
Abstract
Strain hardening of polymer melts is able to improve the uniformity of items in processing operations with elongational deformation. Of particular interest in this aspect is the dependence of strain hardening on elongational rate. In its first part, the paper presents a review on melt strain hardening obtained in uniaxial extensional experiments. Its dependence on elongational rate is of particular interest insofar as besides non-strain-hardening polymers, strain hardening increasing or decreasing with rate can be found. Results on linear polymers like polystyrene (PS), polypropylene (PP), high-density polyethylene (HDPE), and linear low-density polylethylene (LLDPE) in dependence on molecular parameters are discussed, as well as those of various blends. Particularly interesting are the strain-hardening features of certain HDPEs and LLDPEs, which could be understood by the assumption of a non-homogeneous chemical structure of the samples. Blends of various compositions of a linear and a long-chain branched PP throw light on the complex relation between branching structure and rate dependence of strain hardening. In the second part of the paper, the different strain-hardening behavior of linear polymers is interpreted by assessing the Rouse times as decisive physical quantity. For blends of certain linear species like HDPE and PP and those of linear with long-chain branched polymers, the existence of separate phases in the molten state is postulated. The assumptions are discussed in the light of the various studies on miscibility of linear and branched polyolefins from the literature.
Graphical Abstract

Similar content being viewed by others
Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Elongational properties of polymer melts have gained much interest in rheology during the last decades. One reason is the fact that some processing operations are dominated by elongational flow, the behavior of which cannot be derived in general from the properties in shear; the other arises from the challenges to find correlations between the elongational behavior and the molecular structure of polymer melts and to set up theoretical descriptions.
The most striking feature of extensional flow is the increase of tensile stress as a function of time or strain, respectively, above the linear relation in experiments at given elongation rates. This transient behavior is defined as melt strain hardening and should not be mixed up with the mechanical effect appearing in the solid state. For modelling and theoretical interpretation, this nonlinear time-dependent effect is less frequently discussed than the steady state of deformation described by the elongational viscosity. But particularly for polymers with broader molar mass distributions, high molar mass components or long-chain branches, it is experimentally very challenging to get reliable steady-state data due to the uniformity of sample geometries requested at the large elongations often necessary (Münstedt and Stary (2013)). Although several experimental methods have been developed over the years (e.g., Münstedt and Schwarzl (2014)) and the measurement of elongational flow has become more convenient, this aspect has not been addressed with high enough rigor. In spite of these deficiencies, most papers on modelling elongational flow deal with the steady-state elongational viscosity and try to understand its various features of increase (extensional thickening) or decrease (extensional thinning) with elongational rate. It should be mentioned that the viscosity behavior and strain hardening can be different even from a qualitative point of view. Thus, in the extensional thinning regime of a material (decrease of the steady-state viscosity in dependence on strain rate), strain hardening may occur.
However, melt strain hardening in the time-dependent regime is worthwhile being discussed for different materials because such data are particularly relevant for certain processing operations involving elongational flow where steady states of deformation are never reached, and they allow qualitative assessments of some aspects of the processing performance of various polymeric materials. Furthermore, in the transient regime, it is not as challenging as in the steady state to attain a sufficient sample uniformity during stretching due to the smaller total deformations applied. The reader interested in the role of strain hardening in applications may find detailed information on this subject in a review article by Münstedt (2018), for example, that provides various references of particular fields.
As well known from the literature, melt strain hardening can be significantly different for various polymers. It is prominent for long-chain branched polyethylenes (LDPE), and it has been found for long-chain branched polypropylenes (LCB-PP) as well. For linear polyethylenes, strain hardening and non-strain hardening were reported, whereas for linear commercial polypropylenes, strain hardening is very rarely seen in the literature. For polystyrenes with an obvious linear molecular structure, melt strain hardening is found that depends on the molar mass distribution. These different appearances are not understood in detail, and hardly reflected in literature are the various dependencies of strain hardening on the elongational rate showing an upswing or a decline with the increasing rate for different types of polymers.
Thus, this paper tries to fill this obvious gap by reviewing the relevant experimental results of melt strain hardening and its different rate dependencies, in particular, and intends to qualitatively correlate them with molecular data as far as possible. The main focus of this review is on polymeric materials being available for applications. Polymers of more complex molecular structures are addressed shortly when their behavior is thought to contribute to the mainly heuristic explanations of the different features. In addition, the survey intends to be an experimental basis for checking models that may help to understand the various features of strain hardening in more detail.
First, polymeric materials of various chemical compositions are reviewed, and then it is tried to heuristically discuss the structural elements responsible for a distinct strain-hardening behavior in the molten state. This procedure is preferred to grouping different products around a special molecular structure because it facilitates the selection of samples within a distinct polymer class most suitable for a special processing operation involving strain hardening.
Experimental results on melt strain hardening of amorphous polymers
Polystyrene
Commercial polystyrenes preferentially synthesized by radical polymerization consist of linear molecules with broad molar mass distributions. Anionic polymerization offers the possibility to obtain samples with various molar masses and very narrow distributions. Such materials are suitable to investigate the effect of high molar mass components on rheological properties. Branched polystyrenes are laboratory products and can be used to systematically study the influence of length and content of branches.
Linear polystyrenes
Comprehensive studies on the elongational behavior of polystyrenes melts with various well-defined molar mass distributions were performed already many years ago (Münstedt (1980)). Some of their essentials with respect to strain hardening are shortly addressed in the following. In Fig. 1, the molar mass distributions of the two anionically polymerized polystyrenes PS I and PS II with Mw = 74 kg/mol and Mw = 39 kg/mol, respectively, and polydispersity indices Mw/Mn close to one are shown.

Relative weight w in arbitrary units as a function of molar mass M for PS I and PS II obtained from size exclusion chromatography (Münstedt (1980))
PS I reveals a small high molar mass tail and PS II even a high molar mass component distinctly separated from the main peak. Such a molecular picture can be often found for anionic polystyrenes and is sometimes not provided by the molecular analysis like size exclusion chromatography.
The detailed rheological characterization of the samples can be found in Münstedt (1980). In Fig. 2, the transient elongational viscosities defined as the time-dependent tensile stress related to the constant elongational rates applied are plotted as functions of time. The two materials do exhibit similar tendencies. As widely known, in the linear range at small elongations or low rates, respectively, the viscosities follow the so-called Trouton relation µ(t) = 3η0(t) where η0(t) is the time-dependent shear viscosity in the linear range of deformation. At larger strains, the elongational viscosity rises from the linear curve, and the increase is more pronounced the higher the rate. This behavior is often found for the strain hardening of polymer melts. Obviously, strain hardening is stronger for PS 2 with the distinct high molar mass component than for PS 1. In contrast to PS 2, the viscosity curves of PS 1 reach steady states under the conditions applied. For a more detailed discussion of strain hardening, the so-called melt strain-hardening coefficient,
is defined, where µ(t) is the time-dependent elongational viscosity in the linear range of deformation

Elongational viscosity µ as function of time t for PS I and PS II at 130 °C and various elongational rates \(\dot{\varepsilon }\) (Münstedt (1980)). The viscosity axes are shifted by the factor of ten to each other
As it is obvious from Fig. 2, the strain-hardening coefficient is a rather complex quantity and depends on elongation, elongational rate, and the molecular structure. That is the reason why comprehensive discussions of its dependencies are scarce in the literature. Regarding the main topic of this paper, it can be said, however, that for the two PS above strain hardening is more pronounced for the sample with the high molar mass component and increases with elongational rate for both polystyrenes. Furthermore, it can be derived that visible strain hardening occurs between the elongational rates of 0.014 and 0.051 s−1 for PS I and 0.001 and 0.003 s−1 for PS II.
From Fig. 2, it is obvious which problems may occur when steady-state elongational viscosities are discussed. For PS I, steady states are achieved and a faint tendency of viscosities becoming higher than the linear value may be derived in the range of elongational rates applied. For PS II, even an indication of steady states is absent up to the maximum Hencky strain of 3 used, at which the uniformity of sample deformation is still good.
Takahashi et al. (1993) compared two polystyrenes, and they report a stronger strain hardening for the sample with the larger polydispersity index Mw/Mn. For both samples, an increase of the strain-hardening coefficient with strain rate was found. Detailed information on the molar mass distributions is not provided and, thus, the role a high molar mass component may play cannot be discussed.
Polystyrenes with definite heterogeneous molar mass distributions were studied by Minegishi et al. (2001). They produced different blends by adding an anionic polystyrene with a molar mass of about 3∙103 kg/mol or 15∙103 kg/mol (UHMW-PS), respectively, to a commercial radical PS with lower molar mass but much higher polydispersity up to concentrations of 1.5 wt%. The experiments show some scatter, but the tendency is obvious that the high molar mass components increase strain hardening with a slight tendency of being more pronounced at larger elongational rates than at lower ones.
These measurements were described by Wagner et al. (2005) using the molecular stress function (MSF) model. However, the lack of experimental data in the range of higher elongations predicted by the model makes it difficult to assess its general validity. Furthermore, it is somewhat surprising that the elongational viscosities of the PS/UHMW-PS blends could be described by the MSF model using only linear viscoelastic data.
For two anionic polystyrenes, the time-dependent elongational viscosities at different elongational rates have been reported by Bach et al. (2003). At low rates, the viscosity curves correspond to the Trouton relation valid in the linear range of deformation, at higher elongational rates an increase of the viscosity above the linear curve is found, whereby strain hardening becomes more pronounced with growing rate.
The elongational behavior of four anionically polymerized polystyrenes with different molar masses and Mw/Mn around 1.0 was characterized by Nielsen et al. (2006). For all four samples, significant strain hardening was found that strongly enhanced with increasing elongational rate. A still more pronounced behavior with the same strain hardening tendency has been reported for blends from the anionic species. Molar mass distributions are not shown, but these would have been of importance for a thorough discussion of the molecular reasons for the strain hardening of the anionic polystyrenes, since small high molar mass tails may be found for such polymers as demonstrated in Fig. 1.
In Fig. 3, the elongational viscosity as a function of time at various elongational rates is shown for blends of a radically polymerized PS with Mn = 95 kg/mol and Mw/Mn = 1.8 and various additions of an anionic PS with Mn = 1800 kg/mol and Mw/Mn = 1.2 (Hepperle (2002)). Obviously, strain hardening increases with the amount of the high molar mass component, and the tendency of a more pronounced hardening at larger rates is the same as for the other blends discussed above and the samples of Fig. 2 with a high molar mass component according to polymerization.

Elongational viscosity μ as a function of time t at various elongational rates \(\dot{\varepsilon }\) and 169 °C for blends of the radically polymerized polystyrene PS-r 95 (Mn = 95 kg/mol, Mw/Mn = 1.8) and 1 and 5 weight percent of the anionic polystyrene PS-a 1800 (Mn = 1800 kg/mol, Mw/Mn = 1.2) (Hepperle (2002)). wr describes the weight percentage of the radically polymerized and wa that of the anionic polystyrene. The curves are shifted by the factor indicated
Branched polystyrenes
Elongational properties of various long-chain branched styrenic laboratory products are reported by Hepperle and Münstedt (2006). For some samples, in Fig. 4, strain hardening at the Hencky strain 3 is displayed in dependence on the elongational rate \(\dot{\varepsilon }\) multiplied by the zero-shear viscosity η0, which corresponds to a temperature invariant plot. The linear PS-r-95 shows the smallest strain hardening that slightly increases with elongational rate. Branching significantly enhances strain hardening. Due to the similar molar masses of the backbones of the samples between 60 and 90 kg/mol and the molar masses of the branches being around 25 kg/mol within the accuracy of their determination, the effect of the number of branches per molecule becomes obvious. Strain hardening is larger and its dependence on elongational rate is more pronounced the higher the specific number of branches ranging from 0.6 to 3.2 per backbone molecule. Like for all the other polystyrenes discussed before, strain hardening increases with elongational rate. Definite data on the onset rate of strain hardening are not available.

