
Abstract
Laser microdissection is a valuable tool for isolating specific cells from mixtures, such as male cells in a mixture with female cells, e.g., in cases of sexual assault. These cells can be stained with Y-chromosome-specific probes. We developed an automatic screening method to detect male cells after fluorescence in situ hybridization in suspension (S-FISH). To simulate forensic casework, the method was tested on female saliva after cataglottis (a kiss involving tongue-to-tongue contact) and on licking traces (swabs of dried male saliva on female skin) even after drying. After isolation of the detected cells, short tandem repeat profiling was performed. Full DNA profiles could consistently be obtained from as little as ten buccal cells. Isolation of five cells resulted in a mean of 98% (SD of 3.4%) of the alleles detected, showing that the developed S-FISH staining had no significant negative influence on DNA recovery and can be used in forensic casework.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Laser capture microdissection (LCM) is a reliable method to isolate pure populations of specific cell types. It has been used for a variety of forensic purposes, such as isolation of spermatozoa in cases of sexual assault [1–4] isolation of human chorionic villi for parentage testing [5], collection of cells from hair follicles [6], and isolation of cells after sex-specific labeling [7–9]. The latter can be of interest for mixtures of male and female cells, e.g., in cases of sexual assault where the offender is azoospermic, in traces from non-genital acts, e.g., kissing, licking, suction injury, and biting, or to detect male cells under the fingernails of the female victim.
Alternatively, Y-chromosome short tandem repeats (Y-STR) can be used to detect the male component in these mixed samples when the DNA of the male contributor is present in a small amount [10]. However, the discriminatory power of Y-STR analysis is much lower compared to autosomal STR analysis.
Fluorescence in situ hybridization (FISH), using a Y-chromosome-specific probe, is used to detect male cells [7–9, 11–15]. Traditionally, FISH is performed after fixation of the cells on a microscope slide. Alternatively, FISH can be performed in suspension (S-FISH) [16, 17] and the cell suspension can then be cytospun on a microscope slide. We observed several advantages of this S-FISH approach. The first and most important advantage is that the cells are less tethered to the microscope slide in comparison to traditional FISH protocols because only one short ethanol fixation step is performed instead of the ethanol series that are traditionally used. As a consequence, the cells detach easier upon laser capture microdissection, resulting in a higher efficiency and the need for fewer cells to be isolated to obtain a full DNA profile. A second advantage is that any type of microscope slide can be used; there is no need for membrane slides. In addition, in contrast with the traditional FISH protocols, where the procedure is not easy to handle [7], this S-FISH procedure requires minimal equipment, training, and handling and the hands-on time is limited to less than 1 h.
The PALM MicroBeam laser microdissection system (P.A.L.M. Microlaser Technologies, Munich, Germany) allows scanning of microscope slides [18]. The system can be supplemented with image analyzing software modules in which the user can write scripts allowing for automated specimen identification and image processing [19], e.g., the AxioVision Commander (Carl Zeiss, Hallbergmoos, Germany). The selected cells can be collected by a defined laser pulse using the laser pressure catapulting (LPC) function. This contact-free cell collection helps avoiding sample contamination in forensic cases.
In the current report, we have developed an S-FISH protocol and an AxioVision Commander script for the automatic detection of male buccal cells in mixtures of male and female cells. To simulate forensic casework, the detection method was tested on a cataglottis sample (i.e., an open mouth kiss involving tongue-to-tongue contact) and on various licking trace samples (i.e., swabs of the female skin with dried male saliva on). As swabs from sexual assault cases are often dried upon arrival at the forensic laboratory, the influence of drying of the cells on the cotton swab was also tested. After LPC of the detected male cells, the ability of obtaining pure DNA profiles was assessed.
Materials and methods
Sample preparation
Fresh saliva was collected from healthy male and female volunteers. Five hundred microliters of phosphate-buffered saline (PBS, Gibco, Paisly, UK) was added to 1 ml of each saliva sample. The cell suspension was then centrifuged at 800×g for 7 min and the supernatant was discarded. The cell pellet was washed once with 1 ml of PBS. After cell counting, mixtures were prepared by combining male and female buccal cells in 1:10 and 1:50 ratios.
