
Abstract
Keloid scars are often described as having an actively growing peripheral margin with a regressing centre. The aim of this study was to examine the possible heterogeneity within keloids and the involvement of different regions within and around keloid scars in the pathogenesis, using an in vitro keloid scar model. In vitro skin models were constructed from keratinocytes and fibroblasts from normal skin and different regions within and around keloid scars: periphery, centre, and (adjacent) surrounding-normal-skin regions. Additionally, fibroblasts were isolated from the superficial-central and deep-central regions of the keloid and combined with central keratinocytes. All keloid regions showed increased contraction compared to normal skin models, particularly in central regions. Myofibroblasts were present in all keloid regions but were more abundant in models containing central-deep keloid fibroblasts. Secretion of anti-fibrotic HGF and extracellular matrix collagen IV gene expression was reduced in the central deep keloid compared to normal skin. No significant differences between peripheral and central regions within keloids were observed for inflammatory cytokine CCL20, CCL27, CXCL8, IL-6 and IL-18 secretion. Parameters for surrounding-normal-skin showed similarities to both non-lesional normal skin and keloids. In conclusion, a simple but elegant method of culturing keloid-derived keratinocytes and fibroblasts in an organotypic 3D scar model was developed, for the dual purpose of studying the underlying pathology and ultimately testing new therapeutics. In this study, these tissue engineered scar models show that the central keloid region shows a more aggressive keloid scar phenotype than the periphery and that the surrounding-normal-skin also shares certain abnormalities characteristic for keloids.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Keloid formation is an unfortunate complication of wound healing in which raised scar tissue proliferates beyond the boundaries of the original lesion [22]. This type of excessive scar tissue develops as an abnormal wound healing response to cutaneous injury [10, 22, 23]. Research on its pathogenesis has yet to uncover the etiology behind keloid formation and consequently our understanding of the mechanisms responsible for keloid development is limited. This is clearly illustrated by the inability of current treatment methods to satisfactorily manage keloids [3, 22]. As keloids develop exclusively in humans [29, 35] and research on their pathogenesis cannot be conducted solely on an intact original specimen, the need for a life-like in vitro model is evident.
Recently, we have demonstrated that keratinocytes and fibroblasts derived from human scars can be used to construct a full thickness skin model in vitro which shows resemblance to the native scar [16]. The keloid scar model shared several abnormalities with the hypertrophic scar model, but more importantly, differences were identified between these two abnormal scar types (hypertrophic scar and keloid). For the construction of these scar models, the scar tissue was used in its entirety. However, clinical observations suggest that keloids are not simply homogenous outgrowths. The distinction most often employed is that between the periphery and the centre of a keloid. The peripheral margin of the keloid is often described as being elevated, more red in color, actively proliferating and invading the surrounding normal skin; while the central region is seen as less elevated, lighter toned and clinically regressive over time [9, 13, 20, 24]. Differences have also been reported between keloid derived fibroblasts from peripheral or central regions when cultured in vitro with respect to lipid membrane composition [19], expression of apoptosis and extracellular matrix (ECM) related gene [24], collagen production [27], growth characteristics [20, 28], cell cycle distribution & regulation [8, 28, 30] and apoptosis-related protein expression [13, 20]. While the majority of published studies support the notion of an active periphery and a more quiescent centre, the opposite has also been reported with the central region thought to be the driving force behind keloid formation [28, 30]. Regardless, both concepts suggest that heterogeneity probably exists within a keloid scar, a finding that should not be ignored by those studying the mechanisms responsible for keloid formation.
As keloids are defined by their invasive growth into adjacent normal skin, it seems likely that the normal skin directly adjacent to keloids may in fact not truly be ‘normal’ and could, therefore, also play a role in keloid pathogenesis. Increased erythema in the normal skin directly adjacent to the keloid scars is often observed, and in a perfusion imaging study, blood flow in keloids and adjacent skin was indeed significantly higher than in nonadjacent normal skin [18]. Itching has also been reported to extend to peri-keloidal normal-appearing skin [14]. Taken together, these clinical observations suggest the skin directly adjacent to keloid scars may also be involved in keloid scar formation.
Given the aforementioned differences within keloid scars and the possible involvement of surrounding skin, we suspect that these different regions may differentially contribute to keloid scar formation. However, to our knowledge this has not yet been studied in a human, in vitro 3D scar model. In this study, we present an in vitro keloid scar model in which the different regions within and around the keloid scar can be studied and compared to unaffected normal skin to gain insight into keloid scar formation. The models serve a dual purpose of studying the underlying pathology and ultimately testing new therapeutics. To this end, the constructed in vitro models were compared to non-lesional skin with respect to the following scar parameters: contraction, epidermal and dermal thickness, expression of epidermal and dermal cell markers (Ki67, keratin 10, involucrin, vimentin, α-SMA), ECM gene expression and wound healing mediator secretion.