Strain-hardening coefficient SH as function of the elongational rate multiplied by the zero-shear viscosity η0 at the total Hencky strain 3 for various branched PS in comparison to the linear radically polymerized PS-r-95 with Mn = 95 kg/mol. In the nomenclature PS-x-pG-y of the samples, x stands for the number average molar mass of the backbone, p for the number of grafted molecules G per backbone, and y for their number average molar mass in kg/mol (Hepperle and Münstedt (2006))
Elongational properties of branched polystyrenes with a molar mass of the backbone of Mw = 275 kg/mol and numbers of branches up to 190 were studied by Abbasi et al. (2017). All the branches had a molar mass of Mw = 44 kg/mol. Strain rate thickening determined from the extrapolated equilibrium values of the elongational viscosity distinctly increased with the number of branches first but leveled off at numbers approaching 100. A clear tendency of strain hardening increasing with the rate was seen for all samples up to elongational rates of at least 0.1 s−1. At higher rates, this dependency became weaker with larger numbers of branches.
Although being far away from becoming polymeric materials with economic importance, some modifications of polystyrene are shortly addressed in the following that show the potential of macromolecular synthesis with respect to a change of elongational properties.
The strain hardening of well-characterized comb polystyrenes was studied by Lentzakis et al. (2013). They prepared a series of samples based on two backbones with weight average molar masses of 275 and 860 kg/mol and approximately the same number of 30 arms per chain with equal lengths in each case, whereby the molar masses varied between 6.5 and 47 kg/mol. The samples were stretched at 170 °C and elongational rates between 0.003 and 10 s−1 up to maximum Hencky strains of about 3, the exact values of which are difficult to derive, however, from the experimental curves shown. The elongational behavior was assessed by the critical elongational rate for the onset of strain hardening and its strength as a function of rate. From the critical elongational rate, the so-called stretch relaxation time was determined as its inverse. All the samples displayed strain hardening with the tendency of becoming more pronounced with increasing rate.
Data on the strain hardening of the linear backbone molecules are not available, but for the samples with arm lengths of 11.7 and 47 kg/mol each, it can be seen that the longer backbone results in a somewhat stronger strain hardening and a distinctly smaller elongational rate for its onset. The molar mass of the arms has a significant effect on the critical rate for strain hardening. This characteristic quantity decreases with the comb molar masses increasing from 6.5 over 11.7 and 25.7 to 47 kg/mol for samples with the backbone of 860 kg/mol. A particularly distinct change becomes visible when the molar mass of the arms exceeds the entanglement molar mass of 17 kg/mol assumed for PS. The inverse of the critical elongational rate was taken for theoretical discussions. The strength of strain hardening for different samples at a distinct elongational rate was found to be more pronounced the smaller the onset rates.
Studies of elongational viscosity of polystyrenes with two definite three-arm star structures were published by Huang et al. (2016). For the symmetric star with branches of 90 kg/mol and the asymmetric star with two branches of 90 kg/mol and one branch of 20 kg/mol, strain hardening increasing with elongational rate was found similar to the linear sample with 180 kg/mol, and at higher rates, the curves were even identical showing a negligible influence of the stars on strain hardening. All measurements performed at 130 °C and elongational rates ranging from 0.0003 to 0.2 s−1 were numerically described by Wagner et al. (2022) using the enhanced relaxation of stretch (ERS) model.
The rheological behavior of polystyrenes with a multi-star architecture at both ends of a backbone with Mw ≈ 100 kg/mol (pom-pom) was studied by Röpert et al. (2022). The molar masses of the side chains varied between 9 and 300 kg/mol but were the same for all the molecules of a particular sample. Their numbers were 11 to 14 per star. Strain hardening was assessed as the ratio of the maximum (extrapolated) elongational viscosity and the linear shear viscosity determined with the Doi-Edwards theory. The elongational viscosities as a function of time at different constant elongational rates were described by the so-called molecular stress function (MSF), which allowed the extrapolation of steady states. Taking these values as the maximum viscosity, the ratios of the steady-state elongational viscosities reached values up to 40 for the arm molar mass of 50 kg/mol and became smaller with declining molar mass. The linear PS with the backbone molar mass of 100 kg/mol showed viscosity ratios increasing with strain rate. The rate dependencies of the viscosity ratios for the pom-pom samples were different but exhibited a similar feature insofar as they increased more or less with growing elongational rate first and then went through a maximum.
In another paper of members of this group, short-chain and long-chain branched polystyrenes of various architectures were synthesized and elongational properties measured besides others.
Extremely large viscosity ratios with factors above 1000 were reported (Faust et al. (2022)). These results have to be considered with some caution, however, because the chemical reactions necessary for the complex architectures may lead to partial cross-linking that affects enhanced viscosity. The linear PS with Mw = 310 kg/mol providing the backbone of the modified branched samples shows a faint growth of the viscosity ratio becoming somewhat stronger with increasing rate. PS combs show distinct viscosity ratios being more pronounced at higher rates. For the complex structures of long-chain and short-chain branches grafted to each other, large viscosity ratios are found with the tendency of weak increases with decreasing elongational rate.
Blends of a pom-pom PS with 22 branches of Mw = 22 kg/mol at each end of the backbone molecule with Mw = 280 kg/mol and a linear polystyrene with Mw = 90 kg/mol were rheologically characterized by Hirschberg et al. (2023). In addition to the viscosity ratios discussed by the authors, the published experimental data were used to determine the strain hardening at the Hencky strain 3 according to Eq. 1. This quantity does not need any extrapolations, and from experience, the sample deformation can be assumed to be uniform enough at this elongation to obtain comparable data. The pom-pom PS showed strain hardening growing with increasing strain rate as expected, but for pom-pom contents of the blends smaller than about 50 wt%, the strain-rate dependence of strain hardening changed to the reverse. This behavior is discussed below together with similar effects observed for blends of linear and long-chain branched polypropylenes.
Summarizing the results on polystyrene, it is obvious that strain hardening becoming stronger with increasing elongational rate is a common feature of most of the linear and long-chain branched samples studied. The rate dependence gets more pronounced with the addition of higher molar mass components or enhancing the branching intensity. Some few exceptions from this behavior are found for the samples with complex multi-star architectures and blends of a pom-pom PS with a linear PS.
Polymethylmethacrylate, styrene-acrylonitrile copolymer, polycarbonate
Elongational experiments on other amorphous polymers are less frequently found in the literature than those on polystyrene. For the matter of completeness, some of them are shortly described.
Studies on the elongational viscosity of a polymethylmethacrylate (PMMA) and a styrene- acrylonitrile copolymer (SAN) were reported by Takahashi et al. (1999). The polydispersity indices are similar to that of the radically polymerized PS discussed above, and the features of strain hardening of the neat products are comparable showing an enhancement with increasing elongational rates. Analogously to the results on polystyrene, the addition of around 1 wt% of a PMMA with a weight average molar mass about ten times higher than that of the PMMA matrix leads to a distinctly more pronounced strain hardening.
An effect comparable to that of PMMA was obtained by adding the high molar mass PMMA to the SAN matrix. Blending 1 wt% PS of a still higher molar mass than the PMMA to the SAN, however, had no effect on its strain hardening. This result is interesting insofar as PMMA is miscible with SAN but PS not, as was shown directly by electron micrographs. The addition of the PMMA component miscible with SAN has an effect comparable to that found for the homologous PMMA matrix, whereas the minute separate phase of the non-miscible PS leaves the behavior of the PMMA unchanged.
A very faint strain hardening of a PMMA was found by Katsikis et al. (2007) at a Hencky strain of 2 and the highest elongational rate of 0.1 s−1 applied. Its weight average molar mass was about half of that of the sample above The results are in qualitative agreement insofar as the measurements by Takahashi were performed at the distinctly lower temperature of 145 °C instead of 180 °C by Katsikis shifting the corresponding rates to smaller values.
Hepperle (2002) studied the elongational behavior of a commercial polycarbonate (PC) with Mw = 30 kg/mol and Mw/Mn = 2. It did not show any strain hardening in the chosen experimental windows of elongational rates between 0.02 and 0.2 s−1 and Hencky strains up to 2. For two branched PCs, strain hardening was found, but its rate dependence cannot clearly be assessed from the measurements performed. Furthermore, nothing is said about the branching structure and, thus, these results do not contribute much to the elucidation of strain hardening.
Experimental results on strain hardening of semi-crystalline polymers
Besides the amorphous polymers discussed above, semi-crystalline polymers play an important role in practice. Widely used are polyethylenes and polypropylenes, which are frequently processed by techniques involving extensional deformations. Furthermore, they are of particular interest with respect to the influence of molecular structure on elongational properties since linear and branched species are in use.
Linear polypropylenes
Elongational properties of polypropylenes are of relevance because they occur in applications like fiber spinning and film extrusion. Moreover, polypropylenes of various molecular structures have to be processed. Thus, as early as 1980, studies of the extensional behavior of polypropylenes were performed, for example, by Minoshima et al. (1980). Eight products with different molar masses and molar mass distributions were stretched at various constant elongational rates in an oil bath rheometer and the stress growth functions measured in dependence on time at a temperature of 180 °C. The samples were roughly divided into three groups with respect to their molar mass distributions: narrow distributions with Mw/Mn ≈ 5, broad distributions of Mw/Mn ≈ 9, and medium distributions in between. The broadly distributed samples were distinguished by strain hardening decreasing with growing elongational rate in the measured range between 0.005 and 1 s−1. For the narrowly distributed specimens, no strain hardening was observed at low elongational rates with a distinct indication of rising values at higher rates. The authors report non-uniform sample deformations due to necking that counteracts strain hardening. Necking may be the reason for the applied total Hencky strains not exceeding 2. The influence of necking on the stress curves is not discussed in detail, however, and thus the measurements should be considered to only provide some qualitative information.
A commercial polypropylene not specified in detail was studied by Ishizuka and Koyama (1980) at 180, 200, and 220 °C and elongational rates between 0.002 and 0.6 s−1. Strain hardening was observed with the tendency of being more pronounced at the higher rates. Total Hencky strains around 3 were reached; sample uniformities were not discussed.
Trzebiatowski and Wilski (1985) published elongational properties of commercial polypropylenes with M w/M n between 5 and 6. At a temperature of 180 °C and elongational rates between 0.02 and 1 s−1, they did not find any strain hardening up to the maximum Hencky strains of 2 applied. It is interesting to note that blending 3 wt% of a PP with Mw = 876 kg/mol to another PP with Mw = 283 kg/mol significantly increased the elongational viscosity as a function of time but did not result in strain hardening either.
However, strain hardening was reported for a PP with Mw/Mn = 6.2 measured at 180 °C by Takahashi et al. (1993). A rate dependence is difficult to derive because of various total elongations, which extend up to Hencky strains of about 4.
In a later paper from the same laboratory (Sugimoto et al. (2001a)), no strain hardening was found for a linear PP with Mw = 540 kg/mol and Mw/Mn = 4.7 stretched at 180 °C up to Hencky strains of 3 at elongational rates between 0.007 and 0.6 s−1.
Results on a linear PP with Mw = 910 kg/mol and Mw/Mn = 4.2 measured at 175 °C and elongational rates between 0.002 and 0.2 s−1 were reported by Hingmann and Marczinke (1994). Up to the total Hencky strains of 2.5 applied, they did not find any indication of strain hardening. A very similar behavior of a commercial linear PP not specified in detail was shown by Spitael and Macosko (2004).
Stange et al. (2005) studied the elongational behavior of a commercial linear polypropylene with Mw = 456 kg/mol and Mw/Mn = 3.9. For a temperature of 180 °C and elongational rates between 0.01 and 1 s−1, strain hardening was not found at Hencky strains measured up to 3. Rather a slight decrease from the viscosity curve in the linear range could be observed at larger elongations, which may be due to a non-uniform sample deformation, however.
Elongational viscosities of three linear PP of various molar masses and molar mass distributions studied by Auhl (2006) are published by Münstedt and Schwarzl (2014). They are displayed in Fig. 5.

Elongational viscosity μ as function of time t for linear polypropylenes with the molecular data given in Table 1 at T = 180 °C and various elongational rates \(\dot{\varepsilon }\) (Auhl (2006)). The full lines represent the time-dependent linear viscosities 3η0(t) obtained from experiments in shear. For the matter of clarity, the curves of PP-A and PP-B are shifted by the factors indicated
Their molecular data are listed in Table 1 and are in the range of the samples discussed before. The molecular mass distribution curves obtained by SEC were smooth as expected for commercial Ziegler–Natta products. As can be seen, all viscosity curves come to lie on the corresponding linear viscosity functions and strain hardening was not observed up to the highest elongational rate and the maximum Hencky strains of 3 measured.
The majority of the results for linear polypropylenes from the literature points to a non-strain-hardening behavior at laboratory conditions. Although not known in all cases, molar masses and their distributions cover a wide range, particularly for the samples, for which strain hardening was not found. Thus, there is good reason to assume that linear PP is not prone to strain hardening within a wide range of molar masses and their distributions.
Blends of linear polypropylene and high molar mass linear polylethylene
Very interesting results were obtained from studies of the strain hardening of linear non-strain-hardening polypropylenes with usual molar masses around several hundred kg/mol and polydispersity indices around 5 containing small amounts (0.4 and 1 wt%) of a linear ultrahigh molar mass polyethylene (UHMWPE) with molar masses in the range of several thousand kg/mol. The materials were synthesized in a series polymerization of ethylene and propylene using a Ziegler-Natta catalyst. As follows from Fig. 6, the blend of the linear PP with 1 wt% of the UHMWPE did show strain hardening increasing with decreasing rate at least down to the applied smallest values around 0.01 s−1 (Sugimoto et al. (2001a)). This rate dependence of strain hardening is opposite to the behavior reported above for the PS blends.

Elongational viscosity μ as a function of time t for a polypropylene containing 1 wt% of a linear polyethylene with an ultra-high molar mass at 180 °C and various elongational rates \(\dot{\varepsilon }\) (Sugimoto et al. (2001a)). The full line describes three times the time-dependent linear shear viscosity η.0(t)
From electron micrographs, it was seen that the UHMWPE and PP are not miscible as expected and the domains of the immiscible PE were not deformed by stretching. Principally, it cannot be excluded, however, that some kind of miscibility exists in the melt and the morphology may be due to phase separation and recovery processes during cooling. But such a process is not very probable if the low molecular mobility due to the high molar masses is considered. The authors argue that the non-deformed second phase and its minute amount make it difficult to relate the pronounced strain hardening to rheological properties of the PE phase. Rather, the assumption was made that at least some of the long molecules of the polyethylene may be compatible with the polypropylene molecules. This argument was seen to be supported by studies of the PP/PE blends in shear (Sugimoto et al. (2001b)) that showed a plateau-like behavior of the storage modulus at small angular frequencies. Surprisingly, the plateau was not changed after the separation of the distinct high molar mass component of HDPE by temperature-rising elution fractionation (TREF). This result is hard to understand considering the dynamic-mechanical behavior of PP that exhibits moduli becoming continuously smaller with decreasing angular frequency.
Long-chain branched polypropylenes
A molecular modification of polypropylene with respect to elongational properties interesting from a fundamental and application point of view is the introduction of long-chain branches. This has been done either by chemical reaction or electron beam irradiation. Hingmann and Marczinke (1994) studied two polypropylenes with 1.5 long-chain branches (PP-2) and three long-chain branches (PP-3) per molecule on the average. The modification was attained by using crosslinking agents in a concentration high enough to initiate branching but low enough to avoid crosslinking. The characteristic molecular data were Mw = 340 kg/mol and Mw/Mn = 2.5 for PP-2 and Mw = 450 kg/mol and Mw/Mn = 3.0 for PP-3, respectively. Both samples show a linear behavior up to a Hencky-strain of about ε = 1, above which distinct strain hardening becomes obvious (Fig. 7). At all the four elongational rates, strain hardening of PP-3 is distinctly larger than for PP-2. Furthermore, strain hardening is more pronounced at the lower elongational rates in the range measured. Steady states are not reached generally but indicated for PP-2 at the strain rate \(\dot{\varepsilon }\) = 0.05 s−1 and above approaching a strain of 3. Surprising is the amount of strain hardening occurring that attains SH ≈ 15 in the case of PP-3 at the lowest elongational rate of 0.01 s−1 and ε ≈ 3 not showing any indication of a plateau value.