A cell pellet from the saliva of a female volunteer after cataglottis with a male volunteer was prepared in the same way. Additionally, the skin of a female volunteer was swabbed with wet sterile cotton swabs 10 min, 1 h, and 8 h, respectively, after it was licked by a male volunteer. The swabs taken after 10 min and 8 h were immediately agitated in 1 ml of PBS to release the cellular material. The swabs taken after 1 h were dried for 24, 48, and 72 h, respectively, before they were agitated in 1 ml of PBS. From all samples, a cell pellet was prepared as described above.
Fluorescence in situ hybridization
The cell pellets were resuspended in 500 µl of hybridization mixture containing 70% deionized formamide (Sigma Aldrich, St. Louis, MO, USA), 10 mM Tris (MP Biomedicals, Solon, OH, USA) pH 7.2, and 0.3 µg/ml Y-chromosome-specific FITC-conjugated peptide nucleic acid probe (Dako, Glostrup, Denmark). DNA denaturation was performed at 82°C for 10 min. Afterwards, the probe was hybridized overnight at room temperature in the dark. To remove excess and non-specifically bound probe, the cell suspension was centrifuged at 800×g for 7 min and incubated twice at room temperature for 10 min in 500 μl of buffer A containing 70% formamide, 0.1% Tween 20 (MP Biomedicals), and 10 mM Tris pH 7.2. The cell pellet was then resuspended in 1 ml of buffer B containing 0.15 M NaCl (MP Biomedicals), 0.1% Tween 20, and 50 mM Tris pH 7.5 and was then centrifuged at 800×g for 7 min. After the last washing step, the cell pellets were resuspended in 1 ml of PBS and counted. From each sample, 50,000 cells were cytospun on a poly-l-lysine (Sigma Aldrich)-coated 0.17 mm thin glass slide (Chem-Lab, Zedelgem, Belgium) by centrifugation (5 min at 400×g, Rotofix 32A, Hettich, Tuttlingen, Germany). The slides were air-dried for 10 min and fixed for 1 min in a 70% ethanol solution (absolute ethanol, Merck BV, Schiphol-Rijk, The Netherlands) in pure water (MilliQ, Millipore, Billerica, MA, USA). After fixing, the slides were mounted, using 15 µl of Vectashield Mounting Medium with 4′,6-diamidino-2-phenylindole (Vector Labs, Burlingame, CA, USA) and a coverslip (Roth, Karlsruhe, Germany) was applied.
Fluorescence scanning
The scanning stage was controlled by the AxioVision 4.6.3 software (Carl Zeiss), using the MosaiX module. Image acquisition was carried out with the AxioVision multichannel fluorescence module and the AxioCam MRm camera.
The cell nuclei, including male and female nuclei, were visualized using Zeiss filter set no. 49 (G 365 nm, FT 495, BP 445/50). The S-FISH signals were visualized using Zeiss filter set no. 38 (BP 470/40, FT 495, BP 525/50). The slides were scanned at ×20 magnification using a Carl Zeiss short distance Plan-Apochromat® objective. From every slide, 100 images were acquired using the scanning mode. The individual images were stored as tiff files.
Segmentation and masking
For automated detection of the S-FISH signal, the image processing AxioVision Commander module was used. All processing, analysis, and evaluation steps were stored in an AxioVision Commander script, which could be run automatically on the stored images.
