
Abstract
Purpose
Idiopathic pulmonary fibrosis (IPF) is a progressive and lethal lung disease of unknown origin whose incidence has been increasing over the latest decade partly as a consequence of population ageing. New anti-fibrotic therapy including pirfenidone and nintedanib have now proven efficacy in slowing down the disease. Nevertheless, diagnosis and follow-up of IPF remain challenging.
Methods
This review examines the recent literature on potentially useful blood molecular and cellular biomarkers in IPF. Most of the proposed biomarkers belong to chemokines (IL-8, CCL18), proteases (MMP-1 and MMP-7), and growth factors (IGBPs) families. Circulating T cells and fibrocytes have also gained recent interest in that respect. Up to now, though several interesting candidates are profiling there has not been a single biomarker, which proved to be specific of the disease and predictive of the evolution (decline of pulmonary function test values, risk of acute exacerbation or mortality).
Conclusion
Large scale multicentric studies are eagerly needed to confirm the utility of these biomarkers.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Idiopathic pulmonary fibrosis is a rare lung disease of unknown origin which leads rapidly to death [1]. Epidemiological studies suggest that the incidence of IPF has been increasing steadily over the last two to three decades [2]. Although the aetiology and the pathophysiology of IPF are still incompletely understood, two anti-fibrotic drugs, pirfenidone and nintedanib, have recently proven to be effective in slowing down disease progression and are now approved as treatments [3, 4]. Clinical management of IPF remains difficult due to a lack of accurate indicators of disease progression, and an absence of simple short-term measures of therapeutic response [5]. Although the median survival is about 2–3 years, there is a wide spectrum of disease courses ranging from slow evolving disease to quick deterioration [1].
Biological markers, often referred as biomarkers, are commonly defined as objectively measured elevated indicators of physiological / pathological processes or pharmacological response to therapeutic interventions [6]. Biomarkers are highly needed in IPF as tools for differential diagnostic, predictor of the progression of the disease and treatment response [7]. Specifically in IPF, an early diagnostic is important to reduce as much as possible the disease progression [8, 9]. Ideally, biomarkers should easily be sampled and analysed for a widespread utility. Therefore, we focused on blood molecules or circulating cells for the simplicity of sampling and processing.
IPF Definition and Diagnostic Criteria
IPF is defined as a chronic progressive fibrosing interstitial pneumonia of unknown origin [1]. The disease is limited to the lung and associated to a typical radiologic (subpleural and basal predominance, reticulations, honey combing with or without bronchiectasis and no atypical images) and histopathologic pattern (marked fibrosis with architectural distortion, subpleural, patchy involvement of lung, fibroblast foci, absence of inconsistent features) [1]. The diagnosis of IPF also requires the exclusion of other forms of interstitial pneumonia including other idiopathic interstitial lung diseases and ILD’s associated to systemic disease, environmental, exposure or medication. In particular, it is important to exclude chronic hypersensitivity pneumonitis as a hidden cause of IPF by measuring specific IgG [10]. The exploration can be continued by performing a bronchoscopy (with bronchoalveolar lavage), a pulmonary biopsy or a cryobiopsy. All cases have to be discussed by a multidisciplinary group composed of a pulmonologist, a specialist in pulmonary rehabilitation, a rheumatologist, a radiologist, a pathologist and a specialist in occupational medicine.
Biomarkers (Table 1)
Here we have performed a systematic search in pubmed by typing the words “biomarkers” and “IPF” and selecting those measured in blood. Publications dates of selected papers range from 1993 until 2016.
Many biomarkers have been studied in BALF and in serum as potential diagnostic or prognostic tools. Their utility for monitoring the disease has to be assessed [11].
Alveolar Epithelial Markers
The main biomarkers associated with the alveolar epithelial cell damage (or dysfunction) are the Krebs von den Lungen-6 (KL-6) antigen and the surfactant protein A and D (SP-A and SP-D). Compared to SP-A and SP-D, serum KL-6 has a highest accuracy for diagnosis of interstitial lung diseases (IPF and ILD associated with connective tissue diseases) [11].
Krebs von den Lungen-6 Antigen (KL-6)
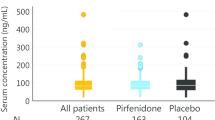
KL-6 is a high molecular weight glycoprotein classified as a human transmembrane mucin 1 (MUC1). That glycoprotein is mainly expressed at the extracellular membrane surface of type II pneumocytes [12–14]. It has been initially studied in non-IPF interstitial lung diseases (ILDs) and appeared to be also elevated in IPF with a possible correlation with an increased risk of IPF associated mortality [14, 15]. KL-6 promotes migration, proliferation and survival of lung fibroblasts [16, 17] and therefore is possibly involved in the IPF pathophysiological process. A serum baseline level above 1000 U/ml seems to be associated with a worse outcome [18, 19]. Our data indicate that serum KL-6 levels are markedly increased in IPF as compared to healthy subjects but we did not find reduced KL-6 levels in the patients treated by pirfenidone or nintedanib [20]. Serum KL-6 could possibly be associated to treatment response but this needs further longitudinal studies to be confirmed.