Strain-hardening coefficient SH as a function of Hencky strain ε for the two long-chain branched PP-2 and PP-3 at 175 °C and various elongational rates \(\dot{\varepsilon }\) (Hingmann and Marczinke (1994)
It should be pointed out that the rate dependence of strain hardening for the two long-chain branched PP is just opposite to that of PS and PMMA reported above, but qualitatively in line with the PP/UHMWPE in Fig. 6.
The introduction of long-chain branches to PP by using various peroxydicarbonates was studied by Lagendijk et al. (2001) and Gotsis et al. (2004). The extensional behavior was measured at the elongational rate of 0.1 s−1 only, and strain hardening was found to increase with the number of branches depending on the kind and concentration of the modifier. The elongational viscosity as function of Hencky strain was successfully described by the two constitutive equations based on the modified Lodge theory and Wagner’s molecular stress function, respectively. Formally, it was concluded that strain hardening should occur for elongational rates larger than the inverse of the longest relaxation time.
Another way of introducing long-chain branching in polypropylene is electron beam irradiation. Results on the elongational flow at constant elongational rates of a commercial branched PP are reported by Kurzbeck et al. (1999). Details of the irradiation process are not available. Using measurements of the intrinsic viscosity as function of the molar mass on fractions obtained by size exclusion chromatography (SEC), long-chain branching was characterized. At molar masses smaller than 50 kg/mol, no branches could be detected, but above that value, their content continuously increased with molar mass up to the fraction with the highest molar mass of 2∙104 kg/mol. The time-dependent elongational viscosity was studied in a wide range of elongational rates between 0.002 and 2 s−1 up to total strains of 3. A pronounced strain hardening was observed increasing strongly with rate first and then levelling off. For a linear PP homopolymer and an ethylene-propylene copolymer, no strain hardening was found under similar experimental conditions.
Studies of electron beam irradiation of PP modified with various polyfunctional monomers are reported by Yoshii et al. (1996). For many modifiers, an increase in melt strength was found. Samples with the most effective monomer hexanediol diacrylate (HDDA) irradiated with a dose of 1 kGy were characterized in more detail. At the three constant elongational rates studied, strain hardening was found but the differences obtained for the low rates applied are not significant enough to determine a rate dependence. From SEC coupled with low-angle laser light scattering (SEC/LALLS), no hint to long-chain branching could be derived. Thus, high molar mass components were postulated as the source of strain hardening but these could not be shown analytically.
In another study, a linear PP was electron beam irradiated at doses of 10, 50, and 80 kGy (Sugimoto et al. (1999)). The extensional behavior was studied at constant elongational rates. For the unmodified PP, no indication of strain hardening was found at \(\dot{\varepsilon }\) of 0.0037 and 0.41 s−1, whereas for the 50 and 80 kGy samples, a distinct hardening could be seen for the rates applied between 0.03 and 0.2 s−1. However, the measurements are not good enough to assess a rate dependence. The molecular characterization by SEC/LALLS showed a degree of branching increasing with irradiation dose. However, the branching structure observed at 10 kGy does not seem to be effective enough to generate strain hardening.
A very detailed study of electron beam irradiated polypropylene was performed by Auhl et al. (2004). A commercial isotactic homopolymer of Mw = 669 kg/mol and Mw/Mn = 4.2 was exposed to doses between 1 and 150 kGy. Mw and Mw/Mn decreased with dose, but in parallel, the number of long-chain branches per chain became higher as shown by size exclusion chromatography coupled with multi-angle laser light scattering (SEC/MALLS). From the analysis, it was obvious that the shorter molecules are less branched than the longer ones. The extensional viscosity was measured at constant elongational rates between 0.01 and 1 s−1. The strain-hardening coefficient as a function of elongational rate is plotted in Fig. 8 for nine differently irradiated samples. The non-irradiated PP does not show any strain hardening within the range of rates chosen. This result is in agreement with those on other linear PP reported above. The three samples PP-60, PP-100, and PP-150 exposed to the highest doses exhibit strain hardening increasing with elongational rate at the total Hencky strain of 2.7. The samples irradiated with lower doses show strain hardening decreasing with rate. These results demonstrate that the general feature of the rate dependence of strain hardening seems to be related to the average number of long-chain branches obtained by irradiation.

Strain-hardening coefficient SH at the Hencky strain ε = 2.7 and T = 180 °C as a function of the elongational rate \(\dot{\varepsilon }\) for polypropylenes electron-beam irradiated with various doses indicated in kGy by the numbers of the sample designations (Auhl et al. (2004))
The results of Fig. 8 are of particular interest, because they may offer a clue for understanding the different rate dependencies of strain hardening occurring for samples of various molecular structures. From a molecular point of view, the irradiated samples are special insofar as their molar mass distribution becomes narrower with irradiation dose and growing degree of branching, respectively, in contrast to other methods of introducing branching. Thus, high molar mass components generated by irradiation can be excluded as a source of strain hardening, and it can be concluded that this behavior found for irradiated polypropylene has to be related to the differences in molecular structure that could be changed by the number of branched molecules, the architecture of the branches, or the branching content.
Blends of linear and long-chain branched polypropylenes
The influence of branching content can be studied by the addition of a definite long-chain branched polypropylene to a linear polypropylene. Such measurements with respect to the rate dependence of elongational viscosity were performed by Stange et al. (2005). Both blend components were commercial products. The characteristic molecular data for the linear PP were Mw = 456 kg/mol and Mw/Mn = 3.9 and for the LCB-PP Mw = 1157 kg/mol and Mw/Mn = 8.7. The extensional experiments were performed at T = 180 °C and constant elongational rates between 0.01 and 1 s−1 up to a total Hencky strain of 2.7. The strain-hardening coefficients at these conditions are displayed in Fig. 9 for selected blends. Data for the linear PP were not given due to a non-uniformity of deformation at relatively small elongations, already (Stange et al. (2005)). The addition of only 2 wt% LCB-PP induced a slight strain hardening at the lower elongational rates. The strain-hardening coefficient increased with LCB-PP content and reached a value of 5.5 at \(\dot{\varepsilon }\)= 0.01 s−1 and even 6.5 at \(\dot{\varepsilon }\) = 1 s−1 for the neat LCB-PP. Interesting is the rate dependence of strain hardening for the blends. From a decrease with growing rate at blend ratios measured up to 50 wt% LCB-PP, it changed to an increase for the LCB-PP. The time-dependent viscosities were modelled with the MSF theory, which allowed an extrapolation up to the steady state (Wagner et al. (2006)). The plateau values obtained showed only weak dependences on the elongational rate with a slight tendency to a decrease with increasing rate. However, these results are not significant enough to relate them to the differences found in strain hardening in Fig. 9.

Strain-hardening coefficient SH as a function of elongational rate for various blends of a linear and a long-chain branched PP at T = 180 °C and Hencky strain 2.7 (Stange et al. (2005)). The broken lines are drawn to guide the eye
The change of the rate dependence of strain hardening from the blends to the LCB is qualitatively comparable to that of the polypropylenes irradiated with various doses shown in Fig. 8 and points to the decisive role of different branching structures of the irradiated molecules for strain hardening.
Blends of a linear and a long-chain branched PP were studied by Ahirwal et al. (2014). The molecular data of the linear PP were Mw = 301 kg/mol and Mw/Mn = 2.9 those of the LCB-PP Mw = 312 kg/mol and Mw/Mn = 4.6. For both components, strain hardening was not observed within the accuracy of the measurements at elongational rates between 0.1 and 1 s−1. Surprisingly, the three blends with 10, 40, and 80 wt% LCB-PP showed strain hardening decreasing with increasing elongational rate.
Unfortunately, an analysis of the branching structure of the samples is not available and, thus, a detailed discussion of the results is not possible.
Extensional viscosities as a function of time at constant elongational rates for blends of a linear and long-chain branched PP prepared by Ziegler–Natta catalysts are reported by Li et al. (2021). Both components were industrial products. They were blended to each other in a twin-screw extruder. The samples were characterized by their molar mass distribution curves and branching indices. The extensional experiments were performed at 200 °C and elongational rates of 0.01, 0.1, and 1 s−1.
Although lacking experimental rigor, it can be concluded from the measurements that the linear PP does not exhibit strain hardening. The LCB-PP shows distinct strain hardening increasing with decreasing rate, and this behavior is found for all blends. Strain hardening occurs from 20 wt% LCB-PP upwards, and it becomes more pronounced with growing LCB content. Furthermore, rheological characterizations in shear and thermodynamic studies of the melting and crystallization behavior were performed the results of which the authors interpret as an indication of miscibility of the linear and branched PP.
These results together with those on the irradiated polypropylenes are of particular interest insofar, as they may offer a clue for understanding the various dependencies of strain hardening on elongational rate due to the variation of branching structure attainable. For elucidating these relations, however, a molecular model should exist, along which the molecular structure could specifically be modified and analyzed.
Polyethylenes
Strain hardening plays a special role for the great variety of commercial polyethylenes insofar as these products are widely used in processing operations involving elongational flow. For example, low-density polyethylenes (LDPE) are preferred materials for film blowing where the uniformity of the wall thickness of films is an important criterion for material and processing performance. It is well known that strain hardening affects this application-relevant property. Linear high-density polyethylenes (HDPE) and linear low-density polyethylenes (LLDPE) are other polyolefins, the processing of which may comprise components of elongational flow.
In spite of several studies of elongational properties of polyethylenes, their fundamental understanding suffers still from some deficiencies, and particularly strain hardening poses a lot of open questions.
Linear polyethylenes of high density (HDPE)
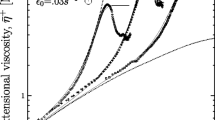
Although apparently very simple in their molecular structure, HDPEs can display very different strain-hardening features. An example is shown in Fig. 10 where the time-dependent elongational viscosities are plotted for the two samples HDPE 1 and HDPE 2. HDPE 1 is a homopolymer with a density of 0.960 g/cm3 at room temperature, and HDPE 2 has a density of 0.954 g/cm3 due to modifications with hexene as a comonomer. The characteristic molecular data are Mw = 152 kg/mol, Mw/Mn = 13 for HDPE 1 and Mw = 173 kg/mol, Mw/Mn = 11.6 for HDPE 2. Although the molar masses and the polydispersity indices of the two HDPE are similar, their viscosity functions in elongation are different. HDPE 1, which was stretched up to Hencky strains of 3, shows a relatively weak strain hardening that becomes smaller with decreasing elongational rate and is not detectable anymore at the two lowest rates of 0.003 and 0.01 s−1. Within the accuracy of the measurements, their time-independent values correspond to 3η0 obtained from shear. The viscosities at the various rates more or less attain plateaus as functions of time indicating the steady states of elongational viscosities. As follows from Fig. 10, these values continuously decrease with elongational rate that means this HDPE is a strain-thinning polymer.

Time-dependent elongational viscosity μ(t) at various constant elongational rates and 150 °C for HDPE 1 and 170 °C for HDPE 2 (Münstedt and Laun (1981))
HDPE 2 exhibits very pronounced strain-hardening features, which are prominent for the three decades of elongational rates measured and show the clear tendency to increase with decreasing rates contrary to HDPE 1. At the largest applied Hencky strains of 3, the viscosity seems to approach a plateau the exact value of which may be difficult to determine, however, due to the effect of non-uniform sample geometries at high deformations superposing strain hardening. The Newtonian viscosity could not be reached neither in elongation nor in shear due to the extremely long relaxation times. In comparison with HDPE 1, this result is very surprising, because the basic molecular data given above are rather similar and, consequently, the rheological behavior should not differ much.
The strain-hardening behavior of HDPE 2 becomes more obvious from Fig. 11 where the strain-hardening coefficient SH = μ(\(\dot{\varepsilon }\),t)/μ(t) as a function of the Hencky strain is plotted for some elongational rates. This plot clearly shows how the strain hardening increases with strain and seems to approach a plateau, which is not reached, however, even at the maximum strain evaluated. The distinct increase of the strain-hardening coefficient with decreasing elongational rate is evident.

Strain-hardening coefficient SH as a function of Hencky strain ε for HDPE 2 at 170 °C and various elongational rates \(\dot{\varepsilon }\) taken from the data of Fig. 10
The strain-hardening feature of HDPE 2 is not unique. Another example is given in Fig. 12 for HDPE 3 of a density of 0.957 g/cm3 with Mw = 222 kg/mol and Mw/Mn = 19.5, the strain hardening of which is still more pronounced than for HDPE 2.

Strain-hardening coefficient SH in dependence on the Hencky strain ε for HDPE 2 and HDPE 3 at two elongational rates
A difference of molecular data can be seen in the higher molar mass and polydispersity index of HDPE 3, from which larger maximum relaxation times should be expected. The assumption that this difference is the source of the increase of strain hardening, particularly pronounced at small rates, would be in contrast to the findings on polystyrene discussed above, which show an increase of strain hardening with growing rates when the molar mass distribution is broadened by the addition of high molar mass components.
Corresponding results on HDPE in the literature are rare, and they suffer from the facts that the measurements often cover a small experimental window only and the molecular structure of the samples is not given in detail. Koyama and Ishizuka (1982) studied the strain-hardening behavior of four commercial HDPEs with various high molar mass tails at the elongational rates of 0.004 and 0.04 s−1. The molar mass distributions were described by the polydispersity indices Mw/Mn of 7, 17, 25, and 43, respectively. The densities were around 0.95 g/cm3 indicating comonomer contents, which were not specified, however. The two samples with the higher polydispersity indices exhibited negligible strain hardening in comparison to the two others with lower polydispersities at least under the experimental conditions chosen. For these, increasing strain hardening with decreasing rate was found. This result is another example that the molar mass distribution is not decisive for strain hardening. Long-chain branching has to be excluded according to the authors.
Of four HDPE with densities smaller than 0.960 g/cm3 studied in elongation by Linster and Meissner (1986), two showed increasing strain hardening with decreasing elongational rate, whereby the measurements of the two others conducted only at one rate can be interpreted of being weakly strain hardening. Neither the densities nor the molar masses and polydispersity indices offer any hint to even qualitative conclusions due to the limited rheological data available.
Kobayashi et al. (1996) report results of an HDPE with Mw = 250 kg/mol and Mw/Mn = 32, but unknown density that showed increasing strain hardening with decreasing rate, and a similar behavior was found by Wagner et al. (1998) for an HDPE with ρ = 0.951 g/cm3, Mw = 104 kg/mol, and Mw/Mn = 5.5.
The uniaxial extension of a blow molding HDPE not further specified was studied by Langouche and Debbaut (1999) with the aim of finding an adequate numerical description. At the three elongational rates of 0.01, 0.1, and 1 s−1 applied, they observed distinctly larger strain hardening for the lower rates.
An HDPE of ρ = 0.960 g/cm3 with Mw = 125 kg/mol and Mw/Mn = 9.8 was studied by Schweizer (2000). He found very weak strain-hardening effects not differing much in a range of elongational rates between 0.01 s−1 and 1 s−1 at Hencky strains of 3.
From the results above, two types of high-density polyethylenes may be classified with respect to strain hardening. For some few samples, the time-dependent elongational viscosity comes close to the linear behavior over a wide range of elongational rates and strain hardening is negligible within the accuracy of the measurements. The others exhibit pronounced strain hardening that increases with decreasing rate. Surprisingly, these features do not show any relation to the molecular structure characterized by molar mass and polydispersity index determined in the usual way by size exclusion chromatography. There seems to be an indication, however, that strain hardening is related to samples with densities smaller than 0.960 g/cm3. The results of strain hardening increasing with decreasing rate obtained on a variety of HDPE samples by different authors are very interesting insofar as they point to the relevance of structural elements that are not determined by the usual analytical methods applied.
A hint to such a supposition becomes obvious from the results on an HDPE named IUPAC 5A showing a unimodal molecular distribution with Mw = 220 kg/mol and Mw/Mn = 2.5 by conventional SEC equipped with a refractive index detector. Its density of 0.950 g/cm3 indicates the existence of comonomers the concentration and type of which are unknown, however. This sample exhibits pronounced strain hardening increasing with decreasing elongational rate as can be seen from Fig. 13. The maxima of the viscosity curves are due to non-uniform sample deformations at higher strains.

