
Abstract
Thermoresponsive colloidal particles were prepared by seeded precipitation polymerization of N-isopropylacrylamide (NIPAM) in the presence of a crosslinking monomer, N,N-methylenebisacrylamide (MBA), using polystyrene latex particles (ca. 50 nm in diameter) as seeds in aqueous dispersion. Phase transitions of the prepared poly(N-isopropylacrylamide), PNIPAM, shells on polystyrene cores were studied in comparison to colloidal PNIPAM microgel particles, in H2O and/or in D2O by dynamic light scattering, microcalorimetry and by 1H NMR spectroscopy including the measurements of spin–lattice (T1) and spin–spin (T2) relaxation times for the protons of PNIPAM. As expected, the seed particles grew in hydrodynamic size during the crosslinking polymerization of NIPAM, and a larger NIPAM to seed mass ratio in the polymerization batch led to a larger increase of particle size indicating a product coated with a thicker PNIPAM shell. Broader microcalorimetric endotherms of dehydration were observed for crosslinked PNIPAM on the solid cores compared to the PNIPAM microgels and also an increase of the transition temperature was observed. The calorimetric results were complemented by the NMR spectroscopy data of the 1H-signal intensities upon heating in D2O, showing that the phase transition of crosslinked PNIPAM on polystyrene core shifts towards higher temperatures when compared to the microgels, and also that the temperature range of the transition is broader.
Similar content being viewed by others

Introduction
The temperature-induced phase transition of poly(N-isopropylacrylamide), PNIPAM, in aqueous solutions, first reported by Heskins and Guillet in 1968 [1], has been an inspiration to an enormous amount of scientific research as well as to many suggestions of application during the recent decades. In dilute aqueous solution, PNIPAM undergoes a coil-to-globule transition at its lower critical solution temperature, LCST, of ca. 32°C, as water molecules bound to the polymer chains are released [2]. The preparation of thermoresponsive colloidal particles was pioneered by Pelton and Chibante [3] in the case of N-isopropylacrylamide, NIPAM, either alone or with styrene by Pelton [4] and by Kawaguchi and coworkers [5–8] in the case of emulsion copolymerization of styrene with various N-substituted acrylamides. In aqueous systems particle production from NIPAM was shown to proceed via a precipitation polymerization mechanism as the reaction was conducted above the LCST and in the presence of crosslinking monomer, N,N-methylenebisacrylamide (MBA) [9]. Since the pioneers, several groups have studied submicron-sized core-shell-like particles prepared of styrene and NIPAM. Pichot et al. [10, 11] focused on studying the effect of polymerization process on particle morphology. The particles were synthesized either by batch or by two-step surfactant-free emulsion copolymerization of styrene, NIPAM, and a cationic amino-containing comonomer with or without an added crosslinking monomer. Characteristics such as particle size, electrokinetic properties, and colloidal stability of the particles were studied [12], as well as covalent and noncovalent binding of an antibody onto the particles [13].
Ballauff and coworkers [14–18] have studied the phase transition of crosslinked PNIPAM as a shell on a polystyrene-based core. They prepared the core particles by conventional emulsion polymerization of styrene and NIPAM (95/5 wt%), and as a second step, they copolymerized NIPAM with the crosslinking monomer, N,N-methylenebisacrylamide, in the presence of the core particles [14, 15]. Phase behavior of the thermosensitive shell in aqueous particle dispersions was studied by combining the data from different scattering methods such as dynamic light scattering (DLS) [15], small-angle neutron scattering (SANS) [16, 17], and small-angle X-ray scattering (SAXS) [14, 17]. The effect of temperature on the rheological properties of the particle dispersions was also studied [18]. Typically, the core particles in aqueous dispersion were of approximately 100 nm in diameter and the crosslinked PNIPAM shell was of 10–50 nm in thickness depending on the temperature.
