
Abstract
The number of “omics” approaches is continuously growing. Among others, epigenetics has appeared as an attractive area of investigation by the cardiovascular research community, notably considering its association with disease development. Complex diseases such as cardiovascular diseases have to be tackled using methods integrating different omics levels, so called “multi-omics” approaches. These approaches combine and co-analyze different levels of disease regulation. In this review, we present and discuss the role of epigenetic mechanisms in regulating gene expression and provide an integrated view of how these mechanisms are interlinked and regulate the development of cardiac disease, with a particular attention to heart failure. We focus on DNA, histone, and RNA modifications, and discuss the current methods and tools used for data integration and analysis. Enhancing the knowledge of these regulatory mechanisms may lead to novel therapeutic approaches and biomarkers for precision healthcare and improved clinical outcomes.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Despite the progress in healthcare in the past half of the century, according to the World Health Organization (WHO), cardiovascular disease (CVD) is still the number one cause of death worldwide. Heart failure is a main cardiovascular condition, both in terms of prevalence and in terms of impact to the population. It has been estimated that in developing countries the prevalence of diagnosed heart failure in the general population is 1–2%, meaning that 64.3 million people globally are currently living with heart failure [12, 41, 97]. Mortality rates in European countries range from about 7–16% in patients with a chronic form of the disease, and 22–37% in patients with acute disease [99]. The socioeconomic burden of heart failure is expected to worsen considerably due to an aging population worldwide. Depending on the form of cardiac stressor and phenotype of pathology, disease-associated remodelling of the heart can include hypertrophy of cardiomyocytes, fibroblast activation, proliferation and transformation into myofibroblasts, endothelial cell dysfunction and immune cell infiltration. All these elements of cellular remodelling can contribute to morphological and functional changes in the heart including interstitial and perivascular fibrosis, cardiomyocyte death, loss of elasticity, cardiac dilatation or stiffening and overall reduced cardiac output. Considering that heart failure encompass complex malfunctions of the heart and blood vessels a deeper understanding of the underlying molecular causes of pathological remodelling of the heart and blood vessels remains imperative in order to develop effective preventive medicine and treatment.
Epigenetics is an exciting field of research that offers new insights into the underlying mechanisms of disease. In particular, epigenetics is essential in understanding cardiac diseases. At its core, epigenetics refers to the heritable changes in gene expression that occur without altering the underlying DNA sequence. The term comes from the Greek words "epi" (above) and "genetics" (birth). This field of research has revealed that lifestyle, environmental and biological factors can influence gene expression, leading to altered cellular behaviour that can have profound health implications [51, 112]. However, the real importance of epigenetic mechanisms is related to the programming of cellular development and the specific identity of the different cell types.
As said, heart failure is a complex disorder, and epigenetics might provide a useful framework for understanding its pathogenesis. Numerous studies have identified epigenetic biomarkers of CVD risk, including changes in DNA methylation and histone modifications [15, 36, 126]. These epigenetic changes are associated with altered gene expression, which may contribute to the development of cardiac disfunction. For example, epigenetic modifications can regulate the expression of genes that are involved in inflammation and atherosclerosis, two processes that are known to aggravate cardiac function [20].
Epigenetics also plays a role in the expression of environmental and lifestyle factors that are associated with heart failure. For example, smoking, diet and physical inactivity can all cause epigenetic changes that are associated with the risk of heart failure development [52].
Risk factors contribute to epigenetic and gene expression changes in cells of the cardiovascular system that culminate in disease, in particular over long periods of time if pathological stressors persist. Gene regulation is not a linear on/off switch, but a dynamic, highly complex, multi-layer process that is responsive to signalling and metabolic cues. Epigenetics is the term used to encompass dynamic, reversible non-genetic mechanisms that regulate chromatin structure and function and the resulting transcriptome. Thus, in order to unravel the complex interplay between epigenetic regulators and to get a whole picture of gene regulation alterations in disease, it is essential to use highly integrative and interconnected network analyses. Current technological revolution and advancement of molecular biology and next-generation sequencing techniques enable us to identify the location and abundance of epigenetic variation such as DNA and histone modifications throughout the mammalian genome, DNA accessibility, higher-order chromatin structure, as well as the resulting transcriptome and modifications thereof. In order to put epigenetic mechanisms of regulation in a wider cardiovascular physiological context, it is now possible to integrate epigenetic datasets with other biological big data such as proteomics and metabolomics, thus creating multi-layer omics profiles. More recent advances in single-cell sequencing technologies have further widened our understanding of molecular and cellular remodelling in disease, as well as brought a number of new challenges along with it.
For instance, the integration of this overwhelming production of data is extremely difficult to achieve without computational tools. Several approaches for multi-omics data integration have been developed, highlighting the advantages of integrating different levels of data with bioinformatics and machine learning approaches. Furthermore, the latest developments in machine learning and artificial intelligence have enabled significant progress in the field. It is now possible to explore inter-relationships between different layers of biological complexity through the construction of multi-modal machine learning systems, also taking into account mechanistically derived biological features [1, 78, 102]. This new approach provides a powerful tool for the investigation of novel players in cardiovascular pathology and can lead to the discovery of novel druggable targets and/or biomarkers. At the single-cell level, it is equally evident that an integrative analysis of several omics data will enable a more comprehensive view of cardiovascular disease molecular features compared to using only one omic layer. This has fuelled the recent development of single-cell multi-omic technologies and related computational tools [65].
In this review, we address how DNA, histone, and RNA modifications influence cardiac physiology and disease with a particular focus on heart failure. We discuss the current methods and tools used to analyse and integrate the gleaned information. Since epigenetics in vascular disease has been covered in recent reviews [106, 131, 139], this article will not review and discuss the role of epigenetics mechanisms in the vascular bed. Also, the relevance of ncRNAs and RNA-based epigenetic mechanisms in cardiac diseases has been discussed in details elsewhere [94].
Overview of mechanisms of gene regulation
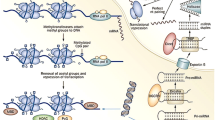
Many