Elongational viscosity μ as a function of time t for HDPE IUPAC 5A at T = 180 °C and different elongational rates \(\dot{\varepsilon }\)
Corresponding creep experiments in elongation at stresses of 15 and 25 kPa shown in Fig. 14 indicate a shoulder and a plateau of elongational rate at longer times superimposed by an increase of rate due to non-uniform sample deformation. These features can be interpreted by the existence of two dominant retardation processes. The viscosities assessed from the shoulders can be assumed to be the same for the two stresses, and they are equal to the Trouton relation. The viscosity determined from the second plateaus is larger for the lower stress. This tendency corresponds to the observed strain hardening increasing with decreasing rate. As reported by Gabriel and Münstedt (2003), a high molar mass component was detected for IUPAC 5A using laser light scattering. The occurrence of a high molar mass component does not explain the rate dependence of the strain hardening, however, because it is just opposite to the polystyrene blends above, exhibiting a more pronounced strain hardening with rising elongational rate if a high molar mass component of the same chemical composition is added.

Elongational rate \(\dot{\varepsilon }\) as a function of time t from creep experiments at 180 °C and two constant stresses σ for HDPE IUPAC 5A.The increase of \(\dot{\varepsilon }\) at longer times is due to the beginning of non-uniform sample deformation
Rather, separate phases have to be postulated that are discussed below.
Linear polyethylenes of low density (LLDPE)
Extensive studies of elongational properties of LLDPEs were performed by Schlund and Utracki (1987a). They stretched 10 samples differing in molar masses Mw between 111 and 256 kg/mol and polydispersity indices Mw/Mn between 7 and 42 at various elongational rates up to break. Of these specimens, only two showed strain hardening, but this behavior could not be correlated with the molecular parameters. Moreover, different tendencies were observed for strain hardening. The one sample exhibited strain hardening increasing with decreasing rate but the other behaved just the opposite. Experimental details like the uniformity of sample deformation being essential for the reliability of the measurements were not discussed, however.
The sensitivity of elongational flow of linear low-density polyethylenes with respect to molecular structure becomes obvious from Fig. 15. The sample mLLDPE 1 was polymerized using a metallocene catalyst and 1-butene as a comonomer. The density is 0.900 g/cm3 and its characteristic molecular data are Mw = 114 kg/mol and Mw/Mn = 2.1 (Gabriel and Münstedt (2003)). At the elongational rates used, this relatively narrowly distributed polyethylene does not show any indication of strain hardening up to the Hencky strain of 3 applied. The role of long-chain branches for strain hardening becomes obvious from the other sample in Fig. 15. LCB-mLLDPE 1 stands for a metallocene-catalyzed LLDPE with Mw/Mn = 2.1, whose small amount of long-chain branches occurring preferentially at higher molar masses was proven by the slight deviation of the radius of gyration in dependence on the absolute molar mass from the curve for the linear mLLDPE (Gabriel and Münstedt (2003)). As it is obvious from Fig. 15, the long-chain branches effect strain hardening the rate dependence of which is hard to assess, however.

Time-dependent elongational viscosity μ of a linear (mLLDPE) and a slightly long-chain branched (LCB- mLLDPE) linear low-density polyethylene with Mw/Mn = 2.1 stretched up to the Hencky strain 3 (Gabriel and Münstedt (2003))
Rheological studies on a series of ethylene/hexene copolymers polymerized with various metallocene catalyst were reported by Malmberg et al. (2002). Some of them show indications of faint long-chain branching as concluded from the kind of dependence of the zero-shear viscosity on the weight average molar mass. Measurements of the molecular structure by light scattering were not performed and, thus, the characterizations remain somewhat vague. In elongational experiments, either no strain hardening or strain hardening increasing with decreasing elongational rate was found. It is difficult, however, to draw any founded conclusions with respect to the structural reasons of the strain hardening observed.
The elongational behavior of LLDPEs and their strain hardening become still more complex when studies published by Gabriel et al. (1998) and Münstedt et al. (1998) are considered. The commercial product with the density ρ = 0.920 g/cm3, Mw = 105 kg/mol, and Mw/Mn = 3 is an octene copolymer polymerized with a Ziegler–Natta catalyst. This sample designated ZN-LLDPE has a strain-hardening characteristic similar to that of some HDPEs discussed before as Fig. 16 demonstrates. The sample stretched up to the Hencky strain 3 showed a significant growth of strain hardening by decreasing the elongational rate from 1 to 0.01 s−1. Creep experiments in elongation reveal an interesting result insofar as the creep curve ε(t) is composed of two parts with different nearly constant slopes as Fig. 17 indicates. This feature becomes more obvious from the elongational rate as a function of time shown in Fig. 17, which clearly exhibits two plateaus with constant elongational rates pointing to the existence of two separate retardation processes within the polyethylene sample. A hint to two retardation processes occurring for the ZN- LLDPE was obtained also from the two steps found in shear creep recovery experiments (Gabriel et al. (1998)). Further analyses in this paper showed two clearly separated melting peaks by differential scanning calorimetry (DSC) and two peaks in the temperature-rising elution fractionation (TREF), whereby the minor fraction with the lower comonomer content had the distinctly higher molar mass. From these results, it may be postulated that the ZN-LLDPE studied consists of two separate fractions of different molar masses. A model based on this assumption and the rheological consequences are discussed below.

Time-dependent elongational viscosity μ of ZN-LLDPE at150 °C and different elongational rates (Münstedt et al. (1998))

Elongational rate \(\dot{\varepsilon }\) and Hencky strain ε as functions of time t for ZN-LLDPE at 150 °C and a stress of σ = 103. Pa (Münstedt et al. (1998))
Low-density long-chain branched polyethylenes (LDPE)
Somewhat less complex and often seen as a typical feature of uniaxial elongational flow is the strain-hardening behavior of long-chain branched low-density polyethylenes (LDPE) polymerized under high pressure. For this type of polyethylene, large degrees of branching with tree-like geometries are widely accepted structures. In Fig. 18, the elongational viscosity as a function of time is plotted at various elongational rates for LDPE 1 with ρ = 0.918 g/cm3 at room temperature, Mw = 467 kg/mol, and Mw/Mn = 25 in comparison to the less branched LDPE 2 with ρ = 0.928 g/cm3, Mw = 256 kg/mol, and Mw/Mn = 10 (Münstedt and Laun (1981)). The curves exhibit a similar feature. Starting from low elongational rates at which the elongational viscosity follows the linear behavior and, thus, strain hardening does not occur, this property becomes more pronounced with the increasing rate for both samples stretched up to a Hencky strain of 4. Although the molar mass distributions are somewhat different, it is obvious that strain hardening is more pronounced for the sample of lower density pointing to a larger efficiency of branching.

