
Abstract
Kikuchi–Fujimoto disease (KFD), also known as histiocytic necrotizing lymphadenitis, is a benign and self-limiting disease typically characterized by the enlargement of regional lymph nodes and accompanied by fever. KFD affects predominantly young adult females of Asian origin and is rarely seen in European countries, where it may cause diagnostic difficulties. Kimura disease is a rare and benign chronic inflammatory soft tissue disorder of unknown origin, characterized by a triad of painless subcutaneous masses in the head or neck region accompanied by regional lymphadenopathy, blood and tissue eosinophilia, and markedly elevated serum immunoglobulin E levels. Although most cases of Kimura disease have originated in China, Japan or Southeast Asia, there have been sporadic case reports from Europe and America. Herein, we review in detail the clinical presentations, complications and current concepts in the pathogenesis, diagnosis and treatment of these diseases.
Similar content being viewed by others


Introduction
Causes of neck lymphadenopathy include a wide variety of disorders from infectious diseases through autoimmune and allergic diseases, and include benign and malignant neoplasms. In most cases, cervical lymph node enlargement is associated with reactive changes in response to bacterial, viral, chlamydial or mycotic pathogens, or with the presence of primary haematological malignancies such as Hodgkin’s disease and non-Hodgkin’s lymphomas.
There are, however, some clinical entities associated with cervical lymphadenopathy that are very rarely seen, particularly in European countries, where they may cause substantial diagnostic difficulties. We present two of those disorders; Kikuchi–Fujimoto disease (KFD) and Kimura’s disease. Both are benign in clinical course but may mimic aggressive malignant conditions and, when misdiagnosed, may lead to potentially harmful and unnecessary evaluations and treatments of patients.
Kikuchi–Fujimoto disease
Definition
Kikuchi–Fujimoto disease (KFD) is a histiocytic, necrotizing lymphadenitis that was described for the first time in Japan almost simultaneously by Kikuchi and Fujimoto in 1972 [1–4]. This is a benign and self-limiting disease characterized by tenderness of regional, predominantly cervical, lymph nodes and accompanied by mild fever, night sweats and flu-like symptoms [3, 4].
Epidemiology
Although KFD has been reported worldwide, it is most commonly seen in Asia; some of the occasionally reported cases in Europe and the United States involve patients of Asian descent [3–7]. The higher prevalence of this disease among Japanese and other Asiatic individuals may be due to HLA class II genes, such as the DPA1*01 allele and DPB1*0202 allele, which are relatively common among Asians [8–12]. The first documented cases of KFD outside of Japan were described by Pileri et al. [13] in 1982, depicting 27 patients in Germany, Iran, Italy, Korea, and Spain. Another large non-Japanese series of 108 patients was reported from the US by Dorfman et al. [14] in 1988. The first Scandinavian case was published in 1994 by Hussein and Hellquist [15]. KFD affects predominantly adults younger than 40 years of age and is less common in the paediatric age group. A striking female preponderance of cases has been emphasised in the literature in the past (female to male ratio 3–4:1) [16], but more recent studies seem to indicate that the actual ratio is closer to 1–2:1 [17, 18]. KFD in children has a different gender predominance compared with the adult population, showing a preference for boys (female to male ratio 1:1.9) [3, 18].
Due to the rarity of KFD, its true incidence in the Western world is difficult to establish, but it has been implicated in 6% of all pathologically abnormal lymph nodes [17, 18].