We have synthesized particles very similar to those reported by Ballauff and coworkers, and studied the phase transition of the crosslinked PNIPAM shell on polystyrene core of ca. 50 nm, in addition to the dynamic light scattering by 1H NMR spectroscopy and by high sensitivity differential scanning microcalorimetry. To our knowledge, there are no reports of such phase transition studies employing the latter two methods for this type of PNIPAM microgel structures. However, few investigations of linear PNIPAM grafted or adsorbed on solid core particles have been published. Zhu and Napper [19] pioneered the experimental studies on coil-to-globule transitions of linear PNIPAM chains attached to the surfaces of polystyrene latex particles. The kinetics of the transition was investigated in the case of chains grafted on polystyrene particles of ca. 100 nm in diameter by DLS and 13C NMR spectroscopy [20]. Studies showed broadening of transitions of interfacial chains compared to free chains in solution. Furthermore, a considerable change in the coil-to-globule transition of interfacial chains was observed as the molecular weight (M w) of the attached chains was varied from 3×105 to 2×106 g/mol. The broadening of the transition towards lower temperatures was more pronounced with the shortest polymer chains in question. The broad overall transition of the interfacial chains was explained to consist of two components. Upon heating, first, the inner region of each chain adopts its globular form, whereas the segments of chains in coronal layer exist in the coil-like conformation until the LCST is reached.
The phase transition of low molecular weight PNIPAM (ca. 5,000 g/mol), covalently bound to the surface of gold clusters of few nanometer in size, have been investigated with microcalorimetry by Shan et al. [21]. The PNIPAM chains in these monolayer protected gold clusters exhibited two separate transition endotherms of dehydration around the LCST. The first transition with a sharp and narrow endothermic peak was observed at lower temperature while the second one with broader peak was observed at higher temperature. As in the case of Zhu and Napper, it was suggested that the inner segments of chains close to the particle surface are densely packed and less hydrated showing the first transition, while the segments in coronal layer are more hydrated showing the second transition. As methods sensitive to dynamic properties in the phase transition on molecular level, both microcalorimetry and NMR spectroscopy have been employed to investigate the high molecular weight (3.5×105 g/mol) PNIPAM adsorbed on silica particles [22]. In this case, the effect of the solid surface was an increased transition temperature and a broadening of the transition. Both effects were more pronounced at low surface coverage. The increase of the transition temperature was concluded to be due to increased motional constraints of the chains on the solid surface. The increase of the transition width was explained by the assumption of motional heterogeneity in the PNIPAM layer. At lower surface coverage, the system is dominated by immobile segments close to silica; while at higher coverage, more mobile segments with a smaller broadening contributed to the transition.
The introduced studies of phase transition show that interfacial PNIPAM exhibits very different and more complex dynamic properties compared to volume systems. This report describes the case of PNIPAM microgel on polystyrene core in comparison to colloidal microgel particles.
Experimental
Materials
Styrene (Merck) and acrylic acid (AA; Merck) were distilled under reduced pressure before use. N-isopropylacrylamide (NIPAM; Acros) was recrystallized from n-hexane and dried in vacuum. Sodium dodecylsulfate (SDS; Merck), potassium peroxydisulfate (KPS; Merck), and N,N-methylenebisacrylamide (MBA; Aldrich) were used as received. Water was deionized using Elga PurelabUltra-equipment. Filter papers (Whatman, 2V) used were of pore size 8 μm. Cellulose membrane tubings (CelluSep T4) with nominal molecular weight cut off of 12,000–14,000 g/mol were used as membranes in particle purification by dialysis.
Particle syntheses
A summary of the conditions of the syntheses and the abbreviations for the particles are shown in Table 1. Polymerizations were carried out in a sealed round-bottom flask equipped with a magnetic stirrer and an oil bath to control the reaction temperature. The seed particles, PS1, and PS2 were prepared by radical polymerization of styrene in aqueous emulsion. Surfactant, SDS, was dissolved in water in the reaction flask and, after which, styrene was added and the flask was sealed with a septum. The mixture was purged with nitrogen and stirred at room temperature for 20 min. The nitrogen inlet and outlet were removed and the flask was placed into a preheated oil bath at 60°C. Polymerization was initiated after 20 min by injecting KPS dissolved in 1 ml of water to the reaction mixture. After which, reaction was allowed to proceed for 3 h with stirring (600–700 rpm). The reaction was stopped by unsealing the flask and cooling the product to room temperature. The product was filtered through a filter paper and purified by dialyzing for 7 days against distilled water that was refreshed daily. The dialyzed aqueous particle dispersion was extracted several times with n-hexane and stored as such in the refrigerator.