As shown in Fig. 1, the AxioVision Commander script was built up in five steps: In the first step, the command “Interactive Contrast” was used to achieve a better view of the image. On the resulting image, adaptive gray value segmentation was performed using the command “Dynamic”. This procedure is particularly suitable for the segmentation of small structures against a varying background. The function initially applies a low-pass filter and then subtracts this low-pass-filtered image from the input image. The resulting image is a binary image. Pixels within the calculated gray level range are set to the maximum gray value 1, while pixels outside it are set to the gray value 0. Thirdly, the command “Scrap” deleted regions smaller than 4 pixels to remove artefacts not originating from an S-FISH signal. In the fourth step, the command “Close” was used. This command performs dilation followed by erosion of the regions. With closing, the original size of the regions is essentially retained, but connections are created between regions that are close together, and gaps in the contours of regions are filled in. In the last step, to be included as a true S-FISH signal, the region had to fulfil four measurement parameter conditions. These parameters were as follows: the area of the region (0.5 to 6.85 µm²), lowest (minimum 77 gray values) and highest (minimum 150 gray values) gray value of the region, and standard deviation of gray values of the region (minimum 10 gray values). The coordinates of the detected regions were stored in a data table which could be used for relocation in the PALM RoboSoftware version 4 (P.A.L.M. Microlaser Technologies). After visual verification of the detected male buccal cells, a catapulting point was set on the detected cells. As the mounting medium and the coverslip need to be removed before LPC, the fluorescent signal is lost. For this reason, the surrounding female cells and debris were outlined to make relocation of the male buccal cells possible under brightfield illumination, as shown in Fig. 2.

Principle of the AxioVision Commander script. a Pseudo-colored image of the cell mixture, showing one male (top) and one female cell (bottom). b Pseudo-colored image after the command “Interactive Contrast”. c Binary image after adaptive gray value segmentation by the command “Dynamic”. d Binary image after removal of small artefacts by the command “Scrap”. e Binary image after dilation and erosion of the detected regions by the command “Close”. f Pseudo-colored image of the buccal cells, the detected S-FISH signal is outlined in yellow

Laser pressure catapulting of detected male buccal cells. a Pseudo-colored image of the cell mixture: a blue catapulting dot is set on the male buccal cells, while the female cells and the debris are outlined in red. b Brightfield image after removal of the mounting medium and coverslip: male and female cells can easily be distinguished by the blue catapulting dots and the red outlining. c Brightfield image after LPC of the central male buccal cell
Laser pressure catapulting
The detected male cells were collected by LPC using a pulsed nitrogen UV-A laser (wavelength 355 nm). The high energy generated by the focused laser light was used to catapult the male cells into the cap of a standard 0.2-ml microfuge tube (Westburg, Leusden, The Netherlands) containing 20 µl of PicoPure DNA extraction buffer (PicoPure DNA extraction kit, Arcturus, Mountain View, CA, USA). This contact-free method helps avoiding contamination of the sample.
DNA extraction
DNA was extracted from the catapulted cells, using the PicoPure DNA extraction kit (Arcturus). The samples were incubated at 65°C for 3 h, centrifuged briefly, and heated to 95°C for 10 min to inactivate proteinase K. If the polymerase chain reaction (PCR) could not be performed immediately, the samples were stored at −20°C.
Amplification and detection
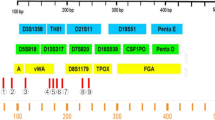
For the amplification of the extracted DNA, the AmpFℓSTR® Profiler Plus® kit from Applied Biosystems (Foster City, CA, USA) was used according to the manufacturer's instructions, except that Hotstar Taq DNA polymerase (Qiagen, Germantown, MD, USA) was used instead of Taq Gold. The total number of cycles was 33. After PCR, the amplified fragments were separated and analyzed by capillary electrophoresis using an ABI 3100 Genetic Analyzer (Applied Biosystems).
Results
Automatic detection of male cells
The 1:10 and the 1:50 mixtures were used to determine whether the developed AxioVision Commander script could be used to automatically detect male buccal cells in a mixture of male and female saliva. From each mixture, 100 images were acquired as shown in Fig. 3. The total amount of nuclei on the scanned images (male and female nuclei) was counted manually. For the 1:10 mixture, 1,757 nuclei were manually counted and 189 male nuclei were automatically detected by the AxioVision Commander script, which means the detected ratio was 1:9.3. For the 1:50 mixture, 1,736 nuclei were manually counted and 40 male nuclei were automatically detected, which means the detected ratio was 1:43.4. The good correlation between the ratio used to make the cytospins and the detected ratio shows the reliability of the developed AxioVision Commander script.

Automatic scanning of the microscope slide and result of the AxioVision Commander script. a Overview image of 100 images acquired from a control slide (1:10 mixture of male and female saliva). b Zoomed in image (×100 zoom)
The method was also evaluated on membrane slides (0.17 mm PET-membrane-covered slides, P.A.L.M. Microlaser Technologies), but this gave rise to difficulties in automated processing of the images due to autofluorescence of the membrane and a non-smooth background structure (data not shown).