Aetiology and pathogenesis
The aetiology of KFD is still unknown. Various infectious agents, including Toxoplasma gondii, Yersinia enterocolitica, human herpes virus 6 and 8, hepatitis B virus, parvovirus B19 and Epstein–Barr virus (EBV) have been postulated to be involved, but so far the specific pathogenic factor has not been definitively identified. Recently, a high frequency of localized persistent parvovirus B19 (B19) in lymph nodes from KFD patients was shown; however, the detailed pathogenic mechanisms governing B19 and KFD are yet to be elucidated [19]. Some authors emphasise the role of immunological mechanisms involved in the pathogenesis of KFD and consider this disease as a rare manifestation of systemic lupus erythematosus (SLE). Previous literature on KFD frequently addressed the link between KFD and SLE, and the reported rate was 1.3–7% in the population of KFD patients [20–24]. Numerous studies have also shown an association between KFD and a wide spectrum of other autoimmune diseases, including Hashimoto’s thyroiditis, polymyositis, mixed connective tissue disease, Still’s disease, autoimmune hepatitis, and antiphospholipid syndrome [25–31]. It has also been suggested that specific necrotizing lesions in KFD may in some cases be due to a vigorous immune response to EBV-infected lymphoid cells [17]. Cases of KFD associated with implanted pacemakers or leaking silicone breast implants as sources of agents triggering hyperimmune reactions have also been published [5, 6, 32, 33]. In the literature, other reported KFD-associated disorders include meningitis [25, 34], breast cancer [35], and buccal cancer [3]. There have been also speculations that KFD may be a manifestation of an early-stage T-cell lymphoma in evolution, and recently, one case of KFD displaying a t (2:16) chromosomal translocation has even been published [11]. However, studies performed by Lin et al. [36] in 56 consecutive KFD cases showed the presence of an oligoclonal pattern of T-cell antigen receptor rearrangement, excluding a possibility of T-cell clonal malignancy and indicating a benign immune reaction. All of those reports may lend further support to the hypothesis that KFD represents an exuberant T cell-mediated immune response to a variety of non-specific stimuli [37, 38].
Clinical features, treatment and prognosis
The onset of KFD can be acute or sub-acute, developing over a period of 2–3 weeks [3–6, 12]. The most commonly involved site is that associated with the cervical lymph nodes (56–98%). The lymph nodes are most often tender, involve the posterior cervical triangle (88.5%) and are generally unilateral. The dimensions of the affected lymph nodes are commonly in the range of 0.5–4 cm, but occasionally lymph nodes may be larger than 8 cm in diameter. In only a few cases has lymphadenopathy been generalized or involved the mediastinal, peritoneal and retroperitoneal regions. Sometimes, patients with KFD are referred with pain, indurations or nodal adherence to surrounding tissue [5, 6]. Occasionally, the sole symptom of the disease is a fever of unknown origin without other associated clinical features [39, 40].
In addition to lymphadenopathy and fever, a variable percentage of patients, ranging from 30 to 50%, show extranodal involvement with upper respiratory symptoms, general malaise, headache, mouth ulcers, hepatosplenomegaly, arthralgias and arthritis [41]. Less common are weight loss, nausea, vomiting, sore throat and night sweats. A wide variety of dermatological lesions have also been observed [42–45]. These lesions usually show histopathological findings described as interface dermatitis and may be a marker of evolution into SLE [21].
KFD is usually a benign and self-limiting disease lasting from 1 to 4 months. Recurrence is low, being described in only 3–4% of cases, and may occur as late as 8–9 years; in one case, recurrence was reported 19 years, after the initial diagnosis [13].
Despite the benign nature of KFD, a fair proportion of patients may experience a worsening in their clinical condition and even a poor outcome. Reported causes of death have included pulmonary haemorrhage, fatal hemophagocytic syndrome, heart failure as well as graft failure following renal, liver and pancreatic transplants [41, 46, 47].
The treatment of KFD is thus symptomatic, with non-steroidal anti-inflammatory drugs used to relieve distressing local and systemic complaints. In more complicated cases, such as patients with recurrent episodes or prolonged symptoms, more aggressive management with systemic steroids is required. Hydroxychloroquine has also shown some benefit in treating this disease. Antibiotics have not been found to be of any benefit [3, 6, 13].
Diagnosis: laboratory and histological features
The results of a wide range of laboratory tests are usually normal in patients with KFD. Patients may have mild anaemia, elevated erythrocyte sedimentation rate (ESR) and increased C-reactive protein (CRP) levels. Leucopoenia with the presence of atypical lymphocytes has been mentioned in 16.6–58.3% of KFD patients [3–5]; hyperleucocytosis, however, has been more rarely observed [4, 5].