Materials and methods
Normal skin (Nskin) was obtained from patients undergoing body contouring surgery to remove excess skin. Keloid scars (Kscar) were obtained from patients undergoing scar removal via excision and were selected by an experienced scar expert (plastic surgeon, author FBN). All scars used were at least 1 year old and had matured (with exception of one keloid donor: 6 months old). See Table 1 for donor characteristics.
Cell culture and construction of skin models
After removal of subcutaneous fat and any other soft tissue until the typical firm and rubbery keloid consistency was reached, keloids were further subdivided into peripheral (P-Kscar), central superficial (Cs-Kscar) and central deep (Cd-Kscar) regions (Fig. 1a–c). Dermal tissue until ± 0.5 cm depth was included for cell isolation, Cs-Kscar and Cd-Kscar samples were obtained from the upper and lower central half, respectively. If present, any extralesional normal skin directly adjacent to the keloid (sNskin) was also included. sNskin extended to approximately ± 0.5 cm beyond P-Kscar (see area between edge of keloid and dotted line in Fig. 1a, b). In contrast to sNskin which was always derived from keloid patients, normal skin (Nskin) was obtained from unaffected control subjects. Keratinocytes and fibroblasts were isolated and cultured essentially as described previously [32, 34]. Skin models were constructed in duplicate from keratinocytes (P2) and fibroblasts (P2-3) essentially as previously described [32] 21 (Fig. 1d). In brief, 4 × 105 Fibroblasts were seeded onto 2.2 × 2.2 cm squares of MatriDerm® (dr. Suwelack Skin & Health Care, Billerbeck, Germany) with FSM-I and cultured submerged in FSM-II for 3 weeks in 0.4 µm pore size transwells (Costar Corning Inc., New York, NY, USA) in a 37 °C, 5% CO2 atmosphere. Keratinocytes (P2) were then seeded on top of the fibroblast-populated MatriDerm® and cultured submerged in KC-I for 3–4 days, prior to culturing at an air–liquid interface in deep-well plates (BD Biosciences, Bedford, MA, USA) in KC-II for an additional 10 days. Upon addition of keratinocytes, SE were cultured in a 37 °C, 7.5% CO2 atmosphere. Medium was changed twice a week. See supplementary table 1 for contents of culture media (KC-I, KC-II, FSM-I, and FSM-II) used.

Construction of skin models. a, b Keloids are dissected into: peripheral region (P), the central superficial (Cs) and central deep regions (Cd); the normal skin directly adjacent to the keloid periphery (sN: surrounding normal skin). Keratinocytes and fibroblasts are isolated from each region (circles indicate where biopsies were taken for cell isolation) and combined to form a peripheral keloid (P-Kscar) model, central superficial keloid (Cs-Kscar) model, central deep keloid (Cd-Kscar) model, surrounding normal skin (sNskin) model (c). Skin models are constructed by first seeding fibroblasts into Matriderm® (d). After 3 weeks, keratinocytes are added and the skin models are then cultured air-exposed for an additional 2 weeks
Wound contraction
Wound contraction is expressed as a reduction in surface area of the skin models at the end of the culture period. Surface area (mm2) was determined in photographs of skin models at the time of harvesting (5 weeks culture), using NIS-Elements AR 2.10 imaging software.
Histological analysis
Paraffin tissue sections (5 µm) were stained with Haematoxylin & Eosin (HE) for histologic evaluation and determination of epidermal and dermal thickness. Epidermal thickness quantified by counting the number of keratinocyte cell layers at three random points in each skin model section (200× magnification). Dermal thickness measured using NIS-elements software to calculate length in µm at five random points per skin model section (100× magnification).
Immunohistochemical staining
Immunohistochemical stains were performed on deparaffinized, formalin-fixed tissue sections to assess epidermal proliferation (Ki67: clone MIB-1, Dakocytomation, Glostrup, Denmark; 1:50), epidermal differentiation (K10: keratin 10, clone DE-K10, Progen, Heidelberg, Germany; 1:500 and involucrin: clone SY5, Novocastra, New Castle, United Kingdom; 1:1000), presence of fibroblasts (vimentin: clone V9, Dakocytomation) and myofibroblasts (α-SMA: clone 1A4, Dakocytomation). Supplementary antigen retrieval treatments were performed prior to incubation with the primary antibody using a 15 min. incubation step with pepsin (K10) and/or heat-induced antigen retrieval with 0.01M citrate buffer pH 6.0 (Ki67, K10, K17, vimentin). Immunohistochemical staining scoring (−) absence of staining; (+) normal staining pattern; (++) increased number of positively stained cells; (+++) strongly increased number of positively stained cells. Ki67 Proliferation index 100 basal cells were counted in three random locations in a tissue section (100× magnification), after which the number of positive cells along this length of the epidermis was counted. The proliferation index was defined as the percentage of Ki67 positive nuclei within these regions.