The two-stage particles were prepared by the following manner. In the synthesis of particle PS1N, NIPAM and MBA were separately dissolved in aqueous seed particle dispersion, PS1, and both solutions were transferred to a reaction flask. The flask was sealed with a septum and polymerization was initiated and carried out by following the procedure described for the synthesis of seed particles. In the synthesis of particle PS1NA, acrylic acid was, in addition, injected to the reaction mixture before sealing the flask. For the synthesis of PS2N, the purified seed particle dispersion, PS2, was diluted with 12.5 ml of water before use in polymerization. In the second stage polymerization, the reaction time was 20 h and the reaction temperature was 60°C. Microgel particles, N1 and N2, were prepared similarly to two-stage particles except of using dilute aqueous solution of SDS as a reaction medium instead of aqueous seed particle dispersion. NIPAM and MBA were separately dissolved in aqueous solutions of SDS (0.1 g/l) and both solutions were transferred into the reaction flask. The flask was sealed with a septum and polymerization was initiated and carried out by following the procedure (N2, KPS) described for the synthesis of seed particles. The reaction time in the microgel particle syntheses was 4 h. The two-stage particles and the microgel particles were purified by dialysis for 7 days against distilled water that was refreshed daily. The purified particle dispersions were stored as such in the refrigerator. Polymer contents of aqueous particle dispersions were obtained by drying weighed samples of aqueous dispersions to equilibrium weight in vacuum.
Characterization methods
FTIR-spectra were measured from freeze-dried particles with PerkinElmer Spectrum One FT-IR-spectrometer, Spectrum One FT-IR -software, and Universal ATR Sampling Accessory. 1H NMR spectra of the particles in an organic solvent were recorded with a 200 MHz Varian Gemini 2000 spectrometer. The samples were prepared from freeze-dried particles by dissolving in deuterated chloroform (30 mg/ml). 1H NMR spectra and relaxation measurements of the particles in D2O were measured using a Varian UNITYINOVA spectrometer operating at 300 MHz for protons equipped with a temperature control unit. The NMR samples were prepared by redispersing 20 mg of freeze-dried particles in 1 ml of D2O and filtered through a filter paper. Measurements were carried out with controlled heating and cooling steps, allowing the samples to equilibrate for 30 min at each incremented temperature. For the 1H T1 relaxation measurements of the polymer protons, a series of ∼30 spectra were collected using the standard inversion recovery sequence varying the delay time from 0.001 to 10 s. The 1H T2 relaxation measurements of the polymer protons were made using the Carr–Purcell–Meiboom–Gill spin–echo sequence using an array of ∼20 values ranging from 0.002 to 1 s. The T1 and T2 relaxation times were obtained by fitting a monoexponential decay to the relaxation data.
The size distributions of particles were obtained with dynamic light scattering (DLS) by using the instrument of Bookhaven Instruments ( BI-200SM goniometer, BI-9000AT digital correlator) equipped with LEXEL 85 1W laser at wavelength 514.5. Scattering was measured at 90° angle and the obtained time correlation functions were analyzed by CONTIN Laplace-inversion program. The polymer concentration in the sample was 0.01 mg/ml obtained by diluting the particle dispersion with deionized water. Temperature of the sample unit was controlled with Lauda RC6 CP-thermostate. HS DSC (high sensitivity differential scanning calorimetry) measurements were performed with a VP-DSC microcalorimeter (Microcal) at an external pressure of ca. 180 kPA. The cell volume was 0.507 ml. Scans were recorded from 10 to 80°C at heating rates 30 and 60°C/h. Before each scan the sample was kept at 10°C for 15 min and each scan was repeated 3 times. Data was analyzed by using the software supplied by the manufacturer. The raw data from HS-DSC was manipulated by subtracting the baseline and with corrections to the sample concentration terms, where the sample concentration was given as molar concentration of NIPAM units in the particle sample. The samples were prepared from dialyzed particle dispersions by diluting with deionized water. Also, some samples prepared from freeze-dried particles were measured in H2O and D2O. Particle concentrations were of 2–20 g/l.