Elongational viscosity μ as a function of time t at various elongational rates up to a Hencky strain of about 4 for two LDPEs with different densities (ρ = 0.918 g/cm3 for LDPE1, ρ = 0.928 g/cm3 for LDPE2 at room temperature). Other characteristic data are given in the text (Münstedt and Laun (1981)). The numbers on the curves describe the elongational rates in s−1. The data of LDPE 1 are shifted by the factor 30
The role of the architecture of long-chain branches for the feature of strain hardening becomes obvious from a comparison of Figs. 15 and 18. As shown by Gabriel and Münstedt (2003), the degree of branching of LCB-mLLDPE 1 is faint and only traceable by SEC–MALLS for high molar masses. Thus, a star-like architecture is probable for LCB-mLLDPE 1, which leads to a less pronounced strain hardening than the tree-like structure of the LDPEs.
Blends of linear and long-chain branched polyethylenes
An LDPE showing strain hardening increasing with elongational rate was blended to a non-strain-hardening LLDPE (Schlund and Utracki (1987b)). By adding 10 wt% of the LDPE, strain hardening was visible already and became more and more pronounced with higher content exhibiting the typical LDPE feature of strain hardening increasing with rate. Similar results are reported by Steffl (2004) who studied the effect of the addition of an LDPE to two different LLDPEs on their elongational behavior. The molecular data of the LDPE were Mw = 130 kg/mol and Mw/Mn = 11, the corresponding values for LLDPE 1 were Mw = 92 kg/mol, Mw/Mn = 5, and for LLDPE 2 Mw = 150 kg/mol, Mw/Mn = 7. At the low elongational rate of 0.1 s−1, the viscosity function followed the linear curve, but distinct strain hardening was measurable at the two largest elongational rates of 0.5 and 1.0 s−1 and a Hencky strain of 3 for each of the LLDPE/LDPE blends with the two non-strain-hardening LLDPE matrices. Strain hardening became stronger with growing LDPE content. Of some interest may be the observation that the strain-hardening effect of LDPE is more pronounced for the LLDPE 2 matrix of higher molar mass. Wagner et al. (2004) studied blends of a commercial LDPE with five long-chain branches per 1000 carbon atoms of the backbone determined by NMR and an LLDPE with 3.6 mol% 1-butene polymerized using a Ziegler-Natta catalyst. The LDPE showed a distinct strain hardening whose exact dependence on elongational rate is difficult to determine, however, because the total elongations applied were different. For the LLDPE stretched to a Hencky strain of 3, a faint indication of strain hardening was visible at the two highest elongational rates of 0.5 and 1 s−1. An addition of only 5 wt% LDPE caused distinct strain hardening becoming more pronounced with increasing rate.
These results are different from those on the blends of a linear and various amounts of a long-chain branched polypropylene that showed a decrease of strain hardening with increasing rate for LCB-PP contents smaller than 50 wt% as seen in Fig. 9.
Polybutadienes
Other semi-crystalline polymers are polybutadienes commercially available with a great variety of molecular parameters. They have a predominantly linear molecular structure. Long-chain branches can be introduced by chemical modifications. Their commercial importance and the processing around room temperature have made polybutadienes interesting for rheological studies also in elongation, and some results on strain hardening are addressed in the following for the matter of completeness.
Linear polybutadienes
Results on polybutadienes with narrow molar mass distributions and blends from them were reported by Berger and Meissner (1992). Blends of PB 1 with Mw = 58 kg/mol and Mw/Mn = 1.05 and PB 3 with Mw = 348 kg/mol and Mw/Mn = 1.08 on the one side, and those of PB 3 and PB 2 with Mw = 161 kg/mol and Mw/Mn = 1.10 on the other were studied in elongation at the two elongational rates of 0.01 and 0.1 s−1. For PB 2, distinct strain hardening was found at the lower rate. At the higher rate, only a faint strain hardening could be seen up to the measured total Hencky strain of 3. The strain hardening at the two rates distinctly increased when PB 3 of higher molar mass was added, but it remained more pronounced at the lower rate for samples up to weight ratios of 0.2 PB 3. For the blend PB 1/PB 3 with a weight ratio of around 0.5, just the reverse rate dependence of strain hardening was found at 0.1 and 0.01 s−1. A convincing explanation of this surprising behavior was not given, however.
Branched polybutadienes
Rheological properties in shear and elongation of a linear and three long-chain branched polybutadienes with different degrees of branching were studied by Kasehagen and Macosko (1998). The characteristic molecular data of the linear anionic precursor were Mw = 137 kg/mol and Mw/Mn = 1.04. Branching was introduced using a hydrosilation reaction with a difunctional crosslinker (Kasehagen et al. (1996)). The differently branched samples were characterized in elongation by measuring the elongational viscosity as a function of time up to Hencky strains of 2 at elongational rates between 0.05 and 1 s−1. All the samples including the linear precursor were found to be strain hardening, whereby this effect became more pronounced with increasing degree of branching. Two observations are of particular interest. Even the linear precursor showed a small strain hardening with the tendency of an increase with rate. This result is similar to that on anionic polystyrene in Fig. 2 and may point to a small high molar mass component not detected by size exclusion chromatography. In contrast, for the weakly branched sample, strain hardening was found to be stronger at lower than at higher rates, and this rate dependency is maintained for the two samples with still higher degrees of branching.
Interesting conclusions were drawn from a numerical description of the measurements in shear and elongation by the so-called K-BKZ time-integral constitutive equation. It is based on the separation of time and deformation effects making use of the relaxation spectrum and the damping function, respectively. Spectra were determined from shear start-up experiments in the linear range, and the damping functions were calculated from corresponding non-linear experiments at higher shear rates. Relaxation spectra and damping functions were found to be related to molecular parameters of the samples as known from the literature.
Based on the K-BKZ constitutive equation, the authors derived a numerical description of the time and rate dependence of the elongational viscosity using the relaxation spectrum and the damping function obtained in shear. Surprisingly, they were able to describe the elongational curves with reasonable accuracy using the damping function in shear. Furthermore, they demonstrated on a sample with distinct strain hardening that the choice of the damping function either obtained for the linear or the strongest branched sample had an only minor effect on the numerical performance. Very interesting are the simulations which show that the addition of a long relaxation time to the spectrum may induce pronounced strain hardening even for a choice of relaxation times and strengths hardly influencing the zero-shear viscosity.
This result may give a hint to the high sensitivity of uniaxial tensile experiments with respect to minute high molar mass components or small branching contents reported in the literature.
All in all, the elongational rate dependence of the polybu-tadienes from the literature leaves some questions open concerning correlations with the molecular structure. This uncertainty may be due to the double bonds in polybutadienes bearing the potential of crosslinking during sample preparation and measurement that may influence elongational flow. Furthermore, nothing is said about the reliability of the experiments and the uniformity of elongation. Thus, the results on polybutadienes have not been considered in the following attempt to interpret the different rate dependencies of strain hardening experimentally observed.
Interpretation of melt strain hardening
General considerations
According to the literature, it is widely accepted that strain hardening for linear polymers is expected when the experimental time of deformation is shorter than the stretching time of a molecule chain characterized by its Rouse time τR. For example, Marrucci and Ianniruberto (2004) discuss the dependence of the steady-state elongational viscosity on elongational rate for a linear polymer in the light of the tube model. They postulate that the steady-state elongational viscosity decreases linearly with the elongational rate \(\dot{\varepsilon }\) from its value in the linear range of deformation for \(\dot{\varepsilon }\) < 1/τd where τd is the terminal relaxation time. They further predict that the elongational viscosity is expected to increase with elongational rate if it becomes larger than the reciprocal Rouse time 1/τR, i.e.,
Under such condition, the chains start to stretch out, and this behavior results in an increase of the resistance to flow that should become more pronounced the larger the rate. This model, although related to the steady state, is applied to the qualitative discussion of the transient effects of strain hardening preferentially considered in the experimental part of this paper.
Determination of the Rouse time for entangled polymer melts
Relation based on the zero-shear viscosity
From the extension of the Rouse time for non-entangled polymers to entangled species follows according to Dealy and Larson (2006) and Menezes and Graessley (1982)
with Mc being the critical molar mass for the effect of entanglements on the zero-shear viscosity η0 and a the exponent of the relation
As well known, K is a temperature-dependent polymer-specific quantity and a = 1 for M ≤ Mc and a = 3.4 to 4 for M > Mc. In the case of polydisperse melts, M stands for the weight average molar mass Mw. Mc is found to lie between 2 Me and 3 Me with Me being the molar mass between entanglements. ρ is the density of the melt at the absolute temperature T and R the universal gas constant.
For the matter of completeness, it should be mentioned that some authors use the factor 12 instead of 6 in Eq. (3).
Rouse-time from dynamic-mechanical experiments
As well known, dynamic-mechanical experiments in the linear range of deformation at high frequencies reflect properties of molecule segments between entanglements. Widely used is the deduction of the molar mass between two entanglements Me from the so-called plateau modulus \({G}_{N}^{0}\) occurring more or less distinctly at higher frequencies. By extending the frequency range beyond this regime, which may be experimentally challenging, however, two features were found that are exploited for the determination of Rouse times. The one is the existence of a second crossover of G′ and G″ and the other is the experimental finding of G′ = α ω0.5 within a distinct range of angular frequencies. By some authors, the reciprocal of the second crossover frequency is postulated to be the longest Rouse time corresponding to the relaxation time τe beyond which entanglements constrain the chain dynamics (see Liu et al. (2007)). Others determine this characteristic time from the frequency at the intersection point of G′ = α ω0.5 with \({G}_{N}^{0}\), that means
From the relation
the Rouse times in dependence on the molar mass M are usually determined for a distinct molecule. In the case of real polymers, one has to deal with a distribution of molar masses and, consequently, with a distribution of Rouse times. For many polymers, this fact makes a discussion of relations between elongational flow and Rouse times very complex and, thus, the weight average molar masses Mw are frequently used to obtain characteristic Rouse times according to Eq. (6).
Osaki et al. (2001) derived the relation
for solutions with c being the polymer concentration and α the slope of G′ scaling with ω0.5 in the high-frequency region. This formula is occasionally applied to polymer melts replacing c by the density ρ.
Linear polymers
Discussions of elongational flow of polymer melts in the light of their Rouse times are scarce in the literature. That is mainly due to the difficulties of their determination and getting reliable data with respect to molecular parameters. Some few data can be found for polystyrenes that are used in the following to discuss the experimental results shown above.
Polystyrenes
In a very recent paper (Zhong et al. (2022)), τe was determined directly from the second crossover point of G′ and G″ as function of angular frequency at 130 °C for the three narrowly distributed polystyrenes PS 545 K, PS 285 K, and PS 185 K with Mw of 545, 285, and 185 kg/mol and polydispersity indices between 1.12 and 1.03. These samples are of special interest insofar as their Rouse times are well defined due to the narrow molar mass distributions. The value of τe = 0.100 s is reported to be independent of molar mass. This independency looks physically reasonable because all the three samples have entanglements with the same molar mass Me that is found for polystyrene in the literature to lie between 13 and 17 kg/mol. Making use of Eq. (6) and assuming Me = 17 kg/mol, Rouse times for the three polystyrenes follow as 102, 28, and 12 s, respectively.
Huang et al. (2013) made use of the four methods based on Eq. (3), Eq. (7), the Likhtman-McLeish theory (Likhtman and McLeish (2002)), and the spectrum analysis according to Baumgaertel et al. (1992) to determine the Rouse times of polystyrenes. They report Rouse times between 700 and 800 s for PS 545 K and around 200 s for PS 285 K at 130 °C. These data are obviously significantly different from 102 and 28 s, respectively, given above for the same two samples.
For the polystyrene PS 185 K with Mw = 185 kg/mol, τR = 0.271 s at 160 °C was determined by Costanzo et al. (2016)) from the spectrum analysis according to Baumgaertel et al. (1992). Using the time–temperature shift factor of aT = 200 as given by Zhong et al. (2022), for example, τR = 54 s follows at 130 °C, which is remarkably different from the Rouse time of 12 s for PS 185 K given above. However, good agreement is found with the above data of Huang for PS 545 K and 285 K if the Rouse times are related to the molar mass of PS 185 K by making use of Eq. (6).
The missing agreement between the Rouse times obtained by various authors is very dissatisfying, particularly for the discussion of their role in strain hardening. The reasons for the discrepancy have not been addressed in the literature. From the comparison of the studies above, it is obvious that the Rouse times determined according to the second crossover of the dynamic moduli are smaller than those from the other methods. Because of the good consistence of the data for PS 545 K and PS 285 K from the four different methods published by Huang et al. (2013), these Rouse times are taken in the following as base for a discussion of the experiments on strain hardening of various polystyrenes.
Comparison of model predictions with experiments
Looking at the elongational behavior of the polystyrenes in Figs. 2 and 3, a region of elongational viscosity decreasing with elongational rate preceding the viscosity increase as predicted by Marrucci and Ianniruberto (2004) is not found. Rather, the viscosities rise directly from their Newtonian values. This behavior is particularly significant for PS II with the distinct high molar mass component and PS-r-95 with a broader molar mass distribution. The molar mass distribution seems to have a remarkable influence on the shape and development of the time-dependent viscosity curve because it is obvious that the strain hardening of PS II with the high molar mass component is more pronounced than that of the narrowly distributed PS I. Furthermore, for this sample, a steady state of the viscosity is reached at smaller Hencky strains than that expected for PS II.
To discuss the critical elongational rate for the onset of strain hardening according to the relation \(\dot{\varepsilon }\)> 1/τR above, the Rouse times of the polystyrenes in Figs. 2 and 3 have to be assessed. For this purpose, Eq. (6) is used. Because the entanglement molar mass Me and the entanglement relaxation time τe can be assumed to be the same for all polystyrenes with molar masses Mw > Mc, one gets
This relation well describes the findings of Huang et al. (2013) on PS 545 K and PS 285 K reported above. Starting from τR = 200 s at 130 °C for PS 285 K with Mw = 285 kg/mol, the Rouse time of PS I with Mw = 74 kg/mol becomes 13.5 s and that of PS II with Mw = 39 kg/mol is 3.7 s.
From Fig. 2, the critical elongational rate for the onset of strain hardening for PS I can be assessed to lie between 0.014 and 0.051 s−1 and that for PS II between 0.001 and 0.003 s−1. The critical elongational rates according to \(\dot{\varepsilon }\) c = 1/τR are 0.07 for PS I and 0.27 s−1 for PS II, respectively. For the narrowly distributed PS I, prediction and measurement are comparable considering the experimental uncertainty of τR of the reference PS 285 K, the approximations with respect to the molar masses underlying Eq. (8) and the experimental range for determining the critical elongational rate. However, a total disagreement has to be stated for PS II. Even the tendency of critical rates experimentally found to be distinctly smaller than for PS I does not become plausible from the simple relations above. This significant discrepancy points to the role the high molar mass component of PS II may play for the strain hardening of polymer melts. Furthermore, it has to be considered that the total molar mass is close to the critical value for entanglement and, thus, not all the molecules may be entangled.
For PS-r-95 with Mw = 150 kg/mol and Mw/Mn = 1.6, the critical elongational rate for strain hardening \(\dot{\varepsilon }_c\) lies between 0.3 and 1 s−1 at 169 °C (Fig. 3). Adding 1 wt% of an anionic polystyrene with Mw = 1 800 kg/mol increases Mw of the matrix by about 10%, but lowers \(\dot{\varepsilon }_c\) by a factor of at least 100 (Fig. 3). This result is in contrast to measurements by Auhl et al. (2009) on blends of two nearly monodisperse polyisoprenes with Mw = 483 kg/mol and Mw = 34 kg/mol. They report an increase of \(\dot{\varepsilon }\)c with growing content of the high molar mass component.
Discrepancies are also found if the Rouse times of PS-r-95 and the narrowly distributed PS 285 K are compared, that were measured at different temperatures, however, and, thus have to be adapted. The range of the critical elongational rate for PS-r-95 measured at 169 °C reduces to values between 0.0006 and 0.002 s−1 at 130 °C by applying the shift factor of 500 from the literature for PS. The Rouse time for the PS-r-95 at 130 °C related to that of PS 285 K from above according to Eq. (8) is 56 s and the corresponding critical elongational rate \(\dot{\varepsilon }_c\) = 0.018 s−1. The discrepancy between prediction and experiment is beyond any experimental uncertainties. This result together with the above finding of a substantial lowering of the critical elongational rate by adding 1 wt% of a high molar mass component that changes the total molar mass only marginally is not consistent with the molar mass dependence of Rouse times according to Eqs. (6) and (8). These results are another hint to the role that molar mass distributions in general and high molar mass components in particular may play for strain hardening.