Enzyme-linked immunosorbent assay (ELISA)
Previously, we have identified a panel of wound healing mediators that are secreted predominantly by the epidermis (IL-1α, TNF-α, CCL5, VEGF), the dermis (TIMP2, HGF), or those significantly increased in the full thickness skin equivalents (CCL2, CXCL1, CXCL8, IL-6, sST2) [25]. CCL27 is found in burn wound exudates and has been implicated in the increased secretion of many of the aforementioned proteins [31]. IL-18 was also included in this panel because it has previously been implicated in keloid formation [4] and is known to be expressed in reconstructed human skin models also [11].
Culture supernatants (1.5 ml KC-II without hydrocortisone) derived from the skin models were collected over a 24-h period at the end of the culture period (5 weeks) to measure the levels of IL-6 and CXCL8 (PeliKine Sanguin Reagents, Amsterdam, The Netherlands); CCL2, CCL5, CCL20, CCL27, CXCL1, HGF and VEGF (R&D System Inc., Minneapolis, MN, USA); and IL-18 (MBL International, Woburn, MA, USA) secreted by the SE, using enzyme-linked immunosorbent assays (ELISA).
Quantitative polymerase chain reaction (qPCR)
For RNA isolation, the epidermis was removed from the dermis using a slide-warmer (40 °C), the dermis was then flash frozen and stored in liquid nitrogen until further processing. Samples were disrupted and homogenized in a TissueLyser, then flash frozen for storage at − 80 °C. RNA isolation was performed using QiaShredder™ kits and RNeasy® Mini kits with on-column DNAse digestion and stored at − 80 °C. The Nanodrop spectrophotometer (Nanodrop technologies, Wilmington, DE, USA) was used to measure total RNA concentration. cDNA was synthesized using the RT2 First Strand Kit, while the RT2 SYBR Green Fluor qPCR Mastermix was used to run the real-time PCR reactions for the following genes (Table 2): COL4A2, HAS1 and MMP3. These three genes were selected because they showed differential expression between Nskin and Kscar in previous work [16]. The geometric mean of two housekeeping genes (ACTB and HPRT1) was used to normalize expression. Unless stated otherwise, all RNA and qPCR reagents were obtained from Qiagen GmbH (Hilden, Germany).
MTT assay
A colorimetric (MTT based) assay was used to quantify cell proliferation and viability (Roche Applied Science, Penzberg, Germany) of the skin models, as described previously [34].
Data analysis
Experiments were performed in duplicate with n ≥ 4 different donors, except for some of the immunohistochemical staining (one of duplicate skin models was stained: keratin 10, vimentin, α-SMA) and qPCR experiments; see Tables 1 and 3 for an overview of donor matching and the number of biological replicates used per experiment. All results in graphs and tables were expressed as the mean ± standard error of the mean, PCR scatter plots showed the median. Normality testing (Shapiro–Wilk test) was performed on the residuals (errors); an ordinary one-way ANOVA with Tukey’s multiple comparisons tests was employed if the residuals passed the normality test (epidermal thickness; dermal thickness; contraction; MTT; ECM gene expression; secretion of VEGF, CCL5, CXCL8), otherwise the Kruskall-Wallis test with Dunn’s multiple comparisons tests (Ki67; α-SMA; secretion of CCL2, CCL20, IL-18, CXCL1, IL-6, HGF, CCL27) was applied. For analysis of the PCR data, gene expression (2−∆Ct) was normalized with the geometric mean of two housekeeping genes (ACTB and HRPT1). Differences were considered significant if p < 0.05 (*), p < 0.01(**) or p < 0.001 (***). It should also be noted that due to our use of stringent statistical analysis for these small sample sizes, our results very likely show underestimation of true statistical significance. A correction for performing multiple comparisons was made using an ANOVA. Additionally, the use of non-parametric tests in the event of non-normal distribution of the residuals, although statistically correct, further reduced power. For this reason, the term ‘trend’ was used when a clear pattern in graph data was observed without significance being reached; the exact p value was listed in the graph if 0.05 < p < 0.08. GraphPad Prism 6 software (GraphPad Software Inc., San Diego, CA, USA) was used to construct all graphs and tables and perform statistical analysis.