Surfactant Protein A and D (SP-A and SP-D)
Surfactant proteins are lipoprotein complexes synthesized and secreted by type II pneumocytes to decrease surface tension at the air–liquid interface. They have also a role in the lung host defence. Serum SP-A and SP-D are known as being elevated in patients with ILD with a greater extend in IPF [21–25]. Some findings also suggest that surfactant lipids may protect against intraluminal fibrogenesis by inducing fibroblast apoptosis and decreasing collagen accumulation [26]. In this context, SP-A and SP-D have been shown to be predictive of survival when assessed at the initial work up of the disease [24, 27]. Their involvement in the IPF physiopathological process is underlined by familial form of IPF associated with mutations of surfactant protein C [28].
Fibrogenesis and Extracellular Remodelling Markers
The main biomarkers identified in IPF focusing on fibrogenesis are matrix metalloproteinases (MMP)-1 and -7, Lysyl oxidase-like 2 (LOXL2) and Periostin.
Matrix Metalloproteinases-1 and -7 (MMP-1 and MMP-7)
Matrix metalloproteinases are a collection of zinc-dependent proteases involved in the breakdown and the remodelling of extracellular matrix components [29]. MMP-1 and MMP-7 seem to be primarily overexpressed in plasma of IPF patients compared to hypersensitivity pneumonitis, sarcoidosis and COPD with a possible usefulness in differential diagnosis [30]. They are also involved in inflammation and seem to take part to the pathophysiological process of pulmonary fibrosis [31, 32]. The most studied is clearly MMP-7 which is known as being significantly increased in epithelial cells both at the gene and protein levels and is considered to be active in hyperplastic epithelial cells and alveolar macrophages in IPF [33]. There is also a significant correlation between higher MMP-7 concentrations and disease severity assessed by forced vital capacity (FVC) and DLCO (%pred) [30]. We therefore can identify MMP-7 as one of the greatest individual biomarkers in IPF.
Concentrations of protein fragments generated by MMP activity are increased in the serum of individuals with IPF compared with healthy controls. Increased neoepitope concentrations were associated with disease progression as defined by death or decline in FVC > 10% at 12 months after study enrolment and the rate of this increase predicted survival [5, 34]. We can hypothesize that as neoepitopes are generated by proteases they could be associated with lung remodelling and fibrotic process.
Lysyl Oxidase-like 2 and Periostin
Lysyl oxidase-like 2 (LOXL2) is expressed in fibrotic lung and is thought to play a crucial role in matrix remodelling and fibrogenesis [35]. Serum level of LOXL2 is correlated to IPF disease progression (assessed by classical regression tree method) [36]. Nevertheless, anti-LOXL2 targeted therapy trial failed to reduce IPF disease progression [37]. Periostin, an extracellular matrix protein that contributes to fibrosis in the lung, is highly elevated in blood of IPF patients as well as in lung tissue [38]. In lung tissue, it is specifically found in the “fibroblast foci”, typically seen in IPF. Plasma level of periostin is also correlated with a composite score reflecting disease progression [39].
Chemokines
CC Chemokine Ligand 18
CC motif chemokine ligand 18 (CCL18) is a small protein derived from alveolar macrophages that acts as a chemo-attractant. CCL18 is mainly secreted by antigen-presenting cells such as monocytes, macrophages and dendritic cells [40]. In the setting of pulmonary fibrosis, alveolar macrophages are believed to be the main source of CCL18 in the lung and play a role in the pathogenesis of pulmonary fibrosis [41]. Serum CCL18 is increased in IPF but is not specific of the disease [41–43]. In IPF, CCL18 is negatively correlated to pulmonary function tests (TLC and DLCO) [42]. In a prospective study, it has been shown that patients with serum level of CCL18 > 150 ng/ml were independently associated with death in IPF (HR 1.98, 95% CI 2.49–25.51, p = 0.005) [44]. Moreover, pirfenidone one of the specific anti-fibrotic therapies in IPF significantly suppressed the expression of CCL18 on macrophages [45]. Therefore, CCL18 could have a potential interest as a prognostic tool in IPF.