Comparing the Rouse time of the high molar mass component of 1800 kg/mol with that of the matrix PS-r-95 with Mw = 150 kg/mol, a factor of about 150 can be expected according to Eq. (8). Then the corresponding critical elongational rate for strain hardening of the high molar mass component should be lower by this factor, which is in the range of the values that may be assessed for the blends with the only small content of the high molar mass component in Fig. 3. This consideration demonstrates the determining role a high molar mass may play for the onset of strain hardening in case of polystyrene blends.
From Fig. 3, it cannot be concluded, however, whether the increase of the high molar mass component from 1 to 5 wt% further shifts the critical elongational rate. But it can be said that the strain hardening itself becomes more pronounced with the amount of the high molar mass component. For all the three samples of Fig. 3, strain hardening increasing with elongational rate is found.
Other amorphous polymers
The strain-hardening features of polymethylmethacrylate (PMMA) and styrene-acrylonitrile copolymer (SAN) of broad molar mass distributions and the effect of small high molar mass components shortly addressed above are similar to those of polystyrene. This behavior may be understood qualitatively according to the considerations for polystyrene, but comparable data on Rouse times are scarce and not convincing. Thus, Narimissa et al. (2021) determined the Rouse times for two PMMA by making use of Eq. (3) and rheological data of Morelly et al. (2019). Surprisingly, the Rouse time of the sample with higher molar mass was found to be distinctly lower than that of smaller molar mass. This discrepancy is explained by the assumption that different tacticities may affect the Rouse times. The principal dilemma of comparing Rouse times from various methods and authors as discussed for PS above becomes also obvious from data of the two PMMA. Morelly et al. (2019) report Rouse times about three times as high as those of Narissima et al. (2021), but their tendency is the same, at least. Morelly et al. calculated the Rouse times according to Eq. (6), whereby τe was set equal to the inverse of the angular frequency of the second crossover of G′ and G″ as shortly described above.
The absence of strain hardening of polycarbonates (PC) with molar masses around Mw = 30 kg/mol and a polydispersity index of 2 found at 200 °C by Hepperle (2002) becomes plausible from the Rouse time calculated according to Eq. (3) making use of the molecular and rheological data given. From the Rouse time of τR ≈ 10−2 s obtained, a critical elongational rate of 100 s−1 follows for the occurrence of strain hardening that is beyond any experimental realization.
Linear polyolefins
Measurements of Rouse times for polyolefins are scarce in the literature. Szanto et al. (2017) determined the entanglement relaxation times τe of various polyethylenes by high-frequency rheometry. They reported values of some few 10−9 s at 190 °C, which were surprisingly found to be not much dependent on the molecular structure of the samples covering HDPE, LDPE, and ethylene/vinylacetate copolymers. The data were compared with those from the literature based on calculations using various models. Differences up to a factor 10 exist for τe. Nevertheless, for qualitative discussions, it may be assumed that τe of PS is larger by about four decades than that of PE compared at 190 °C. This difference becomes smaller, however, if τR is considered. According to Eq. (6), the Rouse times taken at the same Mw scale with \({M}_{e}^{2}\) and thus, the difference of four decades between τe of PS and PE shrinks to around two decades of τR when Me of 17 kg/mol for PS and 1 kg/mol for PE are compared. This rough estimate leads to the conclusion of a distinctly larger critical elongational rate for strain hardening of PE in comparison to PS and agrees with the tendency following from the assessment of Rouse times below making use of Eq. (9).
Regarding PP, whose strain-hardening behavior and that of various modifications are displayed in the experimental part above, values for τe could not be found at all in the literature. Nevertheless, differences of the Rouse times between PS and PP may be assessed using Eq. (3). Comparing two polymer melts at the same temperature and density and assuming that their zero-shear viscosities are described by the well-established power laws
the ratio of their Rouse times for a constant molar mass follows from Eq. (3) as
From a qualitative point of view, the assumption of the same exponent a may be justified by the experimental finding of values between 3.4 and 4 for most polymer melts.
The Rouse times of PP and PS can be assessed as follows. According to Auhl (2006), for PP at 180 °C the relation
holds and for PS at 180 °C according to Hepperle (2002)
with Mw in g/mol. The critical molar masses Mc for entanglement are 33,000 g/mol for PS and 12,000 g/mol for PP (e.g., (Dealy and Larson 2006)) and, thus, Eq. (9) becomes
That means the Rouse time of PP at 180 °C is smaller by about the factor 1000 than that of PS at the same temperature.
These differences should have a remarkable effect on the onset of strain hardening if one follows Eq. (2). For example, our experimental finding, that within the given experimental window, the PS in Fig. 3 shows strain hardening at 170 °C but the polypropylenes in Fig. 5 at 180 °C not, becomes plausible from the different Rouse times assessed above and the critical elongational rates following from them. Obviously, the critical elongational rate for PP is too high to get experimentally verified.
Similar assessments can be made for HDPE. According to Stadler (2007)
is valid for HDPE at 180 °C with Mw in g/mol. Mc ≈ 2 Me can be assumed to be around 2 000 g/mol (e.g., Dealy and Larson (2006)). With these data, the ratio of the Rouse times of HDPE and PS from Eq. (9) becomes 1.3∙10−4 and is obviously still smaller than that between PP and PS. From this value, it is plausible that the necessary rates to obtain strain hardening of linear homologous HDPE are outside the experimental window attainable.
Strain-hardening effects sometimes reported for commercial HDPE and PP are supposed to probably have their origin in deviations from the linear structure of a homopolymer. This topic is discussed below. The feature for HDPE becomes more complicated, however, by the results demonstrated in Figs. 11, 12, and 13 of strain hardening increasing with decreasing elongational rate that has never been observed for linear PS and PMMA.
Conclusions on linear polymers
The model based on the Rouse time is helpful for a qualitative understanding of the experimental findings that the amorphous polymers PS and PMMA studied in this paper show strain hardening but PC not. Furthermore, it becomes plausible that linear PP and HDPE of molar masses comparable to those of PS do not exhibit strain hardening. These differences go back to the distinctly smaller Rouse times assessed above for polyolefins and to critical elongational rates following from them that are outside the experimental windows accessible.
Trying to use the model for a more quantitative interpretation of the studies on polystyrenes with special molar mass distributions shows its limitations, however. Thus, it fails to describe the effect of high molar mass components on the critical elongational rate for strain hardening by considering the total molar mass of the sample because already minute additions of long molecules shift the critical rates to smaller values. This finding points to the role of the molar mass distribution for strain hardening and its interpretation, but the influence is difficult to quantify. These deficiencies are obvious because the Rouse time per definition depends on the molar mass of a molecule and, consequently, using the weight average molar mass of a polymer for its determination does not reflect the contributions of single molecules. But even for narrowly distributed samples, comparable results of the influence of molar mass on the Rouse time are difficult to derive. One reason for this observation can be seen in the different experimental methods used by various authors. The other may be high molar mass components not detected by the molecular characterizations applied.
For the linear polymers exhibiting strain hardening, it can be concluded that its intensity at a distinct elongation increases with elongational rate in the experimental windows applied.
Not understandable from the assumption of Rouse times governing strain hardening is the result on PP/UHMWPE in Fig. 6 showing strain hardening increasing with decreasing rate, although neither for the PP matrix nor for the HDPE component strain hardening should be expected. This behavior is just opposite to the effect of a high molar mass component on the strain hardening expected from the measurements on PS in Fig. 3 for a miscible system. Thus, other strain-hardening mechanisms have to be discussed that may have their origin in some kind of interactions between polyethylene and polypropylene molecules at the phase boundary.
Furthermore, the Rouse-time model does not explain the strain-hardening behavior of the HDPE with small amounts of comonomers and certain Ziegler–Natta polymerized LLDPE increasing with decreasing elongational rate. For an explanation of these features, other mechanisms have to be discussed.
Various strain-hardening mechanisms of heterogeneous systems published in the literature
Molecular interactions at the interface of different phases are reported by Lopez-Barron and Tsou (2017) on blends of a high-density polyethylene (Mw = 70 kg/mol) and an atactic polypropylene (Mw = 306 kg/mol) modified with a poly(ethylene-cb-polypropylene) comb copolymer as a compatibilizer. They found separated phases by atomic force microscopy (AFM) in a 50/50 PE/PP blend with various amounts of the compatibilizer. The PE/PP blend did not show strain hardening, while the addition of the copolymer led to significant strain hardening being distinctly larger at 5 than at 1 wt%. The strain hardening increased with elongational rate first but was found to be smaller at the highest rate of 10 s−1. From the AFM micrographs, a distinct phase deformation could be seen. The authors postulated a mechanism for strain hardening based on the unraveling of the PP arms from the entangled PP phase and their repulsion from the molecules in the PE phase.
Interesting in this aspect is a study by Shabbir et al. (2015) on the effect of hydrogen bonding on elongational flow. They investigated a model system of pure poly(n-butyl acrylate) (PnBA) and copolymers with several amounts of poly(acrylic acid) (PAA). These species are distinguished by different degrees of hydrogen bonding. The authors report strain hardening growing with acrylic acid content corresponding to stronger hydrogen bonding. In all cases, they found strain hardening increasing with decreasing elongational rates down to the lowest rate of 0.001 s−1 measured. As the authors state, this experimental finding is surprising, because according to the Rouse times determined by the method of Baumgaertel et al. (1992), there should not occur any strain hardening for strain rates lower than 13 s−1.
This result may be understood by assuming that hydrogen bonding has an effect on the deformation of molecules in the way that it enhances their resistance to flow. Higher elongational rates result in larger stresses that may overcome those due to hydrogen bonding and make molecular motion possible and decrease strain hardening. Although restricted to special molecular structures, the effect of hydrogen bonding is a hint that molecular processes other than segment stretching according to Rouse may be the source of strain hardening.
Another interesting result throwing light on strain hardening follows from the elongational behavior of a polypropylene modified with 0.5 wt% maleic anhydride (MAPP) and filled with 5 wt% of a dimethyl hydrogenated tallow ammonium nanoclay (Cloisite 20 A) (Park et al. (2006)). The exfoliated nanoclay gave rise to remarkable strain hardening increasing with decreasing elongational rate.
Measurements on the unfilled MAPP were not shown, probably because of its too low viscosity. But considering the well-known non-strain-hardening feature of PP, it is very likely that the strain hardening observed for PP/nanoclay is due to electrostatic interaction forces between the modified polypropylene molecules and the ionic properties of the clay particles with their large surface area due to exfoliation. Again, it can be postulated that the hindrance of molecular mobility and therewith the resistance to flow is more pronounced the smaller the stress and, subsequently, the elongational rate applied. The requirement for such a model is the absence of a percolation network by the particles that seems to be verified by the TEM micrographs shown.
Regarding the influence of particles, it was observed in own studies that the weak strain hardening of a PMMA is not changed by the addition of 1 vol% carbon black, which does not form a percolation network as can be concluded from measurements of the electrical conductivity. This finding being different from those above on MAPP filled with exfoliated nanoclay and the blend of hydrogenated acrylic polymers may be understood by the fact that electrostatic interactions between PMMA molecules and carbon black do not play a role.
Interpretation of strain hardening of the blends of polypropylene with ultrahigh-molecular weight polyethylene
The results on the compatibilized PP/PE blends from above cannot be applied to the PP/UHMWPE blend in Fig. 6 because a compatibilizer was not used. Furthermore, hydrogen bonding does not play a role, and the PP/UHMWPE is free of any fillers that could produce interactions like in the PP/nanoclay described above. Furthermore, a rough estimate of the interfacial tension between the UHMWPE particles and the PP matrix reveals that the tensile stresses applied are not high enough for their deformation. Rather, it is postulated that special effects at the phase boundary come into play.
Thus, it is assumed that entanglements between PP and PE molecules are formed within a small surface layer. They are most effective at small strain rates and gradually lose their efficiency at higher rates similar to the effect of shear thinning being a general feature of entangled polymer melts. Such a process taking place in a small boundary regime may leave the overall shape of the PE phase unchanged as indicated by the TEM micrographs of Sugimoto et al. (2001b). Indications of a relaxation process with long relaxation times can be derived from a plateau-like behavior of the storage modulus at small angular frequencies for PP/UHMWPE reported by these authors, at which a modulus declining with decreasing frequency has to be expected for the PP matrix.
For polyethlylene and polypropylene, it is discussed controversially in the literature, whether their immiscibility found in the solid state can be assumed to exist in the melt. Phase separation of a PE/PP blend at 200 °C was shown by Wignall et al. (1982) making use of small angle neutron scattering (SANS), and by Rajasekaran et al. (1995) the immiscibility of polyethylene and polypropylene was derived theoretically. Thus, it is very probable that two phases with different rheological properties exist in the PP/UHMWPE melt above that support the hypothesis of entanglements in a thin layer being responsible for the strain hardening and its rate dependence observed.
Linear polyethylenes of various compositions
Interesting is the experimental finding of some HDPEs showing a rate dependence of strain hardening comparable to that of PP/UHMWPE. For the assumption of strain-hardening mechanisms of the HDPEs similar to that of PP/UHMWPE, separated phases should exist, however. Such a postulate is not unrealistic due to the following considerations. The densities of the strain-hardening HDPEs are typically smaller than 0.960 g/cm3 due to minute amounts of α-olefin monomers. It may be assumed that these comonomers are not uniformly attached to the polyethylene backbones and, thus, short-chain branched molecules exist that form separate phases within the matrix of linear polylethylene molecules. The assumption is supported by the finding of liquid–liquid phase separations in blends from a linear polyethylene with α-olefin branched polyethylenes for a model system (Crist and Hill (1997)). Adhikari et al. (2005) prepared blends of a commercial HDPE with various ethylene/octene copolymers in a twin-screw extruder and studied their morphologies by scanning force microscopy. They found distinct phase separations for a blend of a HDPE with 25 wt% copolymer, which they correlated with micro- and macromechanical properties.
In most cases, the molecular composition of HDPEs with lowered density is not known. An exception is the HDPE IUPAC 5A showing strain hardening increasing with decreasing elongational rate (Fig. 13). For this material, a small high molar mass component was reported (cf. section “Linear polyethylenes of high density (HDPE)”) that may be related to an ethylene/α-olefin copolymer forming a separate phase within the matrix of linear molecules. Such a morphology could explain the creep experiments in Fig. 14 showing two retardation processes. The shorter retardation time can be assigned to interactions between the matrix molecules because the elongational viscosities calculated for the two stresses applied correspond to the Trouton relation and indicate that strain hardening does not occur as it is found for homologous HDPE. The longer retardation time may result from a strong entanglement network built up in a small boundary layer between the matrix molecules and those of the separated phase having larger molar masses. The viscosity calculated from the second plateau of elongational rate in Fig. 14 is higher for the smaller stress. This result corresponds to strain hardening becoming more pronounced at the lower elongational rate as shown in Fig. 13. The postulated molecular interactions are thought to be restricted to a minute boundary layer because molecular motions within the separated phase are expected to be not strain hardening as seen from the mLLDPE in Fig. 15.
The assumption of phase separation in HDPE of lowered density is supported by the results on the ethylene/octene copolymer ZN-LLDPE in Figs. 16 and 17 that show strain hardening at low rates and the existence of two retardation processes in the creep test. These retardation times may be related to the two separate phases of linear and short-chain branched molecules with different molar masses found by temperature-rising elution fractionation (TREF) analysis (Gabriel et al. (1998)). The reason for phase separation may be seen in a non-uniform comonomer distribution due to the polymerization process with a Ziegler–Natta (ZN) catalyst. Phase separation in an ethylene/octene copolymer synthesized with a ZN-catalyst is reported by van Ruiten and Boode (1992) according to DSC measurements and TEM characterizations. This interpretation is not in contrast to the mLLDPE shown in Fig. 15. The sample does not exhibit strain hardening, but this result can be understood from the uniform distribution of comonomers along backbone molecules with a narrow molar mass distribution as expected from the metallocene catalyst used. Indeed, the TREF analysis by Gabriel (2001) does not show any indication of non-uniform comonomer dispositions.
Branched polystyrenes
A distinct effect of branches on strain hardening is found for comb polystyrenes reported by Lentzakis et al. (2013) with the backbone molar mass Mb, the arm molar mass Ma, and the number of arms per backbone q (see section on branched PS above). They defined a Rouse time according to Eq. (6) as
with Zb = Mb/Me, Za = Ma/Me and τe being the Rouse time of the polymer strand between entanglements.
