
Abstract
Bi-allelic variations in the gap junction protein beta-2 (GJB2) gene cause up to 50% of cases of newborn hearing loss. Heterozygous pathogenic GJB2 variations are also fivefold overrepresented in idiopathic patient groups compared to the normal-hearing population. Whether hearing loss in this group is due to unidentified additional variations within GJB2 or variations in other deafness genes is unknown in most cases. Whole-exome sequencing offers an effective approach in the search for causative variations in patients with Mendelian diseases. In this prospective genetic cohort study, we initially investigated a family of Turkish origin suffering from congenital autosomal recessive hearing loss. An index patient and his normal-hearing father, both bearing a single heterozygous pathogenic c.262G>T (p.Ala88Ser) GJB2 transversion as well as the normal-hearing mother were investigated by means of whole-exome sequencing. Subsequently the genetic screening was extended to a hearing-impaired cohort of 24 families of Turkish origin. A homozygous missense c.5492G>T transversion (p.Gly1831Val) in the Myosin 15a gene, previously linked to deafness, was identified as causative in the index family. This very rare variant is not listed in any population in the Genome Aggregation Database. Subsequent screening of index patients from additional families of Turkish origin with recessive hearing loss identified the c.5492G>T variation in an additional family. Whole-exome sequencing may effectively identify the causes of idiopathic hearing loss in patients bearing heterozygous GJB2 variations.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Hearing loss (HL) is a frequent sensory disorder in humans with an onset at different stages of life. HL occurs in approximately 1:1000 newborns [1] and the number rises to 3.5:1000 in adolescence [2]. A genetic mechanism is causative for the disorder in approximately half of children affected by prelingual HL. In the majority of these cases (70%), HL is not accompanied by further symptoms and is, therefore, termed as non-syndromic (NSHL) [3].
The genetic diagnosis of NSHL remains a challenge in many patients due to the high number of genes involved in the disease. The most common causative gene is GJB2 in the DFNB1 locus that encodes Connexin 26 (gap junction protein, beta-2), a gap junction protein expressed within the stria vascularis, which regulates cochlear development and is essential for maintenance of active cochlear amplification [4]. Variations in GJB2 account for up to 50% of all genetically caused NSHL cases in Caucasians [5].
Although variations in GJB2 almost exclusively cause prelingual autosomal recessive HL, the frequency of heterozygous c.35delG in the newborn hearing-impaired population is over 11% compared to a carrier frequency of below 2% in the normal-hearing Austrian population [6]. Whereas screening within GJB2 heterozygous patients can also lead to the identification of recessive pathogenic variations in additional HL genes [7], we originally hypothesized here that novel alterations affecting the cochlear gap junction network within DFNB1 may be the cause for this discrepancy. Thus, the index patient examined in this study, carrying the heterozygous c.262G>T (p.Ala88Ser) recessive GJB2 transversion [8], has been prescreened for changes in upstream regulatory sequences [9], the basal promoter [10] and alternative transcriptional start sites [11] and deletions in GJB6 [12].
In patients where pathogenic changes in DFNB1 are not found, monosymptomatic HL can be due to genetic defects in any of 90 genes identified to date or alterations in unidentified causative genes [13]. Whereas variations in most non-DFNB1 genes cause hereditary NSHL only in isolated families, some variations have been shown to have a high prevalence in certain populations. For example, pathogenic variations in the TMC1 gene (encoding the transmembrane channel-like protein 1) are found in more than a third of hearing-impaired Jewish patients of Moroccan ancestry [14]. With the exception of variations in GJB2, no such common genetic variants have been identified to date in the Austrian population. Due to practical and financial considerations, routine genetic HL screening in Austria has therefore been confined to screening the coding sequence of GJB2, thereby excluding almost 50% of affected individuals from genetic diagnosis and counseling.
Massively parallel DNA sequencing (MPS) allows the comprehensive variation screening of all known deafness genes simultaneously. This high-throughput DNA sequencing method has proven to produce technically reliable results in a time- and cost-efficient manner and is gradually being integrated into the routine genetic diagnostics of Mendelian diseases, including hereditary HL [14,15,16].