Interleukin-8 (IL-8)
IL-8 is a cytokine, which is highly chemo-attractant for neutrophils and known to be elevated in serum of IPF patients. A study identified a negative correlation between IL-8 and pulmonary function tests (DLCO, TLC, VC) [46, 47] and survival [48] underlying its potential utility as biomarker in IPF.
Growth Factors and Adhesion Molecules
YKL-40
YKL-40 is a chitinase-like protein that regulates cell proliferation and survival and has previously been described in liver fibrosis. YKL-40 has also been well studied in multiple ILDs [13]. Its mechanism of action is not understood yet but seems to be associated with a promitogenic action on lung fibroblasts in animal model and increases macrophages activity in COPD [49, 50]. Moreover, YKL-40 has been found to be increase in fibrotic areas of IPF patients and especially in macrophages and bronchial cells [13]. In a prospective study, YKL-40 is not specific for IPF but a serum level above 79 ng/ml is associated with a worse prognosis (HR 10.9, 95% CI 1.9–63.8, p < 0.01) [51]. Therefore, we think that YKL-40 is useful at diagnosis for prognosis stadification and could be considered.
Insulin-like Growth Factor 2 (IGFBP-2)
IGFBP-2 is a member of a highly conserved family of six insulin-like growth factor (IGF) binding proteins, which has recently been identified in IPF [20, 52]. IGF and IGFBPs are described to be involved in cell proliferation and differentiation. In a cross sectional study on 50 patients suffering from IPF, we have recently shown that IGFBP-1 and IGFBP-2 are increased in newly diagnosed IPF and IGFBP-2 reduced in patients treated with anti-fibrotic therapy though still raised as compared to healthy subjects [20]. However, we did not report any correlation between IGFBP-2 and impaired lung function indices. In contrast to IGFPB-1 and -2, it turned out that IGF-1 and -2 were decreased in serum of untreated IPF. Further, longitudinal studies are needed to evaluate their usefulness as biomarkers in IPF.
ICAM-1 and ICAM-2
Intracellular adhesion molecule-1 and -2 (ICAM-1 and ICAM-2) are also elevated (not specifically) in serum from patient with IPF. ICAM-1 has been found to be overexpressed on pulmonary epithelial cells of patients with IPF [53, 54], whereas ICAM-2 has been inversely associated with DLCO. Its utility in clinical practice is not yet known and has to be explored in longitudinal studies.
VEGF
Vascular endothelial growth factor (VEGF) is a glycoprotein expressed in alveolar epithelial cells [55]. Increased serum VEGF, but not CRP, has been identified in one longitudinal study of 41 IPF patients and potentially associated with the disease severity as reflected by the alveolo-arterial difference of O2. In this study, serum levels of VEGF in severely hypoxemic IPF were similar to those found in non-small cell lung carcinoma [56]. Serum VEGF was associated with increased loss in VC over time. Survival rate at 5 years was decreased in patients whose VEGF level was above 207 pg/ml. Of interest, VEGF is targeted by nintedanib used as a specific IPF therapy [57].
Others
The heat shock protein 70 (HSP 70) is a molecular chaperone that is expressed in response to stress. IgG autoantibodies directed to HSP70 have been associated with a poorer prognostic in IPF [58]. In addition, Fibulin-1 is elevated in IPF and correlated with the disease progression [59]. Plasma leptin level, a biomarker traditionally associated with obesity, is also significantly increased in case of acute exacerbation of IPF. In one longitudinal study of 61 IPF patients, those patients with plasma leptin levels > 13.79 ng/ml at hospital admission had increased mortality [60].
A recent study focusing on biomarkers that might assist in distinguishing IPF from non-IPF-ILDs have identify in the plasma of 149 IPF patients (derivation cohort n = 86, validation cohort n = 63) that a biomarker index composed of SP-D, MMP-7 and osteopontin enhanced diagnostic accuracy for IPF compared to other ILDs (rheumatoid arthritis associated ILD (n = 33); alternative idiopathic ILD (n = 41)) [61]. This three analyte panel of SP-D, MMP-7 and osteopontin enhance the odds of IPF diagnosis when each of them exceed its threshold value (>31 ng/ml, >1.75 ng/ml and >6 ng/ml, respectively) [61].