Serum concentrations of aminotransferases and of LDH are increased in some patients with KFD [48]. Bacteriological and mycological analyses of blood and urine show negative results. Serology titres for CMV, EBV, adenovirus, parvovirus B19, Coxsackie virus, HIV, Borrelia, Mycoplasma, Toxoplasma gondii, Yersinia enterocolitica, Brucella and Bartonella do not indicate any ongoing infection. Immunoglobulin levels, peripheral blood lymphocyte subpopulation analyses, and evaluation of autoimmune conditions including antiphospholipid and antinuclear antibodies, rheumatoid factor, anti-neutrophil cytoplasmic antibodies and complement levels are usually also normal [12, 19, 48, 49].
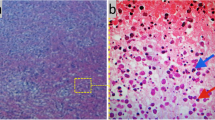
The definite diagnosis of KD can be made reliably only via histopathological study from an open biopsy of the affected lymph nodes. Classic pathological findings in this disease include patchy areas of coagulative necrosis in the cortical and para-cortical areas of enlarged lymph nodes, together with abundant karyorrhectic debris (Fig. 1a). Neutrophils, eosinophils or plasma cells are sparse or totally absent [5, 6, 50, 51]. Cellular infiltration consists of CD68+/CD123+ plasmacytoid histiocytes and transformed lymphocytes (immunoblasts), predominantly of T-cell origin with a predominance of CD8+ over CD4+ T cells (Fig. 1b, c). These T lymphocytes abundantly express perforin, a killer cell-specific cytolytic protein essential for provoking apoptosis in target cells within necrotic areas [52]. In KFD, 25–75% of CD68+ histiocytes co-express myeloperoxidase (MPO) [53]. MPO+/CD68+ blood monocytes may be attracted into lymph nodes because of the lack or paucity of granulocytes and the need of MPO for oxidative processes associated mainly with phagocytosis. The CD68+/CD123+/MPO-negative population of plasmacytoid histiocytes producing type I interferons may, however, be involved in the cytotoxic immune reactions rather than in phagocytic phenomena [51, 53]. Some ultrastructural studies have revealed tubuloreticular structures in the histiocytes, activated T cells, and endothelial cells similar to those usually found in SLE and in viral diseases. The presence of such intracytoplasmic inclusions seems to support the hypothesis that KFD may, in a proportion of cases, reflect a self-limiting SLE-like autoimmune condition induced by virus-infected transformed lymphocytes [19, 49].

a Kikuchi–Fujimoto disease. Extensive areas of coagulative necrosis without granulocytic infiltration in a lymph node. b Kikuchi–Fujimoto disease. Immunohistochemical reactions for CD68 antigen in numerous scattered histiocytes. c Kikuchi–Fujimoto disease. CD8-positive lymphocytes situated on the periphery of necrosis
Differential diagnosis
Clinically and histologically, KFD can be mistaken for SLE or, more importantly, for malignant lymphoma (Hodgkin’s disease, non-Hodgkin’s lymphoma) [54]. Some of these initially misdiagnosed patients have been unnecessarily treated with chemotherapy. Therefore, KFD should be taken into consideration in the diagnostic process in all patients with persistent lymphadenopathy and prolonged fever because misdiagnosis may lead to unnecessary surgery and/or chemotherapy [5–7]. Other conditions that should be excluded in the diagnostic process are: tuberculosis, sarcoidosis, toxoplasmosis, metastatic carcinoma, cat scratch disease, Still’s disease, infectious mononucleosis, AIDS and angioimmunoblastic lymphadenopathy [26–29, 54, 55].
Kimura disease
Definition
Kimura disease is a rare and benign chronic inflammatory soft tissue disorder of unknown origin. The disease was first described in 1937 in the Chinese literature by HT Kimm and C Szeto and termed “eosinophilic hyperplastic lymphogranuloma”. The definitive histological description was published by Kimura et al. in 1948 and thus, the disease has borne his name. Since that time, there has been a gradual increase in the number of reports (as of 1999, about 200 reported cases) [56].