In this study, we applied whole-exome sequencing (WES) on DNA samples from an Austrian family of Turkish origin with monosymptomatic genetic HL containing an index patient bearing a heterozygous c.262G>T pathogenic (p.Ala88Ser) transversion. Extended screening of a hearing-impaired cohort consisting of index patients from 24 families of Turkish origin was subsequently performed to estimate the frequency of the disease-causing variants detected by WES in the Austrian-Turkish HL population.
Patients and methods
Subjects
Study patients were recruited at the Department of Otorhinolaryngology, Head and Neck Surgery at the Medical University of Vienna, Austria, as part of an ongoing screening program for hereditary HL. Informed consent was obtained from all patients and the parents of minors and the study protocol was approved by the Ethics Committee of the Medical University of Vienna (approval number: ECS 198/2004, with annual extensions to date). Full medical and family histories were obtained including the age of onset, exposure to noise or ototoxic medications and presence of additional ear-related complaints such as tinnitus or vertigo. Additionally, all participants underwent a clinical ear inspection and pure tone audiometry to determine the type and degree of HL. The degree of HL was defined as mild (20–40), moderate (41–70), severe (71–95) or profound (>95) in dB [17].
DNA sequencing
Genomic DNA was isolated from frozen peripheral venous blood with a commercial DNA extraction kit (Invisorb blood universal kit 1000, STRATEC Molecular, Berlin, Germany). After screening for GJB2 variations by traditional Sanger sequencing as previously described [8], a deaf index patient with a heterozygous pathogenic GJB2 variant and both unaffected parents were selected for trio analysis by WES. Capture libraries were prepared using the Illumina Nextera® Exome Capture Kit (Illumina Biotechnology, San Diego, CA, USA) according to the manufacturer’s instructions. The enriched samples were then sequenced on a HiSeq 2000 (Illumina) device at the core facility of the Medical University of Vienna. The identified target variants were then validated and tested for co-segregation with the disease in the remaining family members and a patient cohort with Sanger-based sequencing.
Bioinformatics
The resulting reads were mapped to the human reference genome version hg19 using the Burrows–Wheeler read aligner [18]. For variant calling, we applied the Genome Analysis Tool Kit [19]. The variant call format files containing the final single nucleotide, insertion, and deletion variants were further analyzed with the Genomatix GeneGrid interface (Genomatix GmbH, Munich, Germany). Initially, the index patient sample was tested for known deafness-causing variations in all coding sequences and splice sites along with 5 bp of the flanking intronic sequences. Additionally, only variants matching the recessive inheritance pattern of the family were included and variants with an allele frequency in the gnomAD database (http://gnomad.broadinstitute.org) [20] greater than 0.05 were excluded.
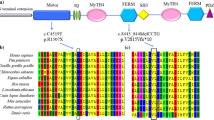
Myo15a sequence comparisons were made with Homo sapiens (NP_057323.3), Pan troglodytes (XP_016787798), Mus musculus (NP_034992.1), Rattus norvegicus (XP_008766109), Canis lupus familiaris (XP_536660), Gallus gallus (XP_414818), Xenopus tropicalis (XP_017952739) and Danio rerio (XP_001919593) peptides. Protein sequences were aligned with the constraint-based multiple alignment tool (http://ncbi.nlm.nih.gov/tools/cobalt/cobalt.cgi).
Results
Clinical presentation of the index family TAR1
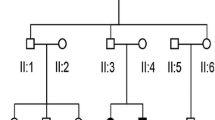
The index Turkish autosomal recessive family (TAR1) examined in this study (Fig. 1a) comprised a 7-year-old patient suffering from congenital profound HL (II/1), normal-hearing parents (I/1, I/2) and a normal-hearing sister (II/2).