Results
Increased epidermal thickness in keloid models
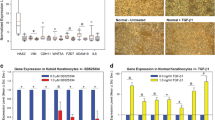
Recently, we have shown that native keloids have increased epidermal thickness which was not related to hyperproliferation but may be related to abnormal involucrin expression [17]. Therefore, we first characterized the epidermal compartments of the keloid models (Fig. 2). All skin models showed a fully differentiated epidermis on a fibroblast-populated matrix. Cd-Kscar showed a significant increase in the number of epidermal keratinocyte layers compared to Nskin (Fig. 2b), with a similar trend occurring for Cs-Kscar (p = 0.0792). It should be noted that the differences described here were minor (1–2 cell layers) and not of the same magnitude as found in vivo (± 5 cell layers). sNskin showed similar results to unaffected Nskin.

Increased contraction, epidermal and dermal thickness in central keloid regions. Absolute surface area of the skin models was measured in duplicate in n = 8 normal skin (Nskin), n = 8 peripheral keloid (P-Kscar), n = 7 central superficial keloid (Cs-Kscar), n = 7 central deep keloid (Cd-Kscar), n = 5 surrounding normal skin (sNskin). a shows macroscopic views of SE at the start and end of culturing; b shows the number of viable epidermal cell layers in the SE; c shows the dermal thickness measured in µm; d shows contraction measured as a reduction in end surface area after 5 weeks of culturing. *p < 0.05, if 0.05 > p < 0.08 then the exact p value is listed in the graph
In line with our previous findings on native tissue biopsies [17], there was no difference in the number of Ki67-positive proliferating basal cells between the different skin models (10–15% of basal cells) and normal suprabasal keratin 10 expression was observed in all the experimental groups (lower panel Fig. 3). We previously reported that while involucrin expression in healthy skin is confined to the stratum granulosum, in native Kscar involucrin is over expressed in all suprabasal layers [17]. In all the skin models described in this study, suprabasal involucrin was observed and was not just limited to the keloid constructs.

Increased α-SMA staining in central keloid regions. Upper panel shows representative pictures of HE, α-SMA and vimentin stainings performed on one of the duplicate skin models in n = 8 normal skin (Nskin), n = 8 peripheral keloid (P-Kscar), n = 7 central superficial keloid (Cs-Kscar), n = 7 central deep keloid (Cd-Kscar), n = 5 surrounding normal skin (sNskin). Magnification 200×, scale bar 100 µm. Lower panel shows the results of immunohistochemical stainings of epidermal markers (Ki67, keratin 10), dermal cellular markers (vimentin, α-SMA) in the skin models. Ki67 is expressed as mean ± SEM; SPB: suprabasal expression; SB: stratum basale; PAN: panepidermal (both SB and SPB); +/−: minimal expression; +: normal expression; ++: increased expression; +++: strongly increased expression; –: absent
Increased contraction and α-SMA expression in central keloid regions
Skin models constructed from all three different keloid scar regions showed a reduction in surface area and therefore increased contraction, compared to Nskin (Fig. 2a, c). sNskin was not significantly more contracted than Nskin. The increased contraction in Kscar was associated with the presence of myofibroblasts (Fig. 3). There was only little α-SMA staining in Nskin, sNskin and P-Kscar, but positive α-SMA staining was clearly present in Cs-Kscar and particularly Cd-Kscar. The difference in α-SMA expression was not the result of a disparity in the cellular contents of the dermis as there was no difference in vimentin staining between the models.
As increased thickness is one of the hallmarks of abnormal scars, we also assessed dermal thickness in the skin models. However, none of the keloid models nor sNskin showed significantly increased dermal thickness compared to Nskin (Fig. 2d).
Keloid models show reduced dermal gene expression of collagen type IV α2
Cd-Kscar showed significantly decreased dermal expression of collagen type IV α2 (COL4A2) compared to Nskin (Fig. 4). sNskin showed intermediate levels of COL4A2 expression between Nskin and Kscar. In contrast to COL4A2, no difference in gene expression of other extracellular matrix genes, matrix metallopeptidase 3 and hyaluronan synthase 1, was observed between the skin models.

Differential dermal expression of ECM related genes. Dermal expression of COL4A2, MMP3 and HAS1 was determined in one of the duplicate skin models in n = 4 normal skin (Nskin), n = 5 peripheral keloid (P-Kscar), n = 4 central superficial keloid (Cs-Kscar), n = 4 central deep keloid (Cd-Kscar), n = 4 surrounding normal skin (sNskin). The scatter plots show the individual data points with the median, with *p < 0.05
Secretion profiles of wound healing mediators
Next, we determined whether soluble wound healing mediators were differentially secreted by the keloid and normal skin models. HGF secretion was significantly decreased in Cd-Kscar compared to Nskin (Fig. 5). No significant differences between the different keloid models, sNskin and Nskin were observed for the other inflammatory cytokines: CCL20, CCL27, CXCL8, IL-6, IL-18, CXCL1, CCL2 and CCL5 (Fig. 5). Cytokine secretion levels were not influenced by differences in viability between the skin models, as MTT values were not significantly different between groups (data not shown).