Epidemiology
While most cases of Kimura disease have originated in China, Japan or Southeast Asia, there have been sporadic case reports from Europe and America. The disease is most prevalent in Asians, uncommon in Caucasians, and rare in Africans. It has been suggested that the common factor is a degree of Asian ancestry. There is a marked male predominance, with a male/female sex ratio of 3.5:7.1. The peak age of onset is during the third decade [57, 58].
Aetiology and pathogenesis
The aetiology of Kimura’s disease is not understood at this time, but it may be related to a disturbance in the normal rate of production of eosinophils and IgE, currently believed to be the product of an interaction between type 1 and 2 T-helper cells [58].
Such an imbalance could result in the excessive production of eosinophilotrophic cytokines such as interleukin 4. Patients with Kimura’s disease have been shown to have high levels of circulating eosinophil cationic proteins and major basic proteins, with heavy concentrations of IgE in their tissues. Proposed theories include persistent antigenic stimulation following arthropod bites, and parasitic, candidal (especially Candida albicans) or viral infections. To date, none of these theories have been substantiated [58, 59].
Clinical features and prognosis
The nature of Kimura’s disease is mostly benign, with a good prognosis. The importance lies in its ability to mimic a number of other benign inflammatory and neoplastic conditions of the head and neck. Kimura’s disease is a chronic inflammatory condition characterized by a triad of:
-
painless subcutaneous masses in the head or neck region, accompanied by regional lymphadenopathy;
-
blood and tissue eosinophilia;
-
markedly elevated serum immunoglobulin E levels.
Clinically, subcutaneous soft tissue masses occur predominantly in the head and neck region and often involve the parotid, submandibular or minor salivary glands. Less frequently, the eyelids, orbit and lachrymal glands may be involved, as well as the hard palate and larynx. Sperm ducts and nerves are rarely affected [59, 60].
The clinical course of Kimura’s disease is usually benign. These observed subcutaneous masses, when left untreated, tend to slowly enlarge and may eventually become disfiguring.
Patients may complain of local or generalized pruritus and sub-acute or chronic dermatitis. Renal involvement, usually extramembranous glomerulonephritis, is found in about half of patients. They may develop isolated proteinuria (about 5% of patents), or demonstrate the full nephrotic syndrome (about 12% of patents) [59, 60].
Approximately 67–100% of these patients develop regional lymphadenopathy, mainly of the cervical lymph nodes. Infrequently, axillary, inguinal or epitrochlear nodes may be affected; in longstanding disease, this lymphadenopathy may become generalized [60, 61]. In the long term, patients seem to do well.
Diagnosis: laboratory and histological features
Laboratory investigations will invariably reveal peripheral eosinophilia and increased serum immunoglobulin E (IgE) levels. They may have elevated erythrocyte sedimentation rates (ESR) and mild hyperleucocytosis; however, hyperleucocytosis has been very rarely observed [58, 59]. In patients with Kimura’s disease eosinophilia is almost always present, while raised IgE is always the case (which can be helpful in the differential diagnosis vs. angiolymphoid hyperplasia with eosinophilia). The histopathological picture of Kimura’s disease is characterized by the presence of tight fibrosis, lymphocytic infiltration with follicle formation, and mixed inflammatory infiltration with eosinophils present. Some authors describe the changes as a component of three elements: cellular, fibrocollagenous and vascular. The cellular element of forming follicles is built mainly from lymphocytes. The fibrocollagenous part is combined from eosinophilic cells and eosinophilic “micro abscesses” (Fig. 2a). Giant cells and plasma cells are also present but in small amounts. Fibrosis is also present, even the in initial period. The vascular component consists of the proliferation of enlarged endothelial cells. There are no atypical cell nuclei and enlarged cytoplasm. In enlarged regional lymph nodes, follicular hypertrophy with increased amounts of eosinophils, with or without fibrosis, is affirmed (Fig. 2b). The deposition of IgE in the germinal centers can be readily demonstrated with immunohistochemistry. The lymph node architecture is preserved [56].