Results and discussion
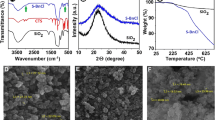
The characteristic peaks of both the polystyrene seed and the crosslinked PNIPAM components were present in the FTIR-spectra (not shown) of the two-stage particles. In the case of PS1NA, only one clear characteristic peak corresponding to carboxylic acid functionality (ca. 1,702 cm−1) could be observed as a tiny shoulder on the strong peak corresponding to the amide functionality of PNIPAM (ca. 1,650 cm−1). The quantitative 1H NMR-spectra (not shown) of the two-stage particles were measured in deuterated chloroform which is a good solvent for the both components and gives a clear solution. The analysis of integrated spectra gave the amounts of NIPAM units of 23, 17, and 44 mol% in the two-stage particles PS1N, PS1NA, and PS2N, respectively. The small amount of crosslinker, MBA, was ignored in the composition calculations because its weak proton signals could not be properly analyzed due to the complete overlap by the proton signals originating from the structurally similar NIPAM units.
The mean hydrodynamic diameters and the size distributions in aqueous dispersions of the particles were obtained from the DLS data. The intensity weighed size distributions at 20°C are shown in Fig. 1. As expected, the particle sizes grew during the second stage polymerization. The largest 2-stage particle, PS2N, was synthesized by using a larger monomer to seed particle mass ratio compared to the syntheses of PS1N and PS1NA. The 2-stage particles show monomodal size distributions that are broader than the size distributions obtained for their seed particles. The mean hydrodynamic diameters of the 2-stage particles vs temperature at range 15–50°C (heating rate ca. 5°C/h) are presented in Fig. 2. The mean hydrodynamic radius vs temperature for PNIPAM microgel particle, N1, is plotted in the same figure for comparison. The microgel particle deswells rapidly within a temperature range 25–35°C and its radius decreases nearly 50% upon heating at the studied temperature range. The 2-stage particles deswell gradually through the whole studied temperature range showing nearly linear deswelling. As an exception, a clear deviation from linearity is observed for PS2N at temperature range 30–40°C. Behavior of PS2N differs from the other particles because its total volume, decreased upon heating, is very small compared to its original size at 20°C. The size distributions in all cases remained monomodal and particle coagulation was not observed during the measurements at and above the phase transition temperature. DLS data upon cooling was also measured (not shown), and for all particles, the deswelling was observed to be reversible. The particles are most likely stabilized against coagulation at elevated temperatures by electrostatic repulsion between the anionic surface groups originating from the polymerization initiator fragments, residual surfactant, and in addition, from the dissociated carboxylic acid groups in the case of PS1NA.

The intensity-weighed size distributions of particles in aqueous dispersions at 20°C

The mean hydrodynamic diameters of the 2-stage particles vs temperature. The mean hydrodynamic radius of the microgel particle, N1, is plotted in the figure for comparison
The phase transition of the crosslinked PNIPAM was studied more closely on molecular level by quantitative 1H NMR spectroscopy in D2O. 1H NMR spectra of the microgel sample N2 in D2O above and below the critical temperature are shown in Fig. 3. Figure 4 shows the curves of the normalized PNIPAM proton to solvent proton (4.8 ppm) spectral intensity ratios vs temperature (22–50°C) for the different protons of the main chain and the side chain N-isopropyl group in microgel particle, N1 (20 mg/ml). The signal intensities of the main chain methylene (ca. 1.5 ppm) and methyne (ca. 2 ppm) protons decrease considerably already at 25–30°C and the signals disappear completely at 50°C. Instead, the methyl protons (ca. 1 ppm) and the lone proton (ca. 4 ppm) of the N-isopropyl group show corresponding rapid response at temperatures closer to the cloud point of PNIPAM at 32–35°C and the side chain proton signals can be detected even above 50°C as small ones. At temperatures above 35°C, we are most likely observing proton signals only from the surface of the collapsed particle.

1H NMR spectra of the microgel, N2, recorded at 22 and 40°C in D2O

Normalized ratios of 1H NMR signal intensity (I) of the PNIPAM protons to the solvent (D2O) protons vs temperature as measured for the protons of the N-isopropyl group (methyl protons ca. 1 ppm and the lone proton ca. 4 ppm) and the protons of the main chain (methylene protons ca. 1.5 ppm and methyne protons ca. 2 ppm) in the microgel sample, N1
Due to the dramatic decrease of the PNIPAM proton signal intensities during the phase transition, the strongest signal of the proton spectra at ca. 1 ppm, corresponding to the methyl protons of the side chain, was chosen for comparing the transitions of crosslinked PNIPAM in different particles. Figure 5 shows the curves of the normalized methyl proton to solvent proton (4.8 ppm) spectral intensity ratios vs temperature (22–50°C) for different particles. It appears that of the 2-stage particles, PS2N shows the closest resemblance to the microgels N1 and N2, although for PS2N the rapid decrease of the methyl proton signal is observed at 35–40°C. It is concluded that the phase transition of crosslinked PNIPAM in this particle shell has shifted towards higher temperatures when compared to the microgels and also that the temperature range of the transition is broader. The 2-stage particle PS1N shows more gradual decrease of methyl proton signal through the whole studied temperature range. Poly(NIPAM-co-acrylic acid) microgel structure in the 2-stage particle PS1NA appears to be quite swollen in D2O even at 50°C due to the presence of the hydrophilic comonomer units.