Inheritance in Turkish autosomal recessive families (TAR) suffering from congenital hearing impairment. The index family TAR1 (a) comprised a single affected individual (II/1) with a normal-hearing sister (II/2) and parents (I/1 and I/2). Individuals highlighted in yellow were selected for whole-exome sequencing. A second family (TAR6) contained two hearing-impaired individuals (II/3 and II/5) with a normal-hearing brother (II/4) and parents (I/3 and I/4). The parents were first cousins (b)
Both the patient and the normal-hearing father were carriers of the heterozygous c.262G>T (p.Ala88Ser) recessive GJB2 transversion. No additional pathogenic allele within GJB2 was found in these family members. HL was severe at frequencies below 500 Hz and profound above 500 Hz (Fig. 2a). The patient (II/1) communicates with a combination of severely dyslalic speech, sign language and lip reading. Hearing aids had been supplied since early infancy in Turkey but were insufficient to allow proper language acquisition. Hearing rehabilitation by means of cochlear implantation was not possible due to insufficient auditory pathway plasticity at the age of first presentation at our department. No other symptoms besides HL were observed in the patient.

Hearing loss in Turkish autosomal recessive family (TAR) members. Unaided, masked pure tone audiograms in dB hearing loss showing affected TAR1 II/1 (a) and TAR6 family members II/3 (b) and II/5 (c). HL in all cases was severe at frequencies below 500 Hz and mostly profound above 500 Hz
Whole-exome sequencing and mapping results
WES was carried out on DNA samples of a deaf index patient (II/1) and both unaffected parents (I/1 and I/2). Sequencing of the capture library of the probands resulted in an average total read number of 6.7 Mbp with a median base coverage of 42×, with 91 and 68% of targeted bases covered by more than 10 or 30 reads, respectively.
Identification of a homozygous missense variation in Myo15a in family TAR1
WES of the index family member II/1 identified a homozygous missense G>T transversion in the Myosin 15a gene (Myo15a) at position c.5492 (NM_016239) that is predicted to lead to a transposition of glycine to valine at position 1831 of the mature Myosin 15a peptide (p.Gly1831Val). This variant was then validated by Sanger-based sequencing and was found in the affected family member (I/1) in a homozygous state (Fig. 3a). Both parents (I/1 and I/2) and the non-affected sister (II/2) were found to be heterozygous carriers of the c.5492G>T variation (Fig. 3b). This very rare variation, which has previously only been described in a single consanguineous family of Turkish origin, is located at an amino acid position highly conserved throughout evolution from mammals to zebra fish (Fig. 4) and is not listed in the gnomAD database [20, 21].

Representative chromatograms showing segregation of the missense c.5492G>T Myo15a transversion. In family TAR1, c.5492G>T (p.Gly1831Val) is found homozygous in the affected son II/1 (a) and heterozygous in the unaffected mother I/1 (b). Codon 1831 is underlined

Cross-species multiple alignments of the Myosin 15a peptide. The glycine residue at position p.1831 is highly conserved from Homo sapiens to Danio rerio sequences
Screening for variations in GJB2 and the c.5492G>T Myo15a variation in a cohort of hearing-impaired patients of Turkish origin
Following identification of the c.5492G>T variation in family TAR1, we screened a collective of patients of Turkish origin with recessive inheritance or sporadic incidence of bilateral congenital HL for variations in the GJB2 gene and for the Myo15a c.5492G>T allele to estimate the frequency of this variant in a HL cohort with a Turkish ancestral background (Table 1). In total, 11 familial and 13 sporadic cases with bilateral congenital HL from 24 families were tested. In one additional family (TAR6), 2 further patients with congenital profound deafness were found to be homozygous for c.5492G>T (Fig. 1b: II/3, II/5) whereas both parents (I/3, I/4) and the non-affected sibling (II/4) were heterozygous carriers of the c.5492G>T transversion. The parents were first cousins and the affected siblings II/3 and II/5, aged 19 and 13 years, respectively, suffered from congenital sensorineural HL that was severe at frequencies below 500 Hz and mostly profound above 500 Hz (Fig. 2b, c).