a Kimura disease. Parotid gland. Dense eosinophilic infiltrations forming “micro abscesses”. b Kimura disease. Hypertrophic follicle with germinal centre in an enlarged lymph node
Pathologically, Kimura’s disease is most difficult to distinguish from angiolymphoid hyperplasia with eosinophilia (ALHE) with which it has been confused in the past since these two conditions were considered to represent the same disease process, but the current consensus is that they represent two ends of a spectrum of similar diseases [57–60, 62]. ALHE is characterized clinically by single to multiple dome-shaped erythematous or hyperpigmented papules or subcutaneous nodules located mainly in the head and neck. In some cases the nodules extend to the dermis or into the muscle. About 1/5 of patients have blood eosinophilia and lymphadenopathy. In contrast to Kimura’s disease, the histology of ALHE is characterized by a prominent proliferation of small, capillary-sized blood vessels with marked irregularity of luminal sizes, often surrounding a muscular artery. The vessels are lined by plump, epithelioid (histiocytoid) endothelial cells. An inflammatory cell infiltrate with numerous eosinophils, mast cells and lymphocytes is present, though the numbers of eosinophils may vary considerably from case to case. Immunohistochemical stains usually show a major population of T lymphocytes with occasional B cells forming lymphoid follicles [62].
Differential diagnosis
The differential diagnosis, while including obvious lesions such as dermatofibrosarcoma protuberans and cylindroma, will ultimately be determined by both the clinical picture and the histopathology. Clinically, malignant lymphoma, parotid tumours, haemangioma, pyogenic granuloma, Mikulicz’s disease and Kikuchi’s disease are all conditions for which Kimura’s disease has been mistaken in the past. Apart from ALHE which is the main differential diagnosis to Kimura’s disease, other conditions to consider include Kaposi’s sarcoma, angiosarcoma, nodal metastasis, reactive lymphadenopathy, eosinophilic lymphoma and angioimmunoblastic lymphadenopathy. Parasitic diseases responsible for subcutaneous masses with an associated lymphadenopathy, such as tissue-invasive helminth infections, toxocariasis, cysticercosis, sparganosis and several forms of invasive miasis may also need to be ruled out [7, 56–59, 62].
Therapy
Three major therapeutic options exist for Kimura’s disease. Resection of the tumour mass may be effective and permanently eradicate the mass if the entire lesion can be removed, but re-growth is common. Local irradiation has also been shown to be effective in shrinking lesions, but it is generally not advocated in younger patients. Systemic corticosteroids and immunosuppressive agents such as cyclosporine A have been used for the treatment of Kimura’s disease as well as for the accompanying nephrotic syndrome [58–63]. Finally, intralesional corticosteroids have been shown to reduce the size of the lesion, but the tumour tends to recur when these drugs are discontinued. In selected patients, it may be advisable to take a conservative approach, treating only if the mass continues to grow or causes significant deformity [59–63].
Conclusions
Kikuchi–Fujimoto and Kimura disease are rare but important causes of lymphadenopathy. KFD should be considered in the differential diagnosis of any patient presenting with unexplained lymphadenopathy associated with non-specific symptoms such as fever and weight loss. Consideration of the diagnosis is particularly important before prescribing potentially inappropriate drug therapy. In some cases, an autoimmune process, particularly SLE, may develop. Therefore, long-term follow-up should be mandatory for KFD patients. Knowledge of Kimura’s disease, its clinical appearance, course and histopathology puts the practitioner in a better position to answer questions from concerned patients and primary caregivers, and to optimize management strategies.