secretion of wound healing mediators. Wound healing mediator secretion of HGF, CCL27, CXCL8, CCL20, IL-6, IL-18, CCL5, CXCL1, VEGF, and CCL2 was determined in duplicate in n = 8 normal skin (Nskin), n = 8 peripheral keloid (P-Kscar), n = 7 central superficial keloid (Cs-Kscar), n = 7 central deep keloid (Cd-Kscar), n = 5 surrounding normal skin (sNskin). Graphs show mean ± SEM, with *p < 0.05
Discussion
In this study, we have used our previously published baseline keloid model [16] to further investigate the underlying keloid pathogenesis. We tested the hypothesis that differences exist within keloids which contribute differentially to keloid formation. Full thickness keloid scar models were constructed using keratinocytes and fibroblasts isolated from different regions within and around a keloid scar. Interestingly, differences were observed in scar phenotype between the different keloid regions: of these, the central deep keloid construct most often resembled the keloid phenotype, while the surrounding normal skin directly adjacent to the keloid showed a mixed normal skin and keloid scar phenotype.
Both the previously published baseline keloid model, as well as the central deep keloid model, showed increased contraction and α-SMA expression, decreased secretion of HGF, as well as decreased dermal expression of collagen type IV [16] compared to Nskin. Minor disparities between the two studies were found with regards to dermal MMP3 and HAS1 expression as well as epidermal thickness. The baseline keloid model comprising the entire keloid showed no increase in epidermal thickness, but reduced COL4A2, HAS1 and MMP3 gene expression in the dermal compartment of the keloid model compared to the normal skin model. In this study, we did observe an increase in epidermal thickness (albeit of a smaller magnitude than the native keloid tissue), but only collagen IV showed significantly reduced gene expression in the central deep keloid model (Cd-Kscar). We did not observe reduced MMP3 or HAS1 gene expression. While the baseline keloid model showed a trend towards increased dermal thickness (p = 0.075), there was no statistically significant difference between the keloid regions and normal skin in this study. Either way, in both the baseline keloid model as well as the current keloid regions models, increased epidermal and dermal thickness are not as excessively present in our in vitro models compared to the in vivo keloids [17]. This may be due to the relatively short culture period (5 weeks) and only two skin cell types currently being present in the models. However, herein now lies the value of our in vitro scar model, as it allows for the relatively easy addition of other cell types (e.g. immune cells and endothelial cells) in the future in a controlled and scalable manner.
Most studies on keloid heterogeneity report an active, proliferating and invasive role for the keloid periphery [1, 5, 12, 19, 24, 27, 33] compared to the quiescent centre. However, our results are in line with studies finding increased activity in the keloid central region [6, 21, 28]. To our knowledge, the only full thickness skin model constructed from keloid derived keratinocytes and fibroblasts from different regions within the keloid, was grafted into a mouse to develop a new keloid animal model [26]. Using superficial or deep keloid fibroblasts with keloid keratinocytes, two different keloid models were constructed and compared to normal skin model. After implantation into athymic mice, both keloid models were shown to be different from normal skin models (abnormal collagen organization) and differences were reported between superficial and deep keloid models. The deep keloid model had a thicker dermis and increased COL1A1 expression, while the superficial keloid model only showed an increased wound area after grafting. Based on the method of cell isolation described, the deep keloid model very likely comprises cells of our central deep keloid construct (Cd-Kscar) and as such, the aforementioned findings correspond to our results. Nonetheless, a human full thickness skin model representing keloid heterogeneity is not currently available. In that regard, our in vitro keloid models described here could serve as an excellent starting point for further research.