For the comb polymer with Mb = 860 kg/mol and Ma = 6.5 kg/mol, a fair agreement between the experimental stretch relaxation time τS = 1/\({\dot{\varepsilon }}_{c}\) and τR according to Eq. (10) with τe = 5∙10−4 s at 170 °C is reported considering the approximations of the model and the uncertainties of the measurements. This agreement becomes worse the larger the molar mass of the arms, and for Ma = 47 kg/mol, τS is higher by a factor of 100 than the corresponding Rouse time. This result points to the growing effect of the arms getting entangled.
Following Lentzakis et al. (2013), the experimental data for the onset of strain hardening of the comb polymers with higher molar mass arms can be approached by introducing a “diluted number of entanglements of a rescaled linear polymer” and the hypothesis that the ratio of the terminal relaxation time to the stretch time is the same as the ratio of the terminal relaxation time to the Rouse time for an equivalent linear polymer with the diluted number of entanglements, whereby the relation for the terminal relaxation time of the equivalent linear polymer was determined according to Likhtman and McLeish (2002). This interpretation contains special assumptions, the general validity of which has to be shown by further studies, however.
A branching architecture still simpler than that of comb polymers represent star polymers. Huang et al. (2016) studied three-star polymers with different arm lengths. For higher elongational rates, they report an elongational behavior being independent of the arm lengths and their symmetry (see section on “Branched polystyrenes”). This observation is surprising insofar as it would lead to the conclusion that neither the molar mass of the arms (20 kg/mol and 90 kg/mol) nor the total molar masses of the molecules (180 kg/mol for the linear and 270 kg/mol for the longest three-star species) have an influence on strain hardening. The authors explain this behavior by the assumption that “the effects from the arms and the branch points appear to be shielded causing all PS melts to behave like linear PS.” However, this explanation is not very elucidating with respect to a better understanding of the processes during elongation of branched molecules, particularly in the light of the discussion of the comb polymers above.
The measurements on the grafted polystyrenes shown in Fig. 4 are difficult to compare with those on the comb polymers because the numbers of the grafted side chains per molecule between 0.6 and 3.2 are distinctly lower. The qualitative conclusion may be drawn, however, that strain hardening of these samples and its dependence on elongational rate become more pronounced by increasing the number of side chains of a molar mass above the entanglement molar mass.
This discussion demonstrates how difficult it is to understand the strain-hardening behavior of comb and star polymers, the branching structures of which are obviously simpler than those of the long-chain branched samples with various architectures exhibiting different features of strain hardening as shown in the experimental part. Still more complex is the behavior found for blends of a pom-pom with a linear PS as reported above. These samples are discussed in the section “Blends of linear and long-chain branched polypropylenes” below.
Long-chain branched polyolefins
Long-chain branched polyethylenes (LDPE) are considered as prototypes of a tree-like architecture with randomly distributed branches. They exhibit a distinct strain hardening increasing with rate as can be seen from Fig. 18. The strain hardening is more pronounced for the LDPE 1 of lower density corresponding to a higher degree of branching. For this sample, even strain hardening running through a maximum as function of elongational rate is observed. This finding may be an experimental artifact, however, due to the short measuring times at high rates and a certain response time of the measuring system. It is evident that for such a branching structure the Rouse mechanism along molecule segments between entanglements does not play a role anymore, but that the branching points have an essential influence on the molecular motion. According to the reptation model, a long-chain branch forms its own tube and has to be withdrawn from it before the backbone may relax. The relaxation times of the branches determine the molecular motion and, thus, it is obvious that the resistance to flow reflected by strain hardening is dependent on strain rate. Strain hardening is assumed to occur when the elongational rate exceeds the inverse of a characteristic relaxation time and to become more pronounced the faster the deformation is applied. Above a certain rate, the sample becomes prone to break and that may be another explanation for the maximum in strain hardening sometimes observed.
A similar feature of strain hardening increasing with elongational rate is found for the commercial LCB-PP in Fig. 9 and the polypropylenes in Fig. 8 electron-beam irradiated with the two highest doses. Surprising is the finding, however, that the samples exposed to lower doses exhibit strain hardening decreasing with rate, and the same tendency is found for the PP/LCB-PP blends in Fig. 9 with 2 to 50 wt% of the long-chain branched PP.
Blends of linear and long-chain branched polypropylenes
Separate phases may play a role for an understanding of the strain hardening of the blends of a linear polypropylene (L-PP) and a randomly long-chain branched polypropylene (LCB-PP) in Fig. 9. The linear component does not exhibit strain hardening, but for the LCB-PP, strain hardening increasing with growing rate is observed as shown above and well known from the literature. Thus, strain hardening decreasing with rate as found for the blends at least up to 50 wt% LCB-PP is very surprising, particularly under the aspect that the longer relaxation times of the LCB-PP extend the relaxation spectrum of the L-PP. In the case of miscibility, the strain-hardening properties of the long-chain branched molecules are expected to come into play and those should lead to an increase with rate similar to the blends of polystyrenes in Fig. 3.
As discussed above for PP/UHMWPE, HDPE with small amounts of comonomers, and a ZN-LLDPE, the observed strain hardening becoming more pronounced at lower rates could be made plausible by assuming a phase separation in the molten state. Such a phase separation may be postulated for long-chain branched PP molecules within a matrix of linear PP molecules. Considering the deformation mechanisms, an essential difference to the linear polyethylene in the PP/UHWMPE exists insofar as a separated LCB-phase would be strain hardening and more easily deformable than that of the UHWMPE. Thus, it seems obvious that at higher rates the elongational behavior is dominated by the linear molecules not exhibiting strain hardening, but with decreasing rate more and more slow relaxation processes related to the long-chain branched molecules are activated. They enhance the resistance to flow and, consequently, strain hardening. The contribution to the overall strain hardening may be assumed to be more pronounced the larger the content of branched molecules. When the branched molecules become the continuous phase, their strain-hardening behavior dominates, and the strain rate dependence changes to that of the LCB-PP.
Postulating a phase separation between linear and long-chain branched PP molecules seems to be somewhat hypothetical because investigations of this kind in the literature making use of DSC measurements are scarce and do not provide a coherent picture. One reason for these experimental difficulties is the small differences of thermal properties between linear and long-chain branched PP. Tabatabaei et al. (2009) investigated rheological and thermal properties of blends of a commercial LCB-PP and two L-PP with various molar masses. They found melting temperatures of the L-PPs being 1 °C to 2.5 °C higher than that of the LCB-PP. These results correspond to those of Auhl (2006) who reports a maximum difference of 4 °C between the melting temperature of the linear syndiotactic PP and the highly branched sample from electron-beam irradiation with the largest dose. Thus, it may be beyond the resolution of thermal characterization methods to detect separate phases within the blends. TREF analysis successfully applied to the structure analysis of a ZN-LLDPE as reported above is not known for polypropylenes.
Moreover, Tabatabaei et al. (2009) report the validity of time–temperature superposition for the dynamic-mechanical experiments of all their various blends and derive miscibility from this finding. This result is not surprising because of the small differences in activation energies between linear and long-chain branched polypropylenes and cannot be taken as a proof for miscibility.
Nevertheless, they conclude a strong evidence for separate phases in melts of linear and branched PP from some of their rheological measurements. Thus, they found the logarithmic mixing rule of the zero-shear viscosity fulfilled for the blends of the LCB-PP with the two linear samples of lower molar mass but not for the other blends of the LCB-PP with the two L-PP of higher molar masses. Furthermore, they were able to numerically describe G′(ω) for the blend of 20 wt% LCB-PP and 80 wt% of the linear component with the highest molar mass using Palierne’s model and assuming a distinct ratio of surface tension to average diameter of a postulated second phase. From this result, immiscibility of the two components was concluded. G′(ω) of the blend with LCB-PP and the linear PP of lowest molar mass could be described by setting the ratio to zero, specific for miscible systems.
A tendency of strain hardening increasing with decreasing rate can be deduced from Tabatabaei’s measurements for the two LCB-PP/L-PP blends with the highest and lowest molar mass and the LCB-PP as well. Thus, it is obviously the same for the miscible and immiscible system, and this finding does not support the assumption that the kind of rate dependence of strain hardening is related to miscibility. These measurements would need some additional experimental support, however. Strain hardening increasing with decreasing rate for the blends would be in accordance with the measurements of the blends in Fig. 9, but this rate dependence found for the long-chain branched PP is not in agreement with that of the LCB-PP in Fig. 9, which shows just the opposite behavior. An explanation of the difference is difficult because the molecular structure of the LCB-PP studied by Tabatabaei et al. (2009 is not given. It should be mentioned, however, that the measurements were performed at comparable total elongations but higher rates than those of Fig. 9.
Strain hardening increasing with decreasing elongational rate for blends of linear and long-chain branched PP is reported by Li et al. (2021). However, they conclude miscibility of their systems from the determination of melting and crystallization temperatures by DSC. But as discussed before, the differences of the characteristic temperatures between linear and branched samples are too small for convincing conclusions.
The assumption of phase separation between linear and branched polypropylenes may be used for making the results on the irradiated polypropylenes in Fig. 8 plausible. At the two lowest doses, only few molecules are branched, and the linear molecules forming the continuous phase do not exhibit strain hardening as known from the purely linear PPs above. Strain hardening occurs when more and more branched molecules of the separated phase would become able to deform, and due to their longer relaxation times, this deformation is more pronounced the smaller the elongational rate. With growing dose and the subsequent enhancement of the branching degree, the strain-hardening effect gets stronger, and it changes its rate dependence when the branched molecules become the continuous phase.
The postulate of phase separation between linear and long-chain branched polypropylene molecules could be taken for a heuristic explanation of the results in Fig. 7 on chemically branched species. The two phases may give rise to strain hardening increasing with decreasing rate in analogy to the polypropylenes discussed before. The effect being more pronounced for the material with the larger average number of branches could be due to an entanglement network efficiency within separated phases being stronger at the higher level of branches.
The discussion of phase separation in blends of linear and long-chain branched polypropylene and their effect on the rate dependence of strain hardening may throw a light on the results of blends from a linear and a pom-pom polystyrene by Hirschberg et al. (2023) reported above. The rate dependence of strain hardening found to be similar to that of the L-PP/LCB-PP blends could become plausible by assuming an immiscibility of the linear and pom-pom PS investigated. However, no studies are known that would support such a hypothesis.
Blends of LLDPE and LDPE
Obviously, the experimental findings of strain hardening of the LLDPE/LDPE blends increasing with rate as described in the section “Blends of linear and long-chain branched polyethylenes” above is different from that of some HDPEs with small amounts of non-uniformly distributed comonomers discussed before. Rather, it is similar to blends of miscible linear polymers with high molar mass components giving rise to longer Rouse times that are thought to determine the onset of strain hardening (see section “Comparison of model predictions with experiments”). However, as discussed above, Rouse times do not come into play for an understanding of stretching of long-chain branched polymers, rather it is determined by their relaxation times dominant in the LLDPE/LDPE blends. This interpretation assumes an at least partial miscibility of the molecules of the two polyethylenes under the conditions of blending and measurement. But such an assumption is in contrast to reports in the literature, which postulate non-miscibility of LDPE and LLDPE.
For example, Schlund and Utracki (1987b) conclude immiscibility of their LLDPE/LDPE blend indirectly from the failure of the mixing rule for the time-dependent linear shear viscosity calculated from the relaxation spectrum. Wagner et al. (2004) derived the conclusion of immiscibility of their LDPE and LLDPE components from the elongational behavior of the LDPE dominant at small additions already and the failure of time–temperature superposition of the moduli in dynamic-mechanical measurements. This argument is not very strong, however, considering the thermorheological complexity found for an LDPE by Kessner et al. (2009), for example.
Two phases were postulated by Wagner et al. (2004). The one is assumed to be composed of highly short-chain branched molecules of low molar mass from the LLDPE and the low molar mass part of the LDPE, the other of the less branched higher molar mass components of the LLDPE and the longer molecules of the LDPE. Direct evidence for the two-phase systems postulated by Wagner et al. (2004) and Schlund and Utracki (1987b) is not provided, however, and thus phase separation remains rather hypothetical.
The complexity of miscibility or non-miscibility of LLDPE/LDPE blends becomes obvious from a paper of Fang et al. (2005). They studied blends of an octene LLDPE and a hexene LLDPE with two LDPE each and found miscibility for the octene LLDPE but not for the hexene LLDPE by the widely used DSC measurements of melting and crystallization temperatures. Moreover, rheological experiments were applied. For an assessment of miscibility, the logarithmic mixing rule for the zero-shear viscosities was considered. The results pointed to a better miscibility of the octene LLDPE than the hexene LLDPE. Furthermore, G′(ω) could successfully be described by the Palierne relation with the assumption of an interfacial tension in the case of the blends of the LDPEs and the hexene copolymer indicating different phases. Complete data of the compositions of the LLDPEs were not given, but from their densities, it may be concluded that the octene content is higher than that of hexene.
Furthermore, for the octene copolymer, a small amount of long-chain branching is mentioned. Thus, the conclusions with respect to the relation between miscibility and molecular structure bear some uncertainties.
With respect to the determination of miscibility of polymer blends from rheological measurements, the logarithmic mixing rule for the zero-shear viscosities is frequently considered. In Wagner et al. (2004), a literature survey on the assessment of miscibility of various polyethylenes by the logarithmic mixing rule for the zero-shear viscosity is listed. Amongst the 13 references on LLDPE/LDPE blends, roughly half of them state immiscibility based on the failure of the mixing rule for the various compositions, but complete molecular data are missing, and, thus, it is not possible to assess the significance of the logarithmic mixing rule.
In a paper by Robledo et al. (2011) on 15 wt% of various LDPEs in a commercial Ziegler–Natta LLDPE, LDPE droplets were postulated from various rheological experiments. Direct evidence for separate domains was not provided, and, thus, the conclusions remain somewhat hypothetical.
Steffl (2004) reported that for LLDPE/LDPE blends not specified in detail one melting point is found by DSC during the first heating of the extruded sample, but two melting points could be distinguished in the second heating after cooling. That observation points to miscibility between LLDPE and LDPE depending on the prehistory of a sample.
The difficulty to get a clear picture about the miscibility of LLDPE/LDPE has to be seen in the fact that this property seems to be sensitively dependent on the molecular structure of the components, the blending process, and the way of characterization. Obviously, the studies from the literature were performed on samples of different structure with various experimental methods making comparisons and stringent conclusions difficult.
As the discussion on blends of short-chain and long-chain branched polyethylenes and the foregoing one on linear and long-chain branched polypropylenes demonstrate, it is not easy to find generally valid explanations of their strain-hardening behavior that go along with all the facets of knowledge on the various properties of the blends coming into play. Nevertheless, the results are interesting from a practical point of view because they show how strain hardening of melts of linear polyolefins can be obtained by the addition of long-chain branched species that improve the uniformity of sample deformation in processing operations dominated by elongational flow (e.g., Münstedt (2018)).
Conclusions
Strain hardening of polymer melts in uniaxial extension is dependent on their chemical and molecular structure and on experimental parameters like elongation and elongational rate. Although not being a time-independent rheological quantity, strain hardening is of relevance with respect to processing operations where extensional flow comes into play. It has frequently been shown and can intuitively be verified that strain hardening supports the uniformity of deformation and, consequently, the quality of processed items. Of particular interest in this aspect is the dependence on elongational rate, which is a dominant processing parameter. For example, in film blowing higher elongational rates may occur than in blow molding. In both cases, the uniformity of wall thickness is a desirable property. Obviously, strain hardening at various elongational rates is an interesting property for the assessment of the processing behavior of polymeric materials.