Profile recovery after laser pressure catapulting
To simulate forensic casework, the detection and isolation method was tested on female saliva after cataglottis and on various cotton swabs of the female skin taken 10 min, 1 h, and 8 h, respectively, after licking by a male volunteer. Different amounts of male buccal cells were collected. The results are presented in Tables 1, 2, and 3.
From the cataglottis sample, a full male DNA profile was recovered in five of the six cases, while in one case an allelic drop out was seen. In three cases, some contaminating female alleles were detected, but in all cases, the male DNA profile was more intense and the male DNA profile was interpreted easily. An example of a DNA profile obtained from five male buccal cells from the cataglottis sample is shown in Fig. 4. The alleles D18S51, D13S317, and D7S820 show some peak imbalance, probably due to the low amount of input DNA.

DNA profile recovery after LPC. Pure male DNA profile derived after catapulting five male buccal cells from the cataglottis sample
In the licking trace samples, where the female skin was swabbed 10 min and 8 h, respectively, after it was licked by a male and the swabs were immediately agitated in PBS to release the cellular material, a full male DNA profile was recovered in 11 of the 13 cases, as shown in Table 2. In the other two cases, two and one allelic drop outs occurred, respectively. No contaminating female alleles were seen in these samples.
Table 3 shows the results of the licking trace samples, where the female skin was swabbed 1 h after it was licked by a male and the swabs were dried for 24, 48, and 72 h, respectively. Full male DNA profiles could be recovered in nine of the 15 cases. Again, no contaminating female alleles were seen. In all samples where at least ten male cells were isolated, a full male DNA profile was found.
Discussion
The results demonstrate that the developed AxioVision Commander script can identify male buccal cells, present in a vast amount of female cells. The S-FISH signals are easily detectable and the background staining is negligible. The slides can be scanned at low magnification, which greatly increases the speed with which male cells can be localized. The coordinates of the single cells assessed as male cells are transferred to an element list of the PALM RoboSoftware. Before removing the mounting medium and coverslip, the detected cells can be verified easily through fluorescence microscopy. A catapulting dot is then set on the male cell, while the surrounding female cells and debris are outlined in another color for ease of relocation using brightfield microscopy.
We demonstrated that the S-FISH protocol has no major adverse effects on the recovery of DNA from buccal cells. In comparison with traditional FISH protocols, where full DNA profiles could be obtained from samples containing at least 30 [8] or 40 [9] male cells, isolation of ten male cells was enough to consistently generate a full male DNA profile using our S-FISH protocol. Isolation of five male buccal cells resulted in a mean of 98% (3.4% standard deviation) of the alleles detected. No differences were seen between fresh licking traces and licking traces that were dried for a longer period of time on the skin or on the sterile cotton swabs. We therefore conclude that the procedure described in this manuscript can also be used on dried samples without significant effects on the results obtained.
Despite the presence of contaminating female alleles in the cataglottis sample, the male DNA profile was interpreted easily. No contaminating female alleles were seen in the licking trace samples. This difference can possibly be attributed to the more abundant presence of DNA containing cells in the female saliva than on the female skin.
It can be concluded that, used in combination with the automatic detection of the S-FISH signal, the LCM method is a fast and sensitive procedure for DNA profiling of the male contributor in forensic samples containing an unfavorable mixture of male and female cells.