Circulating Cells
T-cells are also involved in the pathophysiology of IPF. Semaphorin 7a (Sema 7a), a membrane-bound protein expressed in activated Tcells, also regulates inflammatory responses via stimulation of macrophage chemotaxis and cytokine production chemokine expression, modulation of T-cell function and regulation of collagen production by fibrocytes. Hematopoietic expression of Sema 7a is sufficient to induce fibrosis in TGF-β1–induced murine lung fibrosis [62]. There is an increased expression of Sema7a+ on T-regulatory cells in IPF [62–64]. Its protective or deleterious impact in IPF is not known yet. By contrast, CD28 (a marker of T-cell activation) is less expressed in IPF with a higher incidence of mortality in case of low expression [65]. As it has been shown in several auto-immune disease, there is a lower expression of CD4+CD25+FOXP3+ T reg cells in IPF [66]. Other teams have been focusing on B-cell activation using BLys (B-cell activating factor) and CXCL13, which underlined the potential implication of B-cells in IPF [67].
Circulating CD45+Col-1+ fibrocytes are circulating bone-marrow derived mesenchymal progenitor cells which can differentiate into fibroblast and myofibroblasts [68]. They are increased in IPF compared to healthy subjects. Their circulating level in IPF (>5% of total blood leukocytes) is associated with a worse survival [69, 70].
Further Approach
Genomic, epigenetic and molecular phenotyping are widely used in clinical research. It is getting more and more clear that genetic polymorphisms, whole blood transcriptomic profile, as well as lung microbiome are predicting groups of diverse disease behaviour and response to treatment [71]. Extracellular vesicles containing micro-RNA are known to express surface protein and hold nucleic acid, including microRNAs, and to regulate gene expression in the recipient cells. A recent study identified miR-21-5-p as to be elevated in the serum of IPF and associated to the disease progression and the overall prognosis [72]. Many other miR have also been identified like miR-7, miR-29 and miR-125b [73].
Limitations
One of the limitations of our review is that we have chosen candidate biomarkers in IPF according to our thinking and their potential usefulness. Another limitation is the lack of longitudinal studies for some of the biomarkers, which reduces the clinical impact of the finding because of a lack of assessment of treatment response. We believe that further longitudinal multicentre studies are highly needed to evaluate the real clinical impact of most of the biomarkers in a single or multivariate analysis as diagnostic and prognostic tools.
Conclusion
Diagnosis of IPF remains challenging and requires a multidisciplinary approach. Treatments currently available have a limited efficacy and the overall prognosis of the disease remains poor. Recent advance in pathophysiological process are promising for further therapies; nevertheless, the most important point for now is the early diagnosis as a mean to early treatment. So far, there is no easy to use biomarker available for clinical practice. It is now obviously clear that genetic and epigenetic variations are involved in the pathogenesis giving us different information for the morbi-mortality in IPF. Furthermore, prospective longitudinal studies have started to identify blood biomarkers to be used in clinical practice.
References
Raghu G, Collard HR, Egan JJ, Martinez FJ, Behr J, Brown KK et al (2011) An official ATS/ERS/JRS/ALAT statement: idiopathic pulmonary fibrosis: evidence-based guidelines for diagnosis and management. Am J Respir Crit Care Med 183(6):788–824
Raghu G, Weycker D, Edelsberg J, Bradford WZ, Oster G (2006) Incidence and prevalence of idiopathic pulmonary fibrosis. Am J Respir Crit Care Med 174(7):810–816
