Bril, V. et al. Challenges of clinical trial design when there is lack of clinical equipoise: use of a response-conditional crossover design. J Neurol 259, 348–352 (2012). https://doi.org/10.1007/s00415-011-6200-0
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00415-011-6200-0