
Abstract
A total of 17 Y-specific STR loci were studied in 12 districts of the European part of Russia aiming to ascertain the amount of substructure required for the construction of a representative regional database. All groups exhibited high haplotype diversities but low inter-population variance as measured by an analysis of molecular variance. However, when Western Russia is taken as a whole, the genetic distances to the neighbouring populations were significant. Whereas gradual change in the Y chromosome pool exists between Russia and the Slavic-speaking populations to the West, remarkable discontinuities were observed with neighbouring populations in the East, North and South.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Y chromosome short tandem repeat (STR) haplotypes show a substantial inter-population differentiation both on a worldwide and continental scale [1, 2]. Because Y-STR haplotype frequencies are required to provide statistical estimates of the significance of a match between forensic samples, local databases must be developed taking the demographic history of the investigated population into account. Large metadatabases as the Y Chromosome Haplotype Reference Database (YHRD) pool different local databases based on intrinsic (genetic) and contextual (geographic, linguistic) information [3]. However, assembling different regional pools is only valid if there is no population substructure, i.e. no statistically significant difference between the Y-STR haplotype distributions in different regions [4]. Here, we present haplotype analysis and genetic differentiation tests of a large Russian population consisting of 12 subgroups leading to a sensible decision on the assignment of the analysed population to a framework of worldwide metapopulations.
Population
A total of 545 unrelated males from 12 Western Russian populations previously typed for Y-chromosome single nucleotide polymorphism markers [5] were analysed for 17 Y-STR loci evaluated in forensic routine diagnostics [6]. The sampling was carried out in the administrative centers of the following districts (oblasts): Smolenskaja (Smo, n = 43), Brianskaja (Bri, 43), Ivanovskaja (Iva, 40), Lipezkaja (Lip, 47), Penzenskaja (Pen, 81), Ryazanskaja (Rya, 36), Orlovskaja (Orl, 42), Tverskaja (Tve, 43), Vologodskaja (Vol, 40), Tambovskaja (Tam, 48), Archangelskaja (Arch, 42) and Nowgorodskaja (Now, 40; see Fig. 1). Informed consent and information about the birthplace of the donor and his parents and grandparents were obtained.

Map of Europe depicting the analysed populations from the European part of Russia and neighbouring territories
Materials and methods
Deoxyribonucleic acid (DNA) was extracted from whole blood samples using the QIAmp DNA Blood Mini Kit (Qiagen, Hilden, Germany) following the manufacturer’s recommendations. All samples were genotyped for 17 Y chromosomal STR loci (DYS19, DYS389I, DYS389II, DYS390, DYS391, DYS392, DYS393, DYS385a, DYS385b, DYS437, DYS438, DYS439, DYS448, DYS456, DYS458, DYS635, GATA H4) using the AmpFlSTR®YFiler® PCR Amplification kit (Applied Biosystems, Darmstadt, Germany) according to the manufacturer’s instructions. STR products were analysed on an ABI Prism 3100 AVANT automated sequencer with GeneScan and GenoTyper v. 3.7 (Applied Biosystems). The updated recommendations of the DNA Commission of the International Society of Forensic Genetics for analysis of Y-STR systems were followed [4].
Quality control
Proficiency testing of the German DNA Profiling group (www.gednap.de) and the YHRD (www.yhrd.org) trials was carried out.
Analysis of data
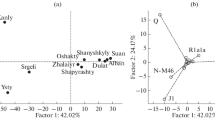
Characteristic parameters for each population, consisting of the number of different haplotypes, the discrimination capacity (D) and the haplotype diversity (h), were calculated (Table 1). Pairwise values of Φ st , an analogue of Wrights Fst that takes the evolutionary distance between individual haplotypes into account [7, 8], were calculated to measure genetic distances between 17 locus haplotypes of 12 Western Russian populations. Subsequently, these populations were treated as one metapopulation and the minimal haplotypes (which includes the nine loci: DYS19, DYS389I, DYS389II, DYS390, DYS391, DYS392, DYS393, DYS385ab) were compared to published data from nine neighbouring regions, namely 271 minimal haplotypes from Belarus [9], 502 from the Caucasus [10], 133 from Estonia, 145 from Latvia and 157 from Lithuania [11], 399 samples from Finland [12], 3,021 from Poland [13–15], 370 from Siberia [16, 17] and 368 from Ukraine [18], with the statistical significance determined by a permutation test (10,000 replicates; Table 2). We used our own implementation of analysis of molecular variance (AMOVA; available at http://rprojekt.org/amova/). The DYS389I allele length was obtained by subtracting the shorter allele from the longer allele at DYS389I/II. The statistics and analytics software package (STATISTICA package; StatSoft Inc.) was used for multi-dimensional scaling (MDS) analysis [19] based on pairwise Φ st values (Fig. 2).