References
Kikuchi M (1972) Lymphadenitis showing focal reticulum cell hyperplasia with nuclear debris and phagocytes. Acta Hematol Jpn 35:379–380
Fujimoto Y, Kojima Y, Yamaguchi K (1972) Cervical subacute necrotizing lymphadenitis. Naika 30:920–927
Lin HC, Su CY, Huang CC, Hwang CF, Chien CY (2003) Kikuchi’s disease: a review and analysis of 61 cases. Otolaryngol Head Neck Surg 128:650–653
Lin HC, Su CY, Huang SC (2005) Kikuchi’s disease in Asian children. Pediatrics 115:92–96
Tsang WYW, Chan JKC, Ng CS (1994) Kikuchi’s lymphadenitis. A morphologic analysis of 75 cases with special reference to unusual features. Am J Surg Pathol 18:219–231
Turner RR, Martin J, Dorfman RF (1983) Necrotizing lymphadenitis. A study of 30 cases. Am J Surg Pathol 7:115–123
Park YW (1995) Evaluation of neck masses in children. Am Fam Physician 51:1904–1912
Kuo TT (1995) Kikuchi’s disease (histiocytic necrotizing lymphadenitis). A clinicopathologic study of 79 cases with an analysis of histologic subtypes, immunohistology, and DNA ploidy. Am J Surg Pathol 19:798–809
Kikuchi M, Takeshita M, Eimoto T (1990) Histiocytic necrotizing lymphadenitis: clinicopathologic, immunologic, and HLA typing study. In: Hanaoka M, Kadin ME, Mikata A (eds) Lymphoid malignancy: immunocytology and cytogenetics. Field and Wood Medical, New York, pp 251–257
Tanaka T, Ohmori M, Yasunaga S et al (1999) DNA typing of HLA class II genes (HLA-DR, -DQ and -DP) in Japanese patients with histiocytic necrotizing lymphadenitis (Kikuchi’s disease). Tissue Antigens 54:246–253
Robertson KE, Forsyth PD, Batstone PJ, Levison DA, Goodlad JR (2007) Kikuchi’s disease displaying a t (2:16) chromosomal translocation. J Clin Pathol 60(4):433–435
Lazzareschi I, Barone G, Ruggiero A, Liotti L, Maurizi P, Larocca LM, Riccardi R (2008) Paediatric Kikuchi–Fujimoto disease: a benign cause of fever and lymphadenopathy. Pediatr Blood Cancer 50(1):119–123
Pileri S, Kikuchi M, Helbron D, Lennert K (1982) Histiocytic necrotizing lymphadenitis without granulocytic infiltration. Virchows Arch A Pathol Anat Histol 395:257–271
Dorfman RF, Berry GJ (1988) Kikuchi’s histiocytic lymphadenitis: an analysis of 108 cases with emphasis on differential diagnosis. Semin Diagn Pathol 5:329–345
Hussein A, Hellquist HB (1994) Necrotizing lymphadenitis of the neck (Kikuchi’s disease). APMIS 102:633–637
Smith KG, Becker GJ, Busmanis I (1992) Recurrent Kikuchi’s disease. Lancet 340:124
Kucukardali Y, Solmazgul E, Kunter E, Oncul O, Yildirim S, Kaplan M (2007) Kikuchi–Fujimoto disease: analysis of 244 cases. Clin Rheumatol 26(1):50–54
Bosch X, Guilabert A, Miquel R et al (2004) Enigmatic Kikuchi–Fujimoto disease. A comprehensive review. Am J Clin Pathol 122:141–152
Zhang WP, Wang JH, Wang WQ, Chen XQ, Wang Z, Li YF, Hu PZ, Zhang W, Wang L, Wang D, Huang GS (2007) An association between parvovirus B19 and Kikuchi–Fujimoto disease. Viral Immunol 20(3):421–428
Martínez-Vázquez C, Hughes G, Bordon J et al (1997) Histiocytic necrotizing lymphadenitis, Kikuchi–Fujimoto’s disease, associated with systemic lupus erythematosus. Q J Med 90:531–533
Paradela S (2008) Interface dermatitis in skin lesions of Kikuchi–Fujimoto’s disease: a histopathological marker of evolution into systemic lupus erythematosus? Lupus 17(12):1127–1135
Vila LM, Mayor AM, Silvestrini IE (2001) Therapeutic response and long-term follow-up in a systemic lupus erythematosus patient presenting with Kikuchi’s disease. Lupus 10:126–128