King TE, Bradford WZ, Castro-Bernardini S, Fagan EA, Glaspole I, Glassberg MK et al (2014) A phase 3 trial of pirfenidone in patients with idiopathic pulmonary fibrosis. N Engl J Med 370(22):2083–2092
Richeldi L, du Bois RM, Raghu G, Azuma A, Brown KK, Costabel U et al (2014) Efficacy and safety of nintedanib in idiopathic pulmonary fibrosis. N Engl J Med 370(22):2071–2082
Maher TM (2013) PROFILEing idiopathic pulmonary fibrosis: rethinking biomarker discovery. Eur Respir Rev 22(128):148–152
Kaarteenaho R, Lappi-Blanco E (2015) Tissue is an issue in the search for biomarkers in idiopathic pulmonary fibrosis. Fibrogenesis Tissue Repair 8(1):3
Zhang Y, Kaminski N (2012) Biomarkers in idiopathic pulmonary fibrosis. Curr Opin Pulm Med 18(5):441–446
Raghu G, Rochwerg B, Zhang Y, Garcia CAC, Azuma A, Behr J et al (2015) An official ATS/ERS/JRS/ALAT clinical practice guideline: treatment of idiopathic pulmonary fibrosis. an update of the 2011 clinical practice guideline. Am J Respir Crit Care Med 192(2):e3–e19
Prasad R, Gupta N, Singh A, Gupta P (2015) Diagnosis of idiopathic pulmonary fibrosis: current issues. Intractable Rare Dis Res 4(2):65–69
Morell F, Villar A, Montero M-Á, Muñoz X, Colby TV, Pipvath S et al (2013) Chronic hypersensitivity pneumonitis in patients diagnosed with idiopathic pulmonary fibrosis: a prospective case-cohort study. Lancet Respir Med 1(9):685–694
Ohnishi H, Yokoyama A, Kondo K, Hamada H, Abe M, Nishimura K et al (2002) Comparative study of KL-6, surfactant protein-A, surfactant protein-D, and monocyte chemoattractant protein-1 as serum markers for interstitial lung diseases. Am J Respir Crit Care Med 165(3):378–381
Kobayashi J, Kitamura S (1995) KL-6: a serum marker for interstitial pneumonia. Chest 108(2):311–315
Furuhashi K, Suda T, Nakamura Y, Inui N, Hashimoto D, Miwa S et al (2010) Increased expression of YKL-40, a chitinase-like protein, in serum and lung of patients with idiopathic pulmonary fibrosis. Respir Med 104(8):1204–1210
Ohshimo S, Ishikawa N, Horimasu Y, Hattori N, Hirohashi N, Tanigawa K et al (2014) Baseline KL-6 predicts increased risk for acute exacerbation of idiopathic pulmonary fibrosis. Respir Med 108(7):1031–1039
Ishikawa N, Hattori N, Yokoyama A, Tanaka S, Nishino R, Yoshioka K et al (2008) Usefulness of monitoring the circulating Krebs von den Lungen-6 levels to predict the clinical outcome of patients with advanced nonsmall cell lung cancer treated with epidermal growth factor receptor tyrosine kinase inhibitors. Int J Cancer 122(11): 2612–2620
Hirasawa Y, Kohno N, Yokoyama A, Inoue Y, Abe M, Hiwada K (1997) KL-6, a human MUC1 mucin, is chemotactic for human fibroblasts. Am J Respir Cell Mol Biol 17(4):501–507
Ohshimo S, Yokoyama A, Hattori N, Ishikawa N, Hirasawa Y, Kohno N (2005) KL-6, a human MUC1 mucin, promotes proliferation and survival of lung fibroblasts. Biochem Biophys Res Commun 338(4): 1845–52
Satoh H, Kurishima K, Ishikawa H, Ohtsuka M (2006) Increased levels of KL-6 and subsequent mortality in patients with interstitial lung diseases. J Intern Med 260(5):429–434
Yokoyama A, Kohno N, Hamada H, Sakatani M, Ueda E, Kondo K et al (1998) Circulating KL-6 predicts the outcome of rapidly progressive idiopathic pulmonary fibrosis. Am J Respir Crit Care Med 158(5 Pt 1):1680–1684
Guiot J, Bondue B, Henket M, Corhay JL, Louis R (2016) Raised serum levels of IGFBP-1 and IGFBP-2 in idiopathic pulmonary fibrosis. BMC Pulm Med 16(1):86
Ishii H, Mukae H, Kadota J, Kaida H, Nagata T, Abe K et al (2003) High serum concentrations of surfactant protein A in usual interstitial pneumonia compared with non-specific interstitial pneumonia. Thorax 58(1):52–57