Normalized ratios of the 1H NMR signal intensity (I) of the methyl protons (ca. 1 ppm) to the solvent (D2O) protons vs temperature in different particles
A noticeable increase of the signal intensities in Fig. 4 is observed when the microgel sample is heated below the transition temperature. Similar increase is also observed for samples PS2N and PS1NA in Fig. 5. The increase of intensities could be interpreted as being due to increasing mobility with increasing temperature below the phase transition temperature. Corresponding observations have been reported by Zhu and Napper [20] for 13C NMR signals from linear PNIPAM chains both in solution and attached to polystyrene latex particles. Increase of the signal intensities was observed to be more pronounced in the case of the interfacial chains.
Dynamic behavior of PNIPAM microgel in the particles was studied by measuring spin–lattice (T1) and spin–spin (T2) relaxation times for the side chain methyl and the main chain methyne protons at temperatures 22–50°C in D2O. The accuracy of the relaxation times is low at temperatures above 35°C due to decreasing proton signals and therefore drawing conclusions from the data obtained at higher temperatures is avoided. Figure 6 shows the temperature dependence of the T1 for protons of the side chain methyl groups in the studied particles. For all samples, T1 increases with temperature showing close to linear dependence. It appears that T1 of the side chain methyl protons is not, at least in this case, highly sensitive to the particle structure because the T1 values measured for different particles at the same temperature deviate from each other only with some tens of milliseconds. Within this small deviation range, the microgel sample N1 gives the highest T1 values at temperatures below 35°C. Larger differences between the particles are observed when looking at the corresponding T1 data for the main chain methyne protons in Fig. 7. In general the 2-stage particles show lower values of T1 for main chain methyne protons than the reference microgels and the sample N1 shows again the highest T1 values. This may be an indication of more mobile structures of PNIPAAm in the 2-stage particles than in the microgel samples. Some samples show increasing T1 values above 35°C but any conclusions about clear overall trends of T1 with temperature during the phase transition cannot be drawn from the data of the relaxation times taking into account the accuracy problems at high temperatures. Figure 8 shows the temperature dependence of the T2 for protons of the main chain methyne groups in the studied particles. At temperatures below 35°C the 2-stage particles show clearly higher T2 values than the microgel particles. These higher T2 values (0.03–0.04 s) are close to the ones (0.03–0.05 s) obtained earlier in our laboratory for a linear PNIPAM (M w 150,000 g/mol, 2 mg/ml in D20 at 27–35°C) in a similar experiment using the same instrumentation (unpublished data, 2005). Higher T2 values refer again to more mobile components existing in 2-stage particle samples and possible explanation is the looser and/or more heterogeneous network structure of PNIPAM compared to the microgel samples. Lower values of T2 for the microgel particles originate most likely from the motional restriction brought by the crosslinking. However, the relaxation decays were monoexponential in all cases, indicating that the signal is coming from the most mobile segments in the systems (the most mobile segments between the crosslinks and dangling chains). This means that signal from the significantly less mobile parts of the polymers is not reflected in the relaxation data due to a very quick relaxation.