Eisner MD, Amory J, Mullaney B, Tierney LJ, Browner WS (1996) Necrotizing lymphadenitis associated with systemic lupus erythematosus. Semin Arthritis Rheum 26:477–482
Quintas-Cardama A, Fraga M, Cozzi SN, Caparrini A, Maceiras F, Forteza J (2003) Fatal Kikuchi–Fujimoto disease: the lupus connection. Ann Hematol 82:186–188
Sato Y, Kuno H, Oizumi K (1999) Histiocytic necrotizing lymphadenitis (Kikuchi’s disease) with aseptic meningitis. J Neurol Sci 163:187–191
Graham LE (2002) Kikuchi–Fujimoto disease and peripheral: a first!. Ann Rheum Dis 61:475
Kawai H, Hasegawa M, Hagiwara S et al (1999) Kikuchi’s disease with leukocytoclastic vasculitis in a 10-year-old girl. Pediatr Int 41:323–326
Cousin F, Grezard P, Roth B et al (1999) Kikuchi disease associated with Still disease. Int J Dermatol 38:464–467
Wilkinson CE, Nichol F (2000) Kikuchi–Fujimoto disease associated with polymyositis. Rheumatology (Oxf) 39:1302–1304
Sarma OP (2001) Unusual systemic disorders associated with interstitial lung disease. Curr Pulm Med 7:291–294
Laeng RH, Stamm B (1994) Kikuchi’s histiocytic necrotizing lymphadenitis driven by activated cytolytic T-cells: an example associated with systemic scleroderma. Histopathology 34:373–374
Taguri AH, McIlwaine GG (2001) Bilateral panuveitis: a possible association with Kikuchi–Fujimoto disease. Am J Ophthalmol 132(3):419–421
Sever L, Leith P, Appenzeller J, Foucar K (1996) Kikuchi’s histiocytic necrotizing lymphadenitis associated with ruptured silicone breast implant. Arch Pathol Lab Med 120:380–385
Debley JS, Rozansky DJ, Miller ML, Katz BZ, Green ME (1996) Histiocytic necrotizing lymphadenitis with autoimmune phenomena and meningitis in a 14-year-old girl. Pediatrics 98:130–133
Agel NM, Peters EE (2000) Kikuchi’s disease in axillary lymph nodes draining breast carcinoma. Histopathology 30:280–281
Lin CW, Chang CL, Li CC, Chen YH, Lee WH, Hsu SM (2002) Spontaneous regression of Kikuchi lymphadenopathy with oligoclonal T-cell populations favors a benign immune reaction over a T-cell lymphoma. Am J Clin Pathol 117(4):627–635
Sierra ML, Vegas E, Blanco-Gonzalez JE et al (1999) Kikuchi’s disease with multisystemic involvement and adverse reaction to drugs. Pediatrics 104:124
Ganga A, Corda D, Gallo Carrabba G et al (1998) A case of carbamazepine-induced lymphadenopathy resembling Kikuchi disease. Eur Neurol 39:247–248
Chuang CH, Yan DC, Chiu CH et al (2005) Clinical and laboratory manifestations of Kikuchi’s disease in children and differences between patients with and without prolonged fever. Pediatr Infect Dis J 24:551–554
Scagni P, Peisino MG, Bianchi M et al (2005) Kikuchi–Fujimoto disease is a rare cause of lymphadenopathy and fever of unknown origin in children report of two cases and review of the literature. J Pediatr Hematol Oncol 27:337–340
Wong CY, Law GT, Shum TT (2001) Pulmonary hemorrhage in a patient with Kikuchi disease. Monaldi Arch Chest Dis 56:118–120
Yasukawa K, Matsumura T, Sato-Matsumura KC et al (2001) Kikuchi’s disease and the skin: case report and review of the literature. Br J Dermatol 144:885–889
Aquel N, Henry K, Woodrow D (1997) Skin involvement in Kikuchi’s disease: an immunocytochemical and immunofluorescence study. Virchows Arch 430:349–352
Kaur S, Thami GP, Mohan H (2001) Kikuchi disease with facial rash and multiforme erythema. Pediatr Dermatol 18:403–405
Kim KJ, Jee MS, Chang SE et al (2003) Kikuchi–Fujimoto disease with papulopustular skin manifestation. Clin Exp Dermatol 28:142–144
O’Neill D, O’Grady J, Variend S (1998) Child fatality associated with pathological features of histiocytic necrotizing lymphadenitis (Kikuchi–Fujimoto disease). Pediatr Pathol Lab Med 18:79–88