A possible explanation for the dichotomy in findings reporting either the periphery or the central keloid region as the driving force behind continued keloid growth may be because different keloid phenotypes exist. Bella et al. [2] suggested that differences in genetic abnormalities may be responsible for heterogeneity between keloids, and distinguished between ‘superficial spreading’ keloids versus ‘raised’ keloids in an African tribe with familial keloids. In this regard, Supp et al. [26] also proposed an interesting model for the development of the ‘bulging’ keloid phenotype in which the deep keloid fibroblasts cause dermal thickening, while the superficial fibroblasts cause an increase in area by spreading the upper dermis (and overlying epidermis). The combination of deep dermal thickening and superficial dermal spreading then ultimately creates a ‘bulging’ keloid. Depending on the keloid phenotype, we propose that the actively growing region may be the periphery or the centre. This could explain why the periphery could very well be the actively expanding region in ‘spreading’ keloids, but not in the ‘bulging’ keloids where growth ensues from the deeper central regions. Retrospective analysis of the pictures of the keloid samples included in our study, in fact, showed that they were all of the ‘bulging’ phenotype. Three of the eight keloids used to construct the various keloid models were multinodular in appearance, consisting of several large dome-shaped nodules fused into a single large keloid. As the centre of each nodule was still more raised than the periphery, these were also considered ‘bulging’ keloids. Given that all our keloid donors were of the ‘bulging’ phenotype, this could explain why only the central deep region was statistically significantly different from normal skin.
In this study, we also included extra-lesional normal skin (sNskin) directly adjacent to but separate from P-Kscar. Even though this surrounding normal skin (sNskin) did not show any statistically significant differences with the other models for the individual parameters studied, when taking all parameters together a clear pattern was emerging. sNskin was usually intermediate between normal skin and keloid in the expression of scar parameters and often similar to the peripheral keloid model (contraction; α-SMA expression; secretion of HGF; expression of COL4A2). Abnormalities in the surrounding normal skin (sNskin) have been previously reported. Lee et al. [15] found that the into the surrounding normal skin. Using expression microarrays, Hahn et al. [7] found that increased expression of many of the genes in keloid-derived keratinocytes and fibroblasts corresponded with similarly increased expression in cells derived from adjacent non-lesional skin. However, we are unable to compare our results to the findings of others because it was often unclear what other authors considered to be surrounding normal skin (sNskin). Definitions of sNskin could also mean the normal skin in the same anatomical location but not necessarily in the direct vicinity of the keloid and alternatively, sometimes the surrounding normal skin was included with the peripheral margin generating what we would consider an sNskin/P-Kscar region.
Notably, from all the parameters studied, significant differences or trends were only obtained in a few parameters. This is indeed a limitation of the study and is most probably a result of the experimental set-up which could not be avoided. Surrounding normal skin was very rarely included with the keloid samples provided to us by the plastic surgeons, and when it was included, the keloid itself was often too small to enable adequate cell isolation from the different keloid regions. Therefore, non-donor paired samples were included in the analysis. Additionally, small sample sizes are inherent to the time-consuming nature of tissue engineering. The problem with statistical analysis of small sample sizes (n < 24) is that one either errs on the side of over- or underestimation, depending on whether a correction is applied for performing multiple comparisons. In this study, we had small sample sizes and corrected for multiple comparisons using a one-way ANOVA, power was further reduced in our study by our use of non-parametric testing when the residuals were not normally distributed. Thus, we have been relatively strict with our statistics to the point of underestimation, but consider this the better option as opposed to risking overestimation.
To conclude, we were able to generate different keloid scar models from keratinocytes and fibroblasts derived from intralesional peripheral, central superficial and central deep regions, as well as extralesional surrounding normal skin. Of these regions, only the central deep keloid regions showed statistically significant differences when compared to normal skin and thus displayed the most aberrant behavior. As all the keloid cells were derived from ‘bulging’ type keloids, this suggests that the central deep keloid region is likely the driving force behind the development of keloids of this ‘bulging’ phenotype. Our study has demonstrated the need for a clear and unambiguous description of the keloid type (e.g. ‘spreading’ or ‘bulging’) and of the exact location within keloid scars from which samples are taken. Additionally, we would encourage the inclusion of the skin adjacent to the keloids, as it is important to find out to what extent this region contributes to keloid scar formation and consequently if it should be targeted for treatment as well. This study is the first demonstration of how the in vitro baseline keloid scar model we have previously established [16] can be utilized not only as a future animal-free drug testing platform but also to further our understanding of the underlying pathogenesis.