Spin–lattice relaxation times (T1) of the methyl protons (side chain) vs temperature for different particles in D2O

Spin–lattice relaxation times (T1) of the methyne protons (main chain) vs temperature for different particles in D2O

Spin–spin relaxation times (T2) of the methyne protons (main chain) vs temperature for different particles in D2O
The calorimetrically obtained transition temperature of PNIPAM-based polymers is often defined as the onset of the DSC transition endotherm, T onset (the intersection of the baseline and the leading edge of the endotherm). The temperature of the maximum heat capacity, T max, is also used to describe the transition temperature. For a sharp peak in a DSC thermogram it is possible to determine T onset very precisely. However, for broad peaks, the determination of T onset is more difficult due to inaccuracy in setting the baseline and, in such cases, it appears to be more reliable to determine the T max in comparative studies. Microcalorimetric endotherms for aqueous dispersions of N1, PS1N, and PS2N are shown in Fig. 9, and some additional information and data is collected in Table 2. The endotherms are presented in this study as excess heat capacities vs temperature. Broader endotherms are observed for crosslinked PNIPAM in the 2-stage particles compared to the reference microgel sample, N1. Especially in the case of PS2N, a clear increase of T max can be observed. This finding is supported by the obtained NMR data of the methyl 1H-signal intensities upon heating in D2O (Fig. 5) showing that the phase transition of crosslinked PNIPAM in this particle structure has shifted towards higher temperatures when compared to the reference microgels, and that the temperature range of the transition is broader. The broadest endotherm (not shown) as indicated by the highest value of T 1/2 (the width of the endotherm at the half-height of the peak) in Table 2 was obtained for the particle containing small amount of acrylic acid. In this case, the transition is evidently broadened by the presence of both the core particle and the hydrophilic groups as was observed clearly from the 1H-signal intensities upon heating (Fig. 5). H2O was used as a solvent in the microcalorimetric measurements. When comparing the NMR results to the calorimetric results, it is useful to keep in mind that H2O and D2O differ in their physical properties and that a “hydrogen’’ bond in D2O is ca. 5% stronger [23]. PNIPAM coils are suggested to be more extented in D2O below the critical temperature and there may be a higher level of ordering of D2O associated with a polymer chain [24]. Kujawa and Winnik [25] reported for linear PNIPAM (of molecular weight ca. 300,000 g/mol) a very similar microcalorimetric endotherm in general features in D2O compared to the corresponding endotherm in H2O. But in comparison to H2O, an increase of T max by 0.6°C and a doubling of the change of heat capacity occurring upon the phase transition were observed in D2O. As control samples in D2O, we measured thermograms (data not shown) of N1 (2 and 20 mg/ml). The differences observed between D2O and H2O in our microgel case were in agreement with the observations of Kujawa and Winnik for the linear PNIPAM.

Microcalorimetric endotherms (heating rate 30°C/h) for aqueous dispersions of the microgel (N1: solid line) and the 2-stage particles (PS1N: dotted line, PS2N: dashed line). For more information, see Table 2
How are the characteristics of the phase transition observed by microcalorimetry connected to the structure of the PNIPAM microgel system? Woodward et al. [26] investigated the volume phase transition of colloidal PNIPAM microgels prepared with varying crosslinker concentrations (0.25–30% of MBA in monomer mixture) by surfactant-free emulsion polymerization. In this case, the system varies from a loose PNIPAM-network to a more compact and rigid particle structure with increasing crosslinking density. DSC analyzes of these aqueous microgel particles showed that the T max and the width of the volume phase transition increase with increasing crosslinker concentration. For example, T max of the calorimetric endotherm increased by about 2°C when the amount of MBA in the monomer mixture was increased from 0.5 to 5%. The calorimetric enthalpy change of the transition decreased with increasing crosslinker concentration. The increased temperature of transition could indicate increased stability of the microgel within the solvent. However, the polarity of the interior of the microgels was studied with fluorescence spectroscopy by using pyrene as a probe. The results indicated that the hydrophobicity sensed by the probe was increasing with increasing crosslinker concentration. The calorimetric results were explained as being due to relatively more rigid structure of the microgel at higher crosslinker concentrations, and, as a result, the volume phase transition broadens and it is pushed towards higher temperatures. Our observations by microcalorimetry (data in Table 2) for the microgel particles, N1 (6 wt% of MBA in monomer feed) and N2 (3 wt% of MBA in monomer feed), appear to be in accordance with their results.