Chan JK, Wong KC, Ng CS (1989) A fatal case of multicentric Kikuchi’s histiocytic necrotizing lymphadenitis. Cancer 63:1856–1862
Samiyoushi Y, Kikuchi M, Minematu T (1994) Analysis of herpesvirus genomes in Kikuchi’s disease. Virchows Arch 424:437–440
Hudnall SD, Chen T, Amr S, Young KH, Henry K (2008) Detection of human herpesvirus DNA in Kikuchi–Fujimoto disease and reactive lymphoid hyperplasia. Int J Clin Exp Pathol 1(4):362–368
Tong TR, Chan OW, Lee KC (2001) Diagnosing Kikuchi disease on fine needle aspiration biopsy: a retrospective study of 44 cases diagnosed by cytology and 8 by histopathology. Acta Cytol 45:953–957
Na DG, Chung TS, Byun HS (1997) Kikuchi disease: CT and MR findings. AJNR Am J Neuroradiol 18:1729–1732
Takakuwa T, Ohnuma S, Koike J, Hoshikawa M, Koizumi H (1996) Involvement of cell-mediated killing in apoptosis in histiocytic necrotising lymphadenitis (Kikuchi–Fujimoto disease). Histopathology 28(1):41–48
Pileri SA, Facchetti F, Ascani S, Sabattini E, Poggi S, Piccioli M, Rondelli D, Vergoni F, Zinzani PL, Piccaluga PP, Falini B, Isaacson PG (2001) Myeloperoxidase expression by histiocytes in Kikuchi’s and Kikuchi-like lymphadenopathy. Am J Pathol 159(3):915–924
Chamulak GA, Brynes RK, Nathwani BN (1990) Kikuchi–Fujimoto disease mimicking malignant lymphoma. Am J Surg Pathol 14:514–552
Baumgartner BJ, Helling ER (2002) Kikuchi’s disease: a case report and review of the literature. Ear Nose Throat J 81:331–335
Borkowski P, Dziubek Z (2001) Lymphadenopathy in infections diseases. Postępy Nauk Medycznych 14(2):14–18
Thomas J, Jayachandran NV, Chandrasekhara PKS, Rajasekhar L, Narisimulu G (2008) Kimura’s disease—an unusual cause of lymphadenopathy in children. Clin Rheumatol 27:675–677
Khan A, Bakhshi GD, Patil KK, Borse HG, Govila AA, Bhandarkar LD (2002) Kimura’s disease: a clinical case. Bombay Hosp J 44(1):118–200
Larroche C, Bletry O (2005) Kimura’s disease. Orphanet Encyclopedia Feb, pp 1–3
Szporek B, Stęplewska K, Cieślik T (2006) Choroba Kimura—opis przypadku. Czas Stomatol 59(10):734–739
Hobeika C, Mohammed T-L, Johnson G (2005) Hanses K. Kimura’s disease. Case report and review of the literature. J Thorac Imaging 20(4):298–300
Henry K (2005) Angiolymphoid hyperplasia with eosinophilia. In: Barnes L, Eveson JW, Reichert P, Sidransky D (eds) World Health Organization classification of tumours. Pathology and genetics of head and neck tumours. IARC Press, Lyon, pp 338–340
Wang DY, Mao JH, Zhang Y, Zhao SA, Chen YF, Liu AM (2009) Kimura disease: a case report and review of the Chinese literature. Nephron Clin Pract 111:55–61
Acknowledgments
Authors thank Dr. Papla B. and Urbańczyk K. from Chair and Department of Pathomorphology, Collegium Medicum, Jagiellonian University Kraków and Dr. Stęplewska Katarzyna from Pathomorphology Department of Silesian Medical University for rendering the histological microscopic photographs.
Conflict of interest statement
Authors have no financial relationship with the organization that sponsored the research. The authors declare that there is no conflict of interest.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Mrówka-Kata, K., Kata, D., Kyrcz-Krzemień, S. et al. Kikuchi–Fujimoto and Kimura diseases: the selected, rare causes of neck lymphadenopathy. Eur Arch Otorhinolaryngol 267, 5–11 (2010). https://doi.org/10.1007/s00405-009-1120-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00405-009-1120-7