Kohno N, Yokoyama A, Hirasawa Y, Kondo K, Fujino S, Abe M et al (1997) Comparative studies of circulating KL-6, type III procollagen N-terminal peptide and type IV collagen 7 S in patients with interstitial pneumonitis and alveolar pneumonia. Respir Med 91(9):558–561
Greene KE, King TE, Kuroki Y, Bucher-Bartelson B, Hunninghake GW, Newman LS et al (2002) Serum surfactant proteins-A and -D as biomarkers in idiopathic pulmonary fibrosis. Eur Respir J 19(3):439–446
Kinder BW, Brown KK, McCormack FX, Ix JH, Kervitsky A, Schwarz MI et al (2009) Serum surfactant protein-A is a strong predictor of early mortality in idiopathic pulmonary fibrosis. Chest 135(6):1557–1563
Barlo NP, van Moorsel CHM, Ruven HJT, Zanen P, van den Bosch JMM, Grutters JC (2009) Surfactant protein-D predicts survival in patients with idiopathic pulmonary fibrosis. Sarcoidosis Vasc Diffus lung Dis 26(2):155–161
Vázquez de Lara L, Becerril C, Montaño M, Ramos C, Maldonado V, Meléndez J et al (2000) Surfactant components modulate fibroblast apoptosis and type I collagen and collagenase-1 expression. Am J Physiol Lung Cell Mol Physiol 279(5):L950–L957
Takahashi H, Fujishima T, Koba H, Murakami S, Kurokawa K, Shibuya Y et al (2000) Serum surfactant proteins A and D as prognostic factors in idiopathic pulmonary fibrosis and their relationship to disease extent. Am J Respir Crit Care Med 162(3 Pt 1):1109–1114
van Moorsel CHM, van Oosterhout MFM, Barlo NP, de Jong PA, van der Vis JJ, Ruven HJT et al (2010) Surfactant protein C mutations are the basis of a significant portion of adult familial pulmonary fibrosis in a dutch cohort. Am J Respir Crit Care Med 182(11):1419–1425
Oikonomidi S, Kostikas K, Tsilioni I, Tanou K, Gourgoulianis KI, Kiropoulos TS (2009) Matrix metalloproteinases in respiratory diseases: from pathogenesis to potential clinical implications. Curr Med Chem 16(10):1214–1228
Rosas IO, Richards TJ, Konishi K, Zhang Y, Gibson K, Lokshin AE et al (2008) MMP1 and MMP7 as potential peripheral blood biomarkers in idiopathic pulmonary fibrosis. PLoS Med 5(4):e93
Vij R, Noth I (2012) Peripheral blood biomarkers in idiopathic pulmonary fibrosis. Transl Res 159(4):218–227
Dancer RCA, Wood AM, Thickett DR (2011) Metalloproteinases in idiopathic pulmonary fibrosis. Eur Respir J 38(6):1461–1467
Fujishima S, Shiomi T, Yamashita S, Yogo Y, Nakano Y, Inoue T et al (2010) Production and activation of matrix metalloproteinase 7 (matrilysin 1) in the lungs of patients with idiopathic pulmonary fibrosis. Arch Pathol Lab Med 134(8):1136–1142
Jenkins RG, Simpson JK, Saini G, Bentley JH, Russell A-M, Braybrooke R et al (2015) Longitudinal change in collagen degradation biomarkers in idiopathic pulmonary fibrosis: an analysis from the prospective, multicentre PROFILE study. Lancet. Respir Med 3(6):462–472
Chien J, Shao L, Lyman S, Adamkewicz J, Smith V, O’Riordan T (2014) Elevated serum LOXL2 levels are associated with rapid disease progression in idiopathic pulmonary fibrosis (IPF). Eur Respir J 40(Suppl 56)
Chien JW, Richards TJ, Gibson KF, Zhang Y, Lindell KO, Shao L et al(2014) Serum lysyl oxidase-like 2 levels and idiopathic pulmonary fibrosis disease progression. Eur Respir J 43(5):1430–1438
Patrick O’Brien (2016) Gilead Terminates Phase 2 Study of Simtuzumab in Patients With Idiopathic Pulmonary Fibrosis | Gilead [Internet]. Available from: http://www.gilead.com/news/press-releases/2016/1/gilead-terminates-phase-2-study-of-simtuzumab-in-patients-with-idiopathic-pulmonary-fibrosis
Okamoto M, Hoshino T, Kitasato Y, Sakazaki Y, Kawayama T, Fujimoto K et al (2011) Periostin, a matrix protein, is a novel biomarker for idiopathic interstitial pneumonias. Eur Respir J 37(5):1119–1127