Wu et al. [9] studied the effect of MBA on the swelling ability of PNIPAM microgels. MBA has higher reactivity in radical polymerization compared to NIPAM. It is assumed that there is a concentration gradient of the crosslinker in these microgels and the crosslinking density is decreasing from the particle core towards the surface. Similar conclusion was reached by Guillermo et al. [27] when studying the heterogeneous structures of thermosensitive microgels of poly(N-isopropylmethacrylamide) produced with MBA. Assuming the existence of a core-shell-like structure, it was suggested that the relative size of the rigid core is increased upon increasing crosslinker concentration. This type of microgel particles are heterogeneous systems in which the crosslinking density determines whether the response to temperature is dominated by the phase transition of linear parts in the system or the volume phase transition of the gel structure. In our case, the feed of the crosslinking monomer was kept constant (6 wt% of the monomers in the reaction batch) in the syntheses of the 2-stage particles and the microgel N1. But how does the presence of the polystyrene seed particles affect to the structure of the forming PNIPAM network? It appears that the PNIPAM network is firmly attached to the core particles because the samples showed good endurance to heating–cooling cycles during the measurements. Precipitation of PNIPAM to the surface of the seed particles evidently occurs in the used synthesis conditions, but the effect of chain transfer from PNIPAM chains to the seed polymer and resulting chain growth from the core surface is unknown. However, the precipitation polymerization of NIPAM in the presence of the crosslinking monomer produces a system locked to a network structure in a shrunken state. Close packing of PNIPAM globules onto the surface of the seed particles during the synthesis must have an effect on the swelling abilities of the produced network.
Ballauff and coworkers [14–18] analyzed the similar system of crosslinked PNIPAM shell on poly(styrene 95 wt%-co-NIPAM 5 wt%) core particles by DLS, SANS, and SAXS in aqueous media. It was shown that the PNIPAM shell in swollen state exhibited static and dynamic inhomogeneities, but clear evidences of radial distribution of crosslinks or inhomogeneities in the network were not found. Investigations by DLS revealed that a small number of chains extended beyond the network yielding a hairy surface. The volume phase transition of the shell was found to be continuous and the degree of shrinking was found to be much less than what is observed for macroscopic networks of a similar degree of crosslinking. This was suggested to be due to the fact that macroscopic networks shrink along three directions whereas the shell networks on a rigid core can only shrink in the dimension along the surface normal. In our case, the 2-stage particles showed continuous, nearly linear deswelling and, in the case of PS2N, it appears that the swelling and the collapse of PNIPAAm network is significantly hindered as shown in Fig. 2. Evidently, severe motional restrictions affect to the volume phase transition of the network. On the other hand, the relaxation data refers to higher mobility in comparison to the microgel samples with high capacity to swell. A logical explanation could be, taking into account the results on linear polymers [20–22], a densely packed and fairly immobile PNIPAM layer close to the seed particle surface accompanied by highly mobile coronal PNIPAM layer. Because the obtained relaxation times showed clearly only one main component, we were monitoring only the most mobile PNIPAM segments of the samples in the T1 and T2 measurements. In the case of PS1N, containing a thinner layer of crosslinked PNIPAM, the broad transition and the low enthalpy of dehydration refer to a more pronounced effect of the core particle through the whole PNIPAM layer.
Analogously, to linear PNIPAM attached to solid particles, the factors affecting to the phase transition of crosslinked PNIPAM on a solid particle appear to be the distance of the polymer segment from the core (the thickness of the PNIPAM layer) and the local concentration/packing of the polymer segments close to the core surface. Increased motional restrictions are brought by the crosslinking in our case. Certain analogy can also be observed when comparing the calorimetric results obtained in our case of crosslinked PNIPAM on a solid polystyrene particle to the results of Woodward et al. [26] which deals with the effect of increasing amount of crosslinker to the temperature and the width of the volume phase transition. Although in their case, the rigid core was prepared by dense crosslinking of PNIPAM instead of using a solid polystyrene particle.
Conclusion
According to our results from microcalorimetry and 1H NMR spectroscopy, the phase transition of crosslinked PNIPAM-shell on solid polystyrene particle shifts towards higher temperatures when compared to the results obtained for PNIPAM microgels and, also, that the temperature range of the transition is broader. The overall transition appears to be affected by the thickness of the PNIPAM layer. The broadest transition was observed for the particle containing small amount of acrylic acid. In this case, the transition is evidently broadened by the presence of both the core particle and the hydrophilic groups. The results from dynamic light scattering showed a surprisingly small change of hydrodynamic size with temperature in the case of a thick PNIPAM layer referring to strong motional restrictions. However, the relaxation times of PNIPAM protons refer to components with higher mobility existing in 2-stage particle samples and possible explanation is a looser and/or more heterogeneous structure of PNIPAM network compared to the microgel samples. The relaxation decays were monoexponential in all cases, including the microgels, indicating that we were monitoring only the most mobile segments in all the systems.