Multi-dimensional scaling (MDS) plot of the Western Russian metapopulation and nine reference populations, from pairwise Φ st values. Stress value = 0.002. Acronyms are as follows: Rus–Russians, Bel–Belarus, Ukr–Ukrainians, Lat–Latvians, Lit–Lithuanians, Est–Estonians, Cau-Caucasus, Pol–Polish, Fin-Finns, Sib–Siberia
Results and discussion
A total of 545 samples from 12 Western Russian populations were investigated in this study and 494 different 17 locus haplotypes (D = 0.906, h = 0.9994) were detected. Two haplotypes occurred six times, one occurred five times, two occurred four times, three occurred three times, 25 twice and 461 were unique (Supplementary Table S1). The eight most frequent haplotypes occurring 34 times in this sample are closely related and belong (with the exception of one haplotype belonging to hg N3-Tat) to haplogroup R1a1-M17 (printed in bold in Table S1). This typical Eastern European haplogroup is the most frequent in all analysed populations with frequencies ranging between 0.31 and 0.56, followed by hg I-M170 (0.09–0.31) and N3-Tat (0.06–0.29) [5]. We observed high haplotype diversities (h) in all 12 populations ranging from 0.993 to 1.000 (Table 1). Three duplications (12 and 13 at locus GATA H4, 15 and 16 at DYS19 and 20 and 21 at DYS448), one “null” allele at DYS19 and several intermediate-sized alleles were observed.
No genetic substructure was found among the Russian populations. All pairwise comparisons were non-significant (p > 0.05). In contrast, significant variation between populations was observed in the comparison of Western Russia, treated as one homogeneous metapopulation, with neighbouring groups (Table 2). Because only reduced haplotype formats were available for such reference populations, we performed AMOVA based on 545 minimal 9-locus haplotypes from Western Russia with the previously published 5,366 haplotypes from 11 neighbouring regions (in clockwise direction: Ukraine, Belarus, Lithuania, Latvia, Estonia, Finland, Siberia and the Caucasus region) [9–18]. All pairwise Φ st comparisons between Russia and these neighbours (with the exception of Russia vs. Belarus) were significant with values ranging between 0.0089 (Russia vs. Poland) and 0.3688 (Russia vs. eight Altaic- and Uralic-speaking groups from Siberia). MDS plot based on pairwise Φ st values shows a closely related core group consisting of Slavic-speaking populations (Russia, Ukraine, Belarus and Poland) with an elevated distance to Baltic populations and a large span to linguistically different groups from Estonia, Finland, Siberia and the Caucasus (Fig. 2). From these analyses, we conclude that autochthonous Russian-speaking populations residing for centuries in the European part of Russia can be pooled to form a representative regional reference database for assessment of Y chromosomal matches in forensic analyses. However, on a level of quite low but significant Φ st values, Western Russian populations can be grouped together with a much larger metapopulation defined within the forensic YHRD. This genetically distinct “Eastern European” metapopulation (n = 5,993, YHRD release 22 from 2007-08-10) comprises 56 Balto-Slavic-speaking populations from Eastern Europe [2]. Because the population frequency of a given haplotype is positively correlated with the combined frequency of closely related (surrounding) haplotypes in the population of origin [20], forensic databases collecting haploid markers have to be tested for substructure. This and other studies demonstrate that AMOVA is a sensitive method to detect the extent of inter-population differences accrued from the genetic and demographic history of the populations. The assignment of each population sample to a set of populations sharing a common linguistic, demographic, geographic and genetic background (metapopulations) facilitates the statistical evaluation of haplotype matches due to a significant enlargement of sample sizes.
The data obtained in this study from Russian populations were submitted to the Y Chromosome Haplotype Reference Database (www.yhrd.org) and they were assigned to the Eurasian–European–Eastern European metapopulation [3].