Naik PK, Bozyk PD, Bentley JK, Popova AP, Birch CM, Wilke CA et al(2012) Periostin promotes fibrosis and predicts progression in patients with idiopathic pulmonary fibrosis. AJP Lung Cell Mol Physiol 303(12):L1046–L1056
Hieshima K, Imai T, Baba M, Shoudai K, Ishizuka K, Nakagawa T et al (1997) A novel human CC chemokine PARC that is most homologous to macrophage-inflammatory protein-1 alpha/LD78 alpha and chemotactic for T lymphocytes, but not for monocytes. J Immunol 159(3):1140–1149
Prasse A, Pechkovsky D V, Toews GB, Jungraithmayr W, Kollert F, Goldmann T et al (2006) A vicious circle of alveolar macrophages and fibroblasts perpetuates pulmonary fibrosis via CCL18. Am J Respir Crit Care Med 173(7):781–792
Prasse A, Pechkovsky DV, Toews GB, Schäfer M, Eggeling S, Ludwig C et al (2007) CCL18 as an indicator of pulmonary fibrotic activity in idiopathic interstitial pneumonias and systemic sclerosis. Arthritis Rheum 56(5):1685–1693
Luzina IG, Tsymbalyuk N, Choi J, Hasday JD, Atamas SP (2006) CCL18-stimulated upregulation of collagen production in lung fibroblasts requires Sp1 signaling and basal Smad3 activity. J Cell Physiol 206(1):221–228
Prasse A, Probst C, Bargagli E, Zissel G, Toews GB, Flaherty KR et al (2009) Serum CC-Chemokine ligand 18 concentration predicts outcome in idiopathic pulmonary fibrosis. Am J Respir Crit Care Med 179(8):717–723
Saito Y, Azuma A, Matsuda K, Kamio K, Abe S, Gemma A (2016) Pirfenidone exerts a suppressive effect on CCL18 expression in U937-derived macrophages partly by inhibiting STAT6 phosphorylation. Immunopharmacol Immunotoxicol 38(6):464–71
Tsoutsou PG, Gourgoulianis KI, Petinaki E, Germenis A, Tsoutsou AG, Mpaka M et al (2006) Cytokine levels in the sera of patients with idiopathic pulmonary fibrosis. Respir Med 100(5):938–945
ZIEGENHAGEN MW, ZABEL P, ZISSEL G, SCHLAAK M, MÜLLER-QUERNHEIM J (1998) Serum level of interleukin 8 is elevated in idiopathic pulmonary fibrosis and indicates disease activity. Am J Respir Crit Care Med 157(3):762–768
Richards TJ, Kaminski N, Baribaud F, Flavin S, Brodmerkel C, Horowitz D et al (2012) Peripheral blood proteins predict mortality in idiopathic pulmonary fibrosis. Am J Respir Crit Care Med 185(1):67–76
Ober C, Chupp GL (2009) The chitinase and chitinase-like proteins: a review of genetic and functional studies in asthma and immune-mediated diseases. Curr Opin Allergy Clin Immunol 9(5):401–408
Létuvé S, Kozhich A, Arouche N, Grandsaigne M, Reed J, Dombret M-C et al (2008) YKL-40 is elevated in patients with chronic obstructive pulmonary disease and activates alveolar macrophages. J Immunol 181(7):5167–5173
Korthagen NM, van Moorsel CHM, Barlo NP, Ruven HJT, Kruit A, Heron M et al (2011) Serum and BALF YKL-40 levels are predictors of survival in idiopathic pulmonary fibrosis. Respir Med 105(1):106–113
Guiot J, Henket M, Corhay JL, Moermans C, Louis R (2017) Sputum biomarkers in IPF: Evidence for raised gene expression and protein level of IGFBP-2, IL-8 and MMP-7. PLoS ONE 12(2):e0171344.
Shijubo N, Imai K, Aoki S, Hirasawa M, Sugawara H, Koba H et al (1992) Circulating intercellular adhesion molecule-1 (ICAM-1) antigen in sera of patients with idiopathic pulmonary fibrosis. Clin Exp Immunol 89(1):58–62
Shijubo N, Imai K, Shigehara K, Hirasawa M, Tsujisaki M, Hinoda Y et al (1995) Soluble intercellular adhesion molecule-1 (ICAM-1) in sera and bronchoalveolar lavage (BAL) fluids of extrinsic allergic alveolitis. Clin Exp Immunol 102(1):91–97
Monacci WT, Merrill MJ, Oldfield EH (1993) Expression of vascular permeability factor/vascular endothelial growth factor in normal rat tissues. Am J Physiol 264(4 Pt 1):C995–C1002
Ando M, Miyazaki E, Ito T, Hiroshige S, Nureki S, Ueno T et al (2010) Significance of serum vascular endothelial growth factor level in patients with idiopathic pulmonary fibrosis. Lung 188(3):247–252.