Discussion
In the index family under study, the normal-hearing father and the affected son, suffering from non-syndromic autosomal recessive HL, were carriers of a known c.262G>T pathogenic variant in the GJB2 gene (Table 1). No second variation within the GJB2 gene had been identified in previous genetic screening of the patient. Another possible molecular cause for hearing loss in the index patient could have been a digenic inheritance pattern associated with a second variation in the GJB6 gene that have been described previously [14, 22]. However, digenic GJB2/GJB6 hereditary hearing loss had been excluded in the patient as described previously [12].
Identification of the homozygous known pathogenic transversion c.5492G>T in Myo15a as the underlying causative variation highlights the importance of involving deaf individuals with heterozygous GJB2 variations in screening for other causative genes. Extending screening for c.5492G>T to a cohort of index patients from 24 families of Turkish descent with congenital HL resulted in a genetic diagnosis in an additional family.
Myosins form a large family of ATP-dependent motor proteins that interact with actin and are essential for a plethora of cellular motility functions. Myosin 15a is a large protein of 3530 amino acids encoded by 66 exons and containing a 1233-residue proline-rich N-terminal domain, an ATPase motor domain, a neck domain with two IQ motifs and a long (1587 amino acids) tail with two myosin tail homology 4 (MyTH4) domains, two band 4.1/ezrin/radixin/moesin (FERM) domains, an Src-homology-3 (SH3) domain and a C-terminal PDZ class I ligand [23, 24].
Myosin 15a is expressed in cochlear outer and inner hair cells and accumulates in the tips of stereocilia. It has been shown to be essential for delivering compounds to the tips that are required for stereocilia elongation during development and for stereocilia maintenance [24,25,26]. In mice, a variation in the motor domain of Myosin 15a causes the deaf-circling shaker 2 (sh2) phenotype [27] and in humans, variations in Myo15a are the cause for DFNB3 type deafness [28]. All known pathogenic variations in Myo15a cause prelingual HL [29].
The missense transversion in exon 22 at c.5492G>T (p.Gly1831Val) replaces a highly conserved glycine residue in the Myosin 15a motor domain with valine. Molecular modeling has previously predicted that p.Gly1831Val may inhibit the ability of Myosin 15a to perform the power stroke, which is essential for myosin movement along actin filaments [21].
The Myo15a c.5492G>T variant identified is a very rare missense variation so far only identified in a single family with congenital HL in Turkey [21] and is not listed in over 277,000 alleles in the gnomAD database. Our results confirm the pathogenicity of this variant. All three individuals with the homozygous c.5492G>T variation identified in this study and the family described previously show a similar phenotype with profound congenital HL, indicating that this variation has a high penetrance. Families TAR1 and TAR6 had no obvious blood-relationship and none of the family members recalled any positive family history of ancestors that suffered from HL. The occurrence of a homozygous single nucleotide change in three unrelated families can be explained by traditional consanguineous mating in the present population and is most likely the result of a founder allele from a common ancestor. Since the c.5492G>T variation was not present in any of the other 23 families tested in this study, it is likely to play a minor role in the pathogenesis of HL in the general Turkish population.
WES allowed a rapid and cost-effective diagnosis in family TAR1. Traditional homozygosity mapping and candidate gene sequencing would have been a very laborious and costly approach due to the large size of the Myo15a gene encoded by 66 exons and would have been of uncertain success due to the small number of available individuals in the index family. The strength of WES in comprehensive screening of large genes and rapid diagnosis even in small families is, therefore, underlined by our results.
Conclusion
WES and subsequent screening for a known Myo15a c.5492G>T (p.Gly1831Val) missense variant provided a genetic diagnosis in 2 of 25 Turkish families with congenital autosomal recessive NSHL. This is the first study to apply WES to genetic deafness screening in Austria. The strength of WES in diagnosing causative variations even in small families and the feasibility to include WES in routine genetic deafness testing in Austria is illustrated by these results. The importance of including patients with heterozygous pathogenic GJB2 variations in further genetic testing pipelines is also emphasized by this study.