Abbreviations
- Nskin:
-
Normal skin
- Kscar:
-
Keloid scar
- P:
-
Peripheral
- Cs:
-
Central superficial
- Cd:
-
Central deep
- ECM:
-
Extracellular matrix
- LAMA1:
-
Laminin alpha 1
- COL4A2:
-
Collagen type IV alpha 2
- ITGA5:
-
Integrin alpha 5
- HAS1:
-
Hyaluronan synthase 1
- MMP1:
-
Matrix metalloproteinase 1
- MMP3:
-
Matrix metalloproteinase 3
References
Aiba S, Tagami H (1997) Inverse correlation between CD34 expression and proline-4 hydroxyase immunoreactivity on spindle cells noted in hypertrophic scars and keloids. J Cutan Pathol 24:65–69. https://doi.org/10.1111/j.1600-0560.1997.tb01098.x
Bella H, Heise M, Yagi KI et al (2011) A clinical characterization of familial keloid disease in unique African tribes reveals distinct keloid phenotypes. Plast Reconstr Surg 127:689–702. https://doi.org/10.1097/PRS.0b013e3181fed645
Butler PD, Ly DP, Longaker MT, Yang GP (2008) Use of organotypic coculture to study keloid biology. Am J Surg 195:144–148. https://doi.org/10.1016/j.amjsurg.2007.10.003
Do DV, Ong CT, Khoo YT et al (2012) Interleukin-18 system plays an important role in keloid pathogenesis via epithelial-mesenchymal interactions. Br J Dermatol 166:1275–1288. https://doi.org/10.1111/j.1365-2133.2011.10721.x
Gao Z, Wu X, Song N et al (2010) Differential expression of growth differentiation factor-9 in keloids. Burns 36:1289–1295. https://doi.org/10.1016/j.burns.2010.02.009
Giugliano G, Pasquali D, Notaro A et al (2003) Verapamil inhibits interleukin-6 and vascular endothelial growth factor production in primary cultures of keloid fibroblasts. Br J Plast Surg 56:804–809. https://doi.org/10.1016/S0007-1226(03)00384-9
Hahn JM, Glaser K, McFarland KL et al (2013) Keloid-derived keratinocytes exhibit an abnormal gene expression profile consistent with a distinct causal role in keloid pathology. Wound Repair Regen 21:530–544. https://doi.org/10.1111/wrr.12060
Javad F, Marriage F, Bayat A (2012) Perturbation of cell cycle expression in keloid fibroblast. Skinmed 10:152–159
Kischer C (1984) Comparative ultrastructure of hypertrophic scars and keloids. Scan Electron Microsc (Pt 1):423–431
Köse O, Waseem A (2008) Keloids and hypertrophic scars: are they two different sides of the same coin? Dermatologic Surg 34:336–346. https://doi.org/10.1111/j.1524-4725.2007.34067.x
Kosten IJ, Buskermolen JK, Spiekstra SW et al (2015) Gingiva equivalents secrete negligible amounts of key chemokines involved in langerhans cell migration compared to skin equivalents. J Immunol Res 2015:1–11. https://doi.org/10.1155/2015/627125
Kurokawa N, Ueda K, Tsuji M (2010) Study of microvascular structure in keloid and hypertrophic scars: Density of microvessels and the efficacy of three-dimensional vascular imaging. J Plast Surg Hand Surg 44:272–277. https://doi.org/10.3109/2000656X.2010.532923
Ladin DA, Hou Z, Patel D et al (1998) P53 and apoptosis alterations in keloids and keloid fibroblasts. Wound Repair Regen 6:28–37
Lee SS, Yosipovitch G, Chan YH, Goh CL (2004) Pruritus, pain, and small nerve fiber function in keloids: A controlled study. J Am Acad Dermatol 51:1002–1006. https://doi.org/10.1016/j.jaad.2004.07.054
Lee WJ, Park JH, Shin JU et al (2015) Endothelial-to-mesenchymal transition induced by Wnt 3a in keloid pathogenesis. Wound Repair Regen 23:435–442. https://doi.org/10.1111/wrr.12300
Limandjaja GC, Van Den Broek LJ, Breetveld M et al (2018) Characterization of in vitro reconstructed human normotrophic, hypertrophic, and keloid scar models. Tissue Eng Part C Methods 24:242–253. https://doi.org/10.1089/ten.tec.2017.0464
Limandjaja GC, van den Broek LJ, Waaijman T et al (2017) Increased epidermal thickness and abnormal epidermal differentiation in keloid scars. Br J Dermatol 176:116–126. https://doi.org/10.1111/bjd.14844
Liu Q, Wang X, Jia Y et al (2016) Increased blood flow in keloids and adjacent skin revealed by laser speckle contrast imaging. Lasers Surg Med 48:360–364. https://doi.org/10.1002/lsm.22470
Louw L, van der Westhuizen J, Duyvene de Wit L, Edwards G (1997) Keloids: peripheral and central differences in cell morphology and fatty acid compositions of lipids. Adv Exp Med Biol 407:515–520
Lu F, Gao J, Ogawa R et al (2007) Biological differences between fibroblasts derived from peripheral and central areas of keloid tissues. Plast Reconstr Surg 120:625–630. https://doi.org/10.1097/01.prs.0000270293.93612.7b
Luo S, Benathan M, Raffoul W et al (2001) Abnormal balance between proliferation and apoptotic cell death in fibroblasts derived from keloid lesions. Plast Reconstr Surg 107:87–96