The broadening of the transition in the core-shell particles compared to microgels is interpreted as being due to more heterogeneous structure of the PNIPAM network. The increase of the transition temperature is concluded to be due to increased motional constraints of the polymer segments on the solid surface.
References
Heskins M, Guillet JM (1968) Macromol Sci Chem A2:1441
Schild HG (1992) Prog Polym Sci 17:163
Pelton RH, Chibante P (1986) Colloids Surf 20:247
Pelton RH (1988) J Polym Sci A Polym Chem 26:9
Hoshino F, Fujimoto T, Kawaguchi H, Ohtsuka Y (1987) Polym J 19:241
Hoshino F, Kawaguchi H, Ohtsuka Y (1987) Polym J 19:1157
Kawaguchi H, Hoshino F, Ohtsuka Y (1986) Makromol Chem Rapid Commun 7:110
Ohtsuka Y, Kawaguchi H, Sugi Y (1981) J Appl Polym Sci 26:1637
Wu X, Pelton RH, Hamielec AE, Woods DR, McPhee W (1994) Colloid Polym Sci 272:467
Duracher D, Sauzedde F, Elaissari A, Perrin A, Pichot C (1998) Colloid Polym Sci 276:219
Duracher D, Sauzedde F, Elaissari A, Perrin A, Pichot C, Nabzar L (1998) Colloid Polym Sci 276:920
Nabzar L, Duracher D, Elaissari A, Perrin A, Chauveteau G, Pichot C (1998) Langmuir 14:5062
Taniguchi T, Duracher D, Delair T, Elaissari A, Pichot C (2003) Colloids Surf B: Biointerfaces 29:53 DOI: 10.1016/S0927-7765(02)00176-5
Dingenouts N, Norhausen Ch, Ballauff M (1998) Macromolecules 31:8912 DOI: 10.1021/ma980985t
Kim JH, Ballauff M (1999) Colloid Polym Sci 277:1210
Dingenouts N, Seelenmeyer S, Deike I, Rosenfeldt S, Ballauff M, Lindner P, Narayanan T (2001) Phys Chem Chem Phys 3:1169 DOI: 10.1039/b009104i
Seelenmeyer S, Deike I, Rosenfeldt S, Norhausen Ch, Dingenouts N, Ballauff M, Narayanan T, Lindner P (2001) J Chem Phys 114:10471
Senff H, Richtering W, Norhausen Ch, Weiss A, Ballauff M (1999) Langmuir 15:102 DOI: 101021/la980979q
Zhu PW, Napper DH (1994) J Colloid Interface Sci 164:489
Zhu PW, Napper DH (1996) Colloids Surf A 113:145
Shan J, Chen J, Nuopponen M, Tenhu H (2004) Langmuir 20:4671 DOI: 10.1021/la0363938
Schönhoff M, Larsson A, Welzel PB, Kuckling D (2002) J Phys Chem B 106:7800 DOI: 10.1021/jp0155381
Nemethy G, Sheraga HA (1964) J Chem Phys 41:680
Wang X, Wu C (1999) Macromolecules 32:4299 DOI: 10.1021/ma9902450
Kujawa P, Winnik FM (2001) Macromolecules 34:4130 DOI: 10.2021/ma002082h
Woodward NC, Chowdhry BZ, Snowden MJ, Leharne SA, Griffiths PC, Winnington AL (2003) Langmuir 19:3202 DOI: 10.1021/la020881i
Guillermo A, Cohen Addad JP, Bazile JP, Duracher D, Elaissari A, Pichot C (2000) J Polym Sci B Polym Phys 38:889 DOI: 10.1002/(SICI)1099-0488(20000315)38:6<889::AID-POLB9>3.0.CO;2-L
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License ( https://creativecommons.org/licenses/by-nc/2.0 ), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
Andersson, M., Hietala, S., Tenhu, H. et al. Polystyrene latex particles coated with crosslinked poly(N-isopropylacrylamide). Colloid Polym Sci 284, 1255–1263 (2006). https://doi.org/10.1007/s00396-006-1470-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00396-006-1470-2