References
Elliott K, Hill D, Lambert C, Burroughes T, Gill P (2003) Use of laser microdissection greatly improves the recovery of DNA from sperm on microscope slides. Forensic Sci Int 137:28–36
Di Martino D, Giuffre G, Staiti N, Simone A, Le Donne M, Saravo L (2004) Single sperm cell isolation by laser microdissection. Forensic Sci Int 146:151–153
Sanders C, Sanchez N, Ballantyne J, Peterson D (2006) Laser microdissection separation of pure spermatozoa from epithelial cells for short tandem repeat analysis. J Forensic Sci 51:748–757
Vandewoestyne M, Van Hoofstat D, Van Nieuwerburgh F, Deforce D (2009) Automatic detection of spermatozoa for laser capture microdissection. Int J Legal Med 123:169–175
Budimlija Z, Lechpammer M, Popiolek D, Fogt F, Prinz M, Bieber F (2005) Forensic applications of laser capture microdissection: use in DNA-based parentage testing and platform validation. Croat Med J 46:549–555
Di Martino D, Giuffre G, Staiti N, Simone A, Todaro P, Saravo L (2004) Laser microdissection and DNA typing of cells from single hair follicles. Forensic Sci Int 146:155–157
Anslinger K, Mack B, Bayer B, Rolf B, Eisenmenger W (2005) Digoxigenin labelling and laser capture microdissection of male cells. Int J Legal Med 119:374–377
Anslinger K, Bayer B, Mack B, Eisenmenger W (2007) Sex-specific fluorescent labelling of cells for laser microdissection and DNA profiling. Int J Legal Med 121:54–56
Murray C, McAlister C, Elliott K (2007) Identification and isolation of male cells using fluorescence in situ hybridisation and laser microdissection, for use in the investigation of sexual assault. Forensic Sci Int Genetics 1:247–252
Cerri N, Ricci U, Sani I, Verzeletti A, De Ferrari F (2003) Mixed stains from sexual assault cases: autosomal or Y-chromosome short tandem repeats? Croat Med J 44:289–292
Dziegelewski M, Simich J, Rittenhouse-Olson K (2002) Use of a Y chromosome probe as an aid in the forensic proof of sexual assault. J Forensic Sci 47:601–604
Collins K, Rao P, Hayworth R, Schnell S, Tap M, Lantz P, Geisinger K, Pettenati M (1994) Identification of sperm and non-sperm male cells in cervicovaginal smears using fluorescence in situ hybridization: applications in alleged sexual assault cases. J Forensic Sci 39:1347–1355
Cina M, Collins K, Fitts M, Pettenati M (2000) Isolation and identification of male and female DNA on a postcoital condom. Arch Pathol Lab Med 124:1083–1086
Pettenati M, Rao P, Schnell S, Hayworth-Hodge R, Lantz P, Geisinger K (1995) Gender identification of dried human bloodstains using fluorescence in situ hybridization. J Forensic Sci 40:885–887
Prahlow J, Lantz P, Cox-Jones K, Rao P, Pettenati M (1996) Gender identification of human hair using fluorescence in situ hybridization. J Forensic Sci 41:1035–1037
Baerlocher G, Mak J, Tien T, Lansdorp P (2002) Telomere length measurement by fluorescence in situ hybridization and flow cytometry: tips and pitfalls. Cytometry 47:89–99
Steinhaeuser U, Starke H, Nietzel A, Lindenau J, Ullmann P, Claussen U, Liehr T (2002) Suspension (S)-FISH, a new technique for interphase nuclei. J Histochem Cytochem 50:1697–1698
Seidl S (2005) Contact-free isolation of sperm and epithelial cells by laser microdissection and pressure catapulting. Forensic Sci Med Pathol 1:153–158
Niyaz Y, Sägmüller B (2005) Non-contact laser microdissection and pressure catapulting: automation via object-oriented image processing. Med Laser Appl 20:223–232
Acknowledgments
The research was funded by a Ph.D. grant from the Institute for the Promotion of Innovation through Science and Technology in Flanders (IWT-Vlaanderen) to Mado Vandewoestyne. The authors are grateful to Wouter Christiaens (Zeiss, Belgium) and Renate Burgemeister and colleagues (P.A.L.M. Microlaser, Germany) for their help in the development of the AxioVision Commander script.
Disclaimer
The authors declare that they have no competing financial, personal or professional interests.
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License (https://creativecommons.org/licenses/by-nc/2.0), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
Vandewoestyne, M., Van Hoofstat, D., Van Nieuwerburgh, F. et al. Suspension fluorescence in situ hybridization (S-FISH) combined with automatic detection and laser microdissection for STR profiling of male cells in male/female mixtures. Int J Legal Med 123, 441–447 (2009). https://doi.org/10.1007/s00414-009-0341-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00414-009-0341-z