Niessen FB, Spauwen PH, Schalkwijk J, Kon M (1999) On the nature of hypertrophic scars and keloids: a review. Plast Reconstr Surg 104:1435–1458
Robles D, Berg D (2007) Abnormal wound healing: keloids. Clin Dermatol 25:26–32. https://doi.org/10.1016/j.clindermatol.2006.09.009
Seifert O, Bayat A, Geffers R et al (2008) Identification of unique gene expression patterns within different lesional sites of keloids. Wound Repair Regen 16:254–265. https://doi.org/10.1111/j.1524-475X.2007.00343.x
Spiekstra SW, Breetveld M, Rustemeyer T et al (2007) Wound-healing factors secreted by epidermal keratinocytes and dermal fibroblasts in skin substitutes. Wound Repair Regen 15:708–717
Supp DM, Hahn JM, Glaser K et al (2012) Deep and superficial keloid fibroblasts contribute differentially to tissue phenotype in a novel in vivo model of keloid scar. Plast Reconstr Surg 129:1259–1271. https://doi.org/10.1097/PRS.0b013e31824ecaa9
Syed F, Ahmadi E, Iqbal SA et al (2011) Fibroblasts from the growing margin of keloid scars produce higher levels of collagen i and III compared with intralesional and extralesional sites: clinical implications for lesional site-directed therapy. Br J Dermatol 164:83–96. https://doi.org/10.1111/j.1365-2133.2010.10048.x
Tsujita-Kyutoku M, Uehara N, Matsuoka Y et al (2005) Comparison of transforming growth factor-beta/Smad signaling between normal dermal fibroblasts and fibroblasts derived from central and peripheral areas of keloid lesions. In Vivo 19:959–963
Tuan TL, Nichter LS (1998) The molecular basis of keloid and hypertrophic scar formation. Mol Med Today 4:19–24
Tucci-Viegas VM, Hochman B, Frana JP, Ferreira LM (2010) Keloid explant culture: a model for keloid fibroblasts isolation and cultivation based on the biological differences of its specific regions. Int Wound J 7:339–348. https://doi.org/10.1111/j.1742-481X.2010.00698.x
van den Broek LJ, Kroeze KL, Waaijman T et al (2014) Differential response of human adipose tissue-derived mesenchymal stem cells, dermal fibroblasts, and keratinocytes to burn wound exudates: potential role of skin-specific chemokine CCL27. Tissue Eng Part A 20:197–209. https://doi.org/10.1089/ten.tea.2013.0123
van den Broek LJ, Niessen FB, Scheper RJ, Gibbs S (2012) Development, validation, and testing of a human tissue engineered hypertrophic scar model. ALTEX 29:389–402. https://doi.org/10.14573/altex.2012.4.389
Varmeh S, Egia A, McGrouther D et al (2011) Cellular senescence as a possible mechanism for halting progression of keloid lesions. Genes Cancer 2:1061–1066. https://doi.org/10.1177/1947601912440877
Waaijman T, Breetveld M, Ulrich MMW et al (2010) Use of a collagen/elastin matrix as transport carrier system to transfer proliferating epidermal cells to human dermis in vitro. Cell Transplant 19:1339–1348
Yang G, Lim I, Phan T et al (2003) From scarless fetal wounds to keloids: Molecular studies in wound healing. Wound Repair Regen 11:411–418. https://doi.org/10.1046/j.1524-475X.2003.11604.x
Acknowledgements
The authors would like to thank W. van Wieringen for assistance with statistical analysis of the data and S.C. Sampat-Sardjoepersad for practical assistance.
Funding
This study was financed by the Dutch Government: Rijksdienst voor Ondernemend Nederland, project number INT102010.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors have no conflicts of interest to declare.
Ethical approval (Research involving Human Participants and/or Animals)
Tissue collection procedures were performed in compliance with the ‘Code for Proper Secondary Use of Human tissue’ as formulated by the Dutch Federation of Medical Scientific Organization (http://www.fmwv.nl).
Informed consent
The discarded skin was collected anonymously if patients had not objected to use of their rest material (opt-out system). Keloid scars (Kscar) were obtained from patients undergoing scar removal via excision. The discarded scar tissue was coded to enable the collection of additional relevant information (e.g. previous treatment, age of scar) and were included only after obtaining oral informed consent.
Additional information
The work for this study was performed in Amsterdam, North-Holland, the Netherlands.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Limandjaja, G.C., van den Broek, L.J., Waaijman, T. et al. Reconstructed human keloid models show heterogeneity within keloid scars. Arch Dermatol Res 310, 815–826 (2018). https://doi.org/10.1007/s00403-018-1873-1
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00403-018-1873-1