References
Kayser M, Krawczak M, Excoffier L et al (2001) An extensive analysis of Y-chromosomal microsatellite haplotypes in globally dispersed human populations. Am J Hum Genet 68(4):990–1018
Roewer L, Croucher PJ, Willuweit S et al (2005) Signature of recent historical events in the European Y-chromosomal STR haplotype distribution. Hum Genet 116(4):279–291
Willuweit S, Roewer L (2007) Y Chromosome Haplotype Reference Database (YHRD): update. Forensic Sci Int Genetics 1(2):83–87
Gusmao L, Butler JM, Carracedo A et al (2006) DNA Commission of the International Society of Forensic Genetics. Commission of the International Society of Forensic Genetics (ISFG): an update of the recommendations on the use of Y-STRs in forensic analysis. Int J Legal Med 120(4):191–200
Fechner A, Quinque D, Rychkov S et al (2007) Boundaries and clines in the West Eurasian Y-chromosome landscape: insights from the European part of Russia. Am J Phys Anthropol (in press)
Mulero JJ, Chang CW, Calandro LM, Green RL, Li Y, Johnson CL, Hennessy LK (2006) Development and validation of the AmpFlSTR Yfiler PCR amplification kit: a male specific, single amplification 17 Y-STR multiplex system. J Forensic Sci 51:64–75
Excoffier L, Smouse PE, Quattro JM (1992) Analysis of molecular variance inferred from metric distances among DNA haplotypes: Application to human mitochondrial DNA restriction data. Genetics 131:479–491
Roewer L, Kayser M, Dieltjes P, Nagy M, Bakker E, Krawczak M, de Knijff P (1996) Analysis of molecular variance (AMOVA) of Y-chromosome-specific microsatellites in two closely related human populations. Hum Mol Genet 5(7):1029–1033
Rebala K, Mikulich AI, Tsybovsky IS, Sivakova D, Dzupinkova Z, Szczerkowska- Dobosz A, Szczerkowska Z (2007) Y-STR variation among Slavs: evidence for the Slavic homeland in the middle Dnieper basin. J Hum Genet 52(5):406–414
Nasidze I, Schädlich H, Stoneking M (2003) Haplotypes from the Caucasus, Turkey and Iran for nine Y-STR loci. Forensic Sci Int 137:85–93
Lessig R, Edelmann J, Krawczak M (2001) Population genetics of Y-chromosomal microsatellites in Baltic males. Forensic Sci Int 118:153–157
Hedman M, Pimenoff V, Lukka M, Sistonen P, Sajantila A (2004) Analysis of 16 Y STR loci in the Finnish population reveals a local reduction in the diversity of male lineages. Forensic Sci Int 142(1):37–43
Ploski R, Wozniak M, Pawlowski R et al (2002) Homogeneity and distinctiveness of Polish paternal lineages revealed by Y chromosome microsatellite haplotype analysis. Hum Genet 110:592–600
Pawlowski R, Dettlaff-Kakol A (2003) Population data of nine Y-chromosomal STR loci in northern Poland. Forensic Sci Int 131(2–3):209–213
Pepinski W, Niemcunowicz-Janica A, Ptaszynska-Sarosiek I, Skawronska M, Koc-Zorawska E, Janica J, Soltyszewski I (2004) Population genetics of Y-chromosome STRs in a population of Podlasie, northeastern Poland. Forensic Sci Int 144(1):77–82
Pakendorf B, Novgorodov IN, Osakovskij VL, Danilova AP, Protodjakonov AP, Stoneking M (2006) Investigating the effects of prehistoric migrations in Siberia: genetic variation and the origins of Yakuts. Hum Genet 120(3):334–353
Pakendorf B, Novgorodov IN, Osakovskij VL, Stoneking M (2007) Mating patterns amongst Siberian reindeer herders: inferences from mtDNA and Y-chromosomal analyses. Am J Phys Anthropol 133:1013–1027
Kravchenko SA, Slominskii PA, Bets LA, Stepanova AV, Mikulich AI, Limborskaya SA, Livshits LA (2002) Polymorphism of STR Loci of the Y chromosome in three populations of Eastern Slavs from Belarus, Russia and Ukraine. Rus J Genet 38(1):80–86
Kruskal JB (1964) Multidimensional scaling by optimizing goodness of fit to a nonmetric hypothesis. Psychometrika 29:1–27
Roewer L, Kayser M, de Knijff P et al (2000) A new method for the evaluation of matches in non-recombining genomes: application to Y-chromosomal short tandem repeat (STR) haplotypes in European males. For Sci Int 114(1):31–43
Acknowledgements
We thank all donors for making this work possible.
This research was, in part, supported by the Max-Planck Society, Germany.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License ( https://creativecommons.org/licenses/by-nc/2.0 ), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
Roewer, L., Willuweit, S., Krüger, C. et al. Analysis of Y chromosome STR haplotypes in the European part of Russia reveals high diversities but non-significant genetic distances between populations. Int J Legal Med 122, 219–223 (2008). https://doi.org/10.1007/s00414-007-0222-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00414-007-0222-2