References
Morton NE (1991) Genetic epidemiology of hearing impairment. Ann N Y Acad Sci 630:16–31
Morton CC, Nance WE (2006) Newborn hearing screening–a silent revolution. N Engl J Med 354:2151–2164
Smith RJH, Shearer AE, Hildebrand MS, Van Camp G (1993–2016) Deafness and hereditary hearing loss overview. In: Pagon RA, Adam MP, Ardinger HH, Wallace SE, Amemiya A, Bean LJH et al (eds) GeneReviews, Seattle (WA)
Wingard JC, Zhao HB (2015) Cellular and deafness mechanisms underlying connexin mutation-induced hearing loss—a common hereditary deafness. Front Cell Neurosci 9:202
Kelsell DP, Dunlop J, Stevens HP, Lench NJ, Liang JN, Parry G et al (1997) Connexin 26 mutations in hereditary non-syndromic sensorineural deafness. Nature 387:80–83
Frei K, Ramsebner R, Lucas T, Hamader G, Szuhai K, Weipoltshammer K et al (2005) GJB2 mutations in hearing impairment: identification of a broad clinical spectrum for improved genetic counseling. Laryngoscope 115:461–465
Wolf A, Frohne A, Allen M, Parzefall T, Koenighofer M, Schreiner MM et al (2017) A novel mutation in SLC26A4 causes nonsyndromic autosomal recessive hearing impairment. Otol Neurotol 38(2):173–179
Frei K, Szuhai K, Lucas T, Weipoltshammer K, Schofer C, Ramsebner R et al (2002) Connexin 26 mutations in cases of sensorineural deafness in eastern Austria. Eur J Hum Genet 10:427–432
Ramsebner R, Ludwig M, Lucas T, de Jong D, Hamader G, del Castillo I et al (2013) Identification of a SNP in a regulatory region of GJB2 associated with idiopathic nonsyndromic autosomal recessive hearing loss in a multicenter study. Otol Neurotol 34:650–656
Koenighofer M, Lucas T, Parzefall T, Ramsebner R, Schoefer C, Frei K (2015) The promoter mutation c.-259C>T (-3438C>T) is not a common cause of non-syndromic hearing impairment in Austria. Eur Arch Otorhinolaryngol 272:229–232
Parzefall T, Lucas T, Koenighofer M, Ramsebner R, Frohne A, Czeiger S et al (2017) The role of alternative GJB2 transcription in screening for neonatal sensorineural deafness in Austria. Acta Otolaryngol 137(4):356–360
Frei K, Ramsebner R, Lucas T, Baumgartner WD, Schoefer C, Wachtler FJ et al (2004) Screening for monogenetic del(GJB6-D13S1830) and digenic del(GJB6-D13S1830)/GJB2 patterns of inheritance in deaf individuals from Eastern Austria. Hear Res 196:115–118
Van Camp G, Smith RJH (2016) Hereditary hearing loss homepage. hereditaryhearingloss.org
Del Castillo I, Moreno-Pelayo MA, Del Castillo FJ, Brownstein Z, Marlin S, Adina Q et al (2003) Prevalence and evolutionary origins of the del(GJB6-D13S1830) mutation in the DFNB1 locus in hearing-impaired subjects: a multicenter study. Am J Hum Genet 73:1452–1458
Shearer AE, DeLuca AP, Hildebrand MS, Taylor KR, Gurrola J 2nd, Scherer S et al (2010) Comprehensive genetic testing for hereditary hearing loss using massively parallel sequencing. Proc Natl Acad Sci USA 107:21104–21109
Yang Y, Muzny DM, Reid JG, Bainbridge MN, Willis A, Ward PA et al (2013) Clinical whole-exome sequencing for the diagnosis of mendelian disorders. N Engl J Med 369:1502–1511
Stephens D (1996) EU Working Group on Genetics of Hearing Impairment, European Commission Directorate, Biomedical and Health Research Programme Hereditary Deafness, Epidemiology and Clinical Research (HEAR). EU Work Group, pp 8–9
Li H, Durbin R (2009) Fast and accurate short read alignment with Burrows–Wheeler transform. Bioinformatics 25:1754–1760
McKenna A, Hanna M, Banks E, Sivachenko A, Cibulskis K, Kernytsky A et al (2010) The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res 20:1297–1303
Lek M, Karczewski KJ, Minikel EV, Samocha KE, Banks E, Fennell T et al (2016) Analysis of protein-coding genetic variation in 60,706 humans. Nature 536:285–291