Wollin L, Wex E, Pautsch A, Schnapp G, Hostettler KE, Stowasser S et al (2015) Mode of action of nintedanib in the treatment of idiopathic pulmonary fibrosis. Eur Respir J
Kahloon RA, Xue J, Bhargava A, Csizmadia E, Otterbein L, Kass DJ et al (2013) Patients with idiopathic pulmonary fibrosis with antibodies to heat shock protein 70 have poor prognoses. Am J Respir Crit Care Med 187(7):768–775
Jaffar J, Unger S, Corte TJ, Keller M, Wolters PJ, Richeldi L et al (2014) Fibulin-1 predicts disease progression in patients with idiopathic pulmonary fibrosis. Chest 146(4):1055–1063
Cao M, Swigris JJ, Wang X, Cao M, Qiu Y, Huang M et al (2016) Plasma leptin is elevated in acute exacerbation of idiopathic pulmonary fibrosis. Mediators Inflamm 2016:1–7
White ES, Xia M, Murray S, Dyal R, Flaherty CM, Flaherty KR et al (2016) Plasma surfactant protein-D, matrix metalloproteinase-7, and osteopontin index distinguishes idiopathic pulmonary fibrosis from other idiopathic interstitial pneumonias. Am J Respir Crit Care Med 194(10):1242–1251
Reilkoff RA, Peng H, Murray LA, Peng X, Russell T, Montgomery R et al (2013) Semaphorin 7a + regulatory T cells are associated with progressive idiopathic pulmonary fibrosis and are implicated in transforming growth factor-β1–induced pulmonary fibrosis. Am J Respir Crit Care Med 187(2):180–8
Gan Y, Reilkoff R, Peng X, Russell T, Chen Q, Mathai SK et al (2011) Role of semaphorin 7a signaling in transforming growth factor β1-induced lung fibrosis and scleroderma-related interstitial lung disease. Arthritis Rheum 63(8):2484–2494
Kang H-R, Lee CG, Homer RJ, Elias JA (2007) Semaphorin 7 A plays a critical role in TGF-β 1 –induced pulmonary fibrosis. J Exp Med 204(5):1083–1093
Gilani SR, Vuga LJ, Lindell KO, Gibson KF, Xue J, Kaminski N et al (2010) CD28 down-regulation on circulating CD4 T-cells is associated with poor prognoses of patients with idiopathic pulmonary fibrosis. PLoS ONE 5(1):e8959
Kotsianidis I, Nakou E, Bouchliou I, Tzouvelekis A, Spanoudakis E, Steiropoulos P et al (2009) Global impairment of CD4 + CD25 + FOXP3 + regulatory T cells in idiopathic pulmonary fibrosis. Am J Respir Crit Care Med 179(12):1121–1130
Vuga LJ, Tedrow JR, Pandit K V., Tan J, Kass DJ, Xue J et al (2014) C-X-C motif chemokine 13 (CXCL13) is a prognostic biomarker of idiopathic pulmonary fibrosis. Am J Respir Crit Care Med 189(8):966–974
Moeller A, Gilpin SE, Ask K, Cox G, Cook D, Gauldie J et al (2009) Circulating fibrocytes are an indicator of poor prognosis in idiopathic pulmonary fibrosis. Am J Respir Crit Care Med 179(7):588–594
Phillips RJ, Burdick MD, Hong K, Lutz MA, Murray LA, Xue YY et al (2004) Circulating fibrocytes traffic to the lungs in response to CXCL12 and mediate fibrosis. J Clin Invest 114(3):438–446
Mehrad B, Burdick MD, Zisman DA, Keane MP, Belperio JA, Strieter RM (2007) Circulating peripheral blood fibrocytes in human fibrotic interstitial lung disease. Biochem Biophys Res Commun 353(1):104–108
Daccord C, Maher TM (2016) Recent advances in understanding idiopathic pulmonary fibrosis. F1000Research 5
Makiguchi T, Yamada M, Yoshioka Y, Sugiura H, Koarai A, Chiba S et al (2016) Serum extracellular vesicular miR-21-5p is a predictor of the prognosis in idiopathic pulmonary fibrosis. Respir Res 17(1):110
Jiang X, Tsitsiou E, Herrick SE, Lindsay MA (2010) MicroRNAs and the regulation of fibrosis. FEBS J 277(9):2015–2021
Yokoyama A, Kondo K, Nakajima M, Matsushima T, Takahashi T, Nishimura M et al (2006) Prognostic value of circulating KL-6 in idiopathic pulmonary fibrosis. Respirology 11(2):164–168
Ley B, Brown KK, Collard HR (2014) Molecular biomarkers in idiopathic pulmonary fibrosis. AJP Lung Cell Mol Physiol 307(9):L681–L691
Spagnolo P, Tzouvelekis A, Maher TM (2015) Personalized medicine in idiopathic pulmonary fibrosis. Curr Opin Pulm Med 21(5):470–478
DePianto DJ, Chandriani S, Abbas AR, Jia G, N’Diaye EN, Caplazi P et al (2015) Heterogeneous gene expression signatures correspond to distinct lung pathologies and biomarkers of disease severity in idiopathic pulmonary fibrosis. Thorax 70(1):48–56
Herazo-Maya JD, Noth I, Duncan SR, Kim S, Ma S-F, Tseng GC et al (2013) Peripheral blood mononuclear cell gene expression profiles predict poor outcome in idiopathic pulmonary fibrosis. Sci Transl Med 5(205):205ra136–205ra136
Acknowledgements
Thank you to all the co-authors for their help, support and collaboration to the redaction of this review.
Funding
There was no funding for this article.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Guiot, J., Moermans, C., Henket, M. et al. Blood Biomarkers in Idiopathic Pulmonary Fibrosis. Lung 195, 273–280 (2017). https://doi.org/10.1007/s00408-017-9993-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00408-017-9993-5