As shown in the experimental part of this paper, various polymers may exhibit distinct differences in their rate dependence of strain hardening. Such it is found that in the range experimentally accessible linear olefinic homopolymers like HDPE and PP are not strain hardening at all, whereas amorphous linear polymers like polystyrene and polymethylmethacrylate may exhibit distinct strain hardening becoming more pronounced at larger rates. This behavior can be enhanced by the addition of high molar mass components. A similar effect was shown to be obtained for polystyrene by the introduction of long-chain branches.
The strain-hardening behavior of linear polymers can semi-quantitatively be understood by considering the Rouse time as the decisive quantity for chain stretching. For typical PP and HDPE, much smaller Rouse times than for PS and PMMA were determined from rheological data resulting in critical elongational rates for the onset of strain hardening far outside the feasible experimental window.
Relationships between the critical elongational rate for the onset of strain hardening and Rouse times were found to be limited to narrowly distributed samples as shown for polystyrenes. For broader distributions and blends with components of higher molar masses in particular, Rouse times were not useful for an understanding of the strain-hardening behavior. In such cases, the high molar masses seem to play the dominant role for strain hardening, whereas the experimentally determined Rouse time of a sample represents some kind of average quantity for the characterization of the molecule stretchability. This field is still open to intensive experimental and theoretical research.
Strain hardening of linear PP was obtained by adding small amounts of HDPE with ultrahigh molar masses (UHMWPE) and that of HDPE by introducing α-olefin comonomers resulting in samples with a density smaller than 0.960 g/cm3 at room temperature typical of homologous linear polyethylenes. These modified materials are characterized by strain hardening increasing with decreasing strain rate. For an explanation of these surprising effects, the existence of separate phases was assumed and entanglement processes in small layers at the interfaces were postulated. But this hypothesis needs some experimental and theoretical support to become verified.
Strain hardening of polyethylenes and polypropylenes is well known for highly branched materials with a tree-like architecture. They show strain hardening increasing with rate and are widely used in applications. For these species, Rouse modes cannot be considered as the source of strain hardening, rather the relaxation processes due to the branches are thought to be decisive.
The strain-hardening features of LLDPEs as reported in the literature are diverse. The absence of strain hardening and its increase with decreasing rate as well are reported. A satisfying structural picture for an even qualitative understanding could not be established within this review due to the lack of comprehensive molecular data, particularly with respect to the kind and content of short-chain branches. But from the experimental results discussed, some evidence may be derived that the comonomer distributions along the backbones play a decisive role for the strain-hardening features.
Challenging with respect to a deeper understanding are the results on blends of linear and long-chain branched polypropylenes (PP/LCB-PP) on the one side and linear low-density polyethylenes and long-chain branched polyethylenes (LLDPE/LDPE) on the other. Strain hardening is observed, and its intensity may be controlled by the amount of branched polymers. These results directly support the tailoring of materials for specific purposes. Questions arise, however, from the different strain-hardening dependences on elongational rate, at least for the limited amount of measurements available. For an PP/LCB-PP blend, strain hardening was found to increase with decreasing rate in the cases where PP is the major phase, with LCB-PP as the continuous phase it is just the other way round. For some LLDPE/LDPE blends, however, an enhancement of strain hardening with increasing elongational rate is reported if a small amount of LDPE is added to the LLDPE.
The differences lead to the hypothesis that the PE-blends are miscible but the PP-blends not. These assumptions were discussed in the light of compatibility studies on these materials from the literature obtained by various methods. But contrary results are reported, and questions remain open that could only be answered by specific investigations on well-defined samples.
All in all, it has to be stated that the strain-hardening behavior of polymer melts exhibits very many interesting features that are directly relevant for applications, but still offer a wide field for research to get answers to the questions related to a better fundamental understanding of the various facets.
References
Abbasi M, Faust L, Riazi K, Wilhelm M (2017) Linear and extensional rheology of model branched polystyrenes: from loosely grafted combs to bottlebrushes. Macromolecules 50:5964–5977
Adhikari R, Godehardt R, Lebek W, Frangov S, Michler GH, Radusch H-J, Calleja FJB (2005) Morphology and micromechanical properties of ethylene/1-octene copolymers and their blends with high density polyethylene. Polym Adv Technol 16:156–166
Ahirwal D, Filipe S, Neuhaus I, Busch M, Schlatter G, Wilhelm M (2014) Large amplitude oscillatory shear and uniaxial extensional rheology of blends from linear and long-chain branched polyethylene and polypropylene. J Rheol 58:635–658
Auhl D, Stange J, Münstedt H, Krause B, Voigt D, Lederer A, Lappan U, Lunkwitz K (2004) Long- chain branched polypropylenes by electron beam irradiation and their rheological properties. Macromolecules 37:9465–9472
Auhl D (2006) Molekulare Struktur und rheologische Eigenschaften strahlenmodifizierter Polypropylene (Molecular structure and rheological properties of irradiation modified polypropylenes), Doctoral Thesis, University Erlangen-Nürnberg
Auhl D, Chambon P, McLeish TCB, Read DJ (2009) Elongational flow of blends of long and short polymers: effective stretch relaxation time. Phys Rev Lett 103:136001
Bach A, Almdal K, Rasmussen HK, Hassager O (2003) Elongational viscosity of narrow molar mass distribution polystyrene. Macromolecules 36:5174–5179
Baumgaertel M, Schausberger A, Winter HH (1992) The relaxation of polymers with linear flexible chains of uniform length. Rheol Acta 29:400–408
Berger L, Meissner J (1992) Linear viscoelasticity, simple and planar melt extension of linear polybutadienes with bimodal molar mass distributions. Rheol Acta 31:63–74
Costanzo S, Huang Q, Ianniruberto G, Marrucci G, Hassager O, Vlassopoulos D (2016) Shear and extensional rheology of polystyrene melts and solutions with the same number of entanglements. Macromolecules 49:39
Crist B, Hill MJ (1997) Recent developments in phase separation of polyolefin melt blend. J Polym Sci B Polym Phys 35:2329–2353
Dealy JM, Larson RG (2006) Structure and Rheology of Molten Polymers. Hanser, Munich
Fang Y, Carreau PJ, Lafleur PG (2005) Thermal and rheological properties of mLLDPE/LDPE blends. Polym Engn Sci 45:1254–1264
Faust L, Röpert MC, Esfahani MK, Abbasi M, Hirschberg V, Wilhelm M (2022) Comb and branch-on-branch model polystyrenes with exceptionally high strain hardening factor SHF > 1 000 and their impact on physical foaming. Macrom Chem Phys 224:2200214
Gabriel C, Kaschta J, Münstedt H (1998) Influence of molecular structure on rheological properties of polyethylenes, Part I. Creep recovery measurements in shear. Rheol Acta 37:7–20
Gabriel C (2001) Einfluss der molekularen Struktur auf das viskoelastische Verhalten von Polyethylenschmelzen (Influence of the molecular structure on the viscoelastic behavior of polyethylene melts), Doctoral Thesis, University Erlangen-Nürnberg, Shaker Verlag, Aachen
Gabriel C, Münstedt H (2003) Strain hardening of various polyolefins in uniaxial elongational flow. J Rheol 47:619–630
Gotsis AD, Zeevenhoven BL, Tsenoglou C (2004) Effect of long branches on the rheology of polypropylene. J Rheol 48:895–914
Hepperle J (2002) Einfluss der molekularen Struktur auf rheologische Eigenschaften von Polystyrol- und Polycarbonatschmelzen (Influence of the molecular structure on rheological properties of polystyrene and polycarbonate melts), Doctoral Thesis, University Erlangen-Nürnberg, Shaker Verlag Aachen
Hepperle J, Münstedt H (2006) Rheological properties of branched polylstyrenes: nonlinear shear and extensional behavior. Rheol Acta 45:717–727
Hingmann R, Marczinke BL (1994) Shear and elongational flow properties of polypropylene melts. J Rheol 38:573–587
Hirschberg V, Lyu S, Schußmann MG (2023) Complex polymer topologies in blends: shear and elongational rheology of linear/pom-pom polystyrene blend. J Rheol 67:403–415
Huang Q, Mednova O, Rasmussen HK, Alvarez NJ, Skov AL, Almdal K, Hassager O (2013) Concentrated polymer solutions are different from melts: Role of entanglement molecular weight. Macromolecules 46:5026–5035
Huang Q, Agostini S, Hengeller L, Shivokhin M, Alvarez NJ, Hutchings LR, Hassager O (2016) Dynamics of star polymers in fast extensional flow and stress relaxation. Macromolecules 49:6694–6699
Ishizuka O, Koyama K (1980) Elongational viscosity at a constant elongational strain rate of polypropylene melt. Polymer 21:164–170
Kasehagen LJ, Macosko CW, Trowbridge D, Magnus F (1996) Rheology of long-chain randomly branched polybutadiene. J Rheol 40:689–709
Kasehagen LJ, Macosko CW (1998) Nonlinear shear and extensional rheology of long-chain randomly branched polybutadiene. J Rheol 42:1303–1327
Katsikis N, Königer T, Münstedt H (2007) Elongational viscosities of polymethylmethacrylate/nano-clay composites. Appl Rheol 17:52751–1–52751–9
Kessner U, Kaschta J, Münstedt H (2009) Determination of method-invariant activation energies of long-chain branched low-density polyethylene. J Rheol 53:1001–1016
Kobayashi M, Takahashi T, Takimoto J, Koyama K (1996) Influence of glass beads on the elongational viscosity of polyethylene with anomalous strain rate dependence of strain-hardening. Polymer 37:3745–3747
Koyama K, Ishizuka O (1982) Influence of molecular weight distribution on elongational flow of polylethylene. Sen-I Gakkaishi 38:239–245
Kurzbeck S, Oster F, Münstedt H, Nguyen TQ, Gensler R (1999) Rheological properties of two polypropylenes with different molecular structure. J Rheol 43:359–374
Lagendijk RP, Hogt AH, Buitenhuijs A, Gotsis AD (2001) Peroxydicarbonate modification of polypropylene and extensional flow properties. Polymer 42:10035–10043
Langouche F, Debbaut B (1999) Rheological characterization of a high-density polyethylene with a multi-mode differential viscoelastic model and numerical simulation of transient elongational recovery experiments. Rheol Acta 38:48–64
Lentzakis H, Vlassopoulus D, Read DJ, Lee H, Chang T, Driva P, Hadjichristidis N (2013) Uniaxial extensional rheology of well-characterized comb polymers. J Rheol 57:605–625
Li K, Qin Y, Zhao S, Dong J (2021) Blending behavior of high-degree long-chain-branched polypropylene prepared by Ziegler-Natta catalysis with common polypropylene. Ind Eng Chem Res 60:13614–13626
Likhtman A, McLeish TCB (2002) Quantitative theory for linear dynamics of linear entangled polymers. Macromolecules 35:6332–6343
Linster JJ, Meissner J (1986) Melt elongation and structure of linear polyethylene (HDPE). Polym Bull 16:187–194
Liu CY, Keunings R, Bailly C (2007) Direct rheological evidence of monomer density reequilibration for entangled polymer melts. Macromolecules 40:2946–2954
Lopez-Barron CR, Tsou AH (2017) Strain hardening of polyethylene/polypropylene blends via interfacial reinforcement with poly(ethylene-cb-propylene) comb block copolymer. Macromolecules 50:2986–2995
Malmberg A, Gabriel C, Steffl T, Münstedt H, Löfgren B (2002) Long-chain branching of metallocene-catalyzed polyehylenes investigated by low oscillatory shear and uniaxial extensional rheometry. Maromolecules 35:1038–1048
Marrucci G, Ianniruberto G (2004) Interchain pressure effect in extensional flow of entangled polymer melts. Macromolecules 37:3934–3942
Menezes EV, Graessley WW (1982) Nonlinear rheological behavior of polymer systems for several shear flow histories. J Polym Sci Polym Phys 20:1817–1833
Minegishi A, Nishioka A, Takahashi T, Masubishi Y, Takimoto J, Koyama K (2001) Uniaxial elongational viscosity of PS/a small amount of UHMW-PS blends. Rheol Acta 40:329–338
Minoshima W, White JL, Spruiell JE (1980) Experimental investigation of the influence of molecular weight distribution on the rheological properties of polypropylene melts. Polym Eng Sci 20:1166–1176
Morelly SL, Palmese L, Watanabe H, Alvarez NJ (2019) Effect of finite extensibility on nonlinear extensional rheology of polymer melts. Macromolecules 52:915–922
Münstedt H (1980) Dependence of the elongational behavior of polystyrene melts on molecular weight and molecular weight distribution. J Rheol 24:847–867
Münstedt H, Laun HM (1981) Elongational properties and molecular structure of polyethylene melts. Rheol Acta 20:211–221
Münstedt H, Kurzbeck S, Egersdörfer L (1998) Influence of molecular structure on rheological properties of polyethylenes. Part II Elongational Behavior Rheol Acta 37:21–29
Münstedt H, Stary Z (2013) Steady states in extensional flow of strain hardening polymer melts and the uncertainties of their determination. J Rheol 57:1065–1077
Münstedt H, Schwarzl FR (2014) Deformation and Flow of Polymeric Materials. Springer, Heidelberg
Münstedt H (2018) Extensional rheology and processing of polymeric materials. Intern Polymer Processing 33:594–618
Narimissa E, Poh L, Wagner MH (2021) Elongational viscosity scaling of polymer melts with different chemical constituents. Rheol Acta 60:163–174
Nielsen JK, Rasmussen HK, Hassager O, McKinley GH (2006) Elongational viscosity of monodisperse and bidisperse polystyrene melts. J Rheol 50:453–476
Osaki K, Inoue T, Uematsu T, Yamashita Y (2001) Evaluation methods of the longest Rouse relaxation time of an entangled polymer in a semidilute solution. J Polym Sci B Polym Phys 39:1704–1712
Park JU, Kim JL, Kim DH, Ahn KH, Lee SJ (2006) Rheological behavior of polymer/layered silicate nanocomposites und uniaxial extensional flow. Macrom Res 14:318–323
Rajasekaran JJ, Curro JG, Honeycutt JD (1995) Theory for the phase behavior of polyolefin blends: Application to the polyethylene/isotactic polypropylene blend. Macromolecules 28:6843–6853
Robledo N, Vega JF, Nieto J, Martinez-Salazar J (2011) Role of the interface in the melt-rheology properties of linear low-density polyethylene/low-density polyethylene blends: effect of the molecular architecture of the dispersed phase. J Appl Polym Sci 119:3217–3226
Röpert MC, Schußmann MG, Esfahani MK, Wilhelm M, Hirschberg V (2022) Effect of side chain length in polystyrene POM - POMs on melt rheology and solid mechanical fatigue. Macromolecules 55:5485–5496
Schlund B, Utracki LA (1987a) Linear low density polyethylenes and their blends: Part 3. Extensional flow of LLDPE’s. Polym Eng Sci 27:380–386
Schlund B, Utracki L (1987b) Linear low density polyethylenes and their blends: Part 5. Extensional flow of LLDPE blend. Polym Eng Sci 27:1523–1529
Schweizer T (2000) Uniaxial elongational rheometry with little strain hardening linear polyethylene. Proceedings of the XIIth International Congress on Rheology. Cambridge, UK
Shabbir A, Goldansatz H, Hassager O, Van Ruymbeke E, Alvarez NJ (2015) Effect of hydrogen bonding on linear and nonlinear rheology of entangled polymer melts. Macromolecules 48:5988–5996
Spitael P, Macosko C (2004) Strain hardening in polypropylenes and its role in extrusion foaming. Polym Eng Sci 44:2090–2100
Stadler F J (2007) Molecular structure and rheological properties of linear and long-chain branched ethene-α-olefin copolymers. Doctoral Thesis, University Erlangen-Nürnberg, Sierke Verlag, Göttingen
Stange J, Uhl C, Münstedt H (2005) Rheological behavior of blends from a linear and a long-chain branched polypropylene. J Rheol 49:1059–1079
Steffl T (2004) Rheological and film blowing propertied of various low density polyethylenes and their blends, Doctoral Thesis, University Erlangen-Nürnberg, Shaker Verlag, Aachen
Sugimoto M, Tanaka T, Masubuchi Y, Takimoto J, Koyama K (1999) Effect of chain structure on the melt rheology of modified polypropylene. J Appl Polym Sci 73:1493–1500
Sugimoto M, Masubuchi Y, Takimoto J, Koyama K (2001a) Melt rheology of polypropylene containing small amounts of high-molecular weight chain. 2. Uniaxial and Biaxial Extensional Flow. Macromolecules 34:6056–6063
Sugimoto M, Masubichi Y, Takimoto J, Koyama K (2001b) Melt rheology of polypropylene containing small amounts of high molecular weight chain. I. Shear flow. J Polym Sci B Polym Phys 39:2692–2704
Szanto L, Vogt R, Meier J, Auhl D, Van Ruymbeke E, Friedrich C (2017) Entanglement relaxation time of polyethylene melts from high-frequency rheometry in the mega-hertz range. J Rheol 61:1023–1033
Tabatabaei SH, Carreau PJ, Ajji A (2009) Rheological and thermal properties of blends of a long-chain branched polypropylene and different linear polypropylenes. Chem Engn Sci 64:4719–4731
Takahashi M, Isaki T, Takigawa T, Masuda T (1993) Measurement of biaxial and uniaxial flow behavior of polymer melts at constant strain rates. J Rheol 37:827–846
Takahashi T, Takimoto J, Koyama K (1999) Elongational viscosity for miscible and immiscible polymer blends. II. Blends with a small amount of UHMW polymer. J Appl Polym Sci 72:961–969
Trzebiatowski T, Wilski H (1985) Die Dehnviskosität des Polypropylens. Rheol Acta 24:582–587
Van Ruiten J, Boode JW (1992) Mixing and demixing of heterogeneous ethylene/octene copolymers: evidence of bimodal melt demixing. Polymer 33:2548–2552
Wagner MH, Bastian H, Ehrecke P, Kraft M, Hachmann P, Meissner J (1998) Nonlinear viscoelastic characterization of a linear polyethylene (HDPE) melt in rotational and irrotational flow. J Non-Newt Fluid Mech 79:283–296
Wagner MH, Kheirandish S, Yamaguchi M (2004) Quantitative analysis of melt elongational behavior of LLDPE/LDPE blends. Rheol Acta 44:198–218
Wagner MH, Kheirandish S, Koyama K, Nishioka A, Minegishi A, Takahashi T (2005) Modeling strain hardening of polydisperse polystyrene melts by molecular stress function theory. Rheol Acta 44:235–243
Wagner MH, Kheirandish S, Stange J, Münstedt H (2006) Modelling elongational viscosity of blends of linear and long-chain branched polypropylenes. Rheol Acta 46:211–222
Wagner MH, Narimissa E, Huang Q (2022) Analysis of elongational flow of star polymers. Rheol Acta 51:415–425
Wignall GD, Chikd HR, Samuela RJ (1982) Structural characterization of semicrystalline polymer blends by small-angle neutron scattering. Polymer 23:957–964
Yoshii F, Makuuchi K, Kikukawa S, Tanaka T, Saitoh J, Koyama K (1996) High-melt-strength polypropylene with electron beam irradiation in the presence of polyfunctional monomer. J Appl Polym Sci 60:617–623
Zhong Y, Yu L, Huang Q (2022) Investigating the linear viscoelastic behavior at high frequencies in the transition to glassy regime for polystyrene melts and solutions. Rheol Acta 61:689–700
Funding
Open Access funding enabled and organized by Projekt DEAL.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The author declares no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Münstedt, H. Various features of melt strain hardening of polymeric materials in uniaxial extension and their relation to molecular structure: review of experimental results and their interpretation. Rheol Acta 62, 333–363 (2023). https://doi.org/10.1007/s00397-023-01400-4
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00397-023-01400-4