Kalay E, Uzumcu A, Krieger E, Caylan R, Uyguner O, Ulubil-Emiroglu M et al (2007) MYO15A (DFNB3) mutations in Turkish hearing loss families and functional modeling of a novel motor domain mutation. Am J Med Genet A 143A:2382–2389
del Castillo I, Villamar M, Moreno-Pelayo MA, del Castillo FJ, Alvarez A, Telleria D et al (2002) A deletion involving the connexin 30 gene in nonsyndromic hearing impairment. N Engl J Med 346:243–249
Belyantseva IA, Boger ET, Naz S, Frolenkov GI, Sellers JR, Ahmed ZM et al (2005) Myosin-XVa is required for tip localization of whirlin and differential elongation of hair-cell stereocilia. Nat Cell Biol 7:148–156
Liang Y, Wang A, Belyantseva IA, Anderson DW, Probst FJ, Barber TD et al (1999) Characterization of the human and mouse unconventional myosin XV genes responsible for hereditary deafness DFNB3 and shaker 2. Genomics 61:243–258
Belyantseva IA, Boger ET, Friedman TB (2003) Myosin XVa localizes to the tips of inner ear sensory cell stereocilia and is essential for staircase formation of the hair bundle. Proc Natl Acad Sci USA 100:13958–13963
Anderson DW, Probst FJ, Belyantseva IA, Fridell RA, Beyer L, Martin DM et al (2000) The motor and tail regions of myosin XV are critical for normal structure and function of auditory and vestibular hair cells. Hum Mol Genet 9:1729–1738
Probst FJ, Fridell RA, Raphael Y, Saunders TL, Wang A, Liang Y et al (1998) Correction of deafness in shaker-2 mice by an unconventional myosin in a BAC transgene. Science 280:1444–1447
Wang A, Liang Y, Fridell RA, Probst FJ, Wilcox ER, Touchman JW et al (1998) Association of unconventional myosin MYO15 mutations with human nonsyndromic deafness DFNB3. Science 280:1447–1451
Rehman AU, Bird JE, Faridi R, Shahzad M, Shah S, Lee K et al (2016) Mutational spectrum of MYO15A and the molecular mechanisms of DFNB3 human deafness. Hum Mutat 37:991–1003
Acknowledgements
Open access funding provided by Medical University of Vienna. The authors would like to cordially thank all the patients participating in this study. We greatly appreciate the help of Dr. Michael Schuster from the Center for Molecular Medicine (CeMM) of the Austrian Academy of Sciences for advice on data management.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Funding
This study was funded by the MedEl Corporation, Innsbruck, Austria and by funds of the Oesterreichische Nationalbank (Anniversary Fund, project number AP14058ONB).
Conflict of interest
K.F. received funding from the Oesterreichische Nationalbank (Anniversary Fund, project number AP14058ONB). A.F. receives research assistance from the MedEl Corporation, Innsbruck, Austria. Neither funding entities had an influence on the study design, data acquisition or interpretation and were not involved in the writing of the manuscript or the decision to publish this manuscript. All authors declare that they have no conflict of interest.
Ethical approval
All procedures performed in studies involving human participants were in accordance with the ethical standards of the institutional and/or national research committee (Ethics Committee of the Medical University of Vienna; approval number ECS 198/2004, with annual extensions to date) and with the 1964 Helsinki declaration and its later amendments or comparable ethical standards. This article does not contain any studies with animals performed by any of the authors.
Informed consent
Informed consent was obtained from all individual participants included in the study and from parents of minors.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Parzefall, T., Frohne, A., Koenighofer, M. et al. Whole-exome sequencing to identify the cause of congenital sensorineural hearing loss in carriers of a heterozygous GJB2 mutation. Eur Arch Otorhinolaryngol 274, 3619–3625 (2017). https://doi.org/10.1007/s00405-017-4699-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00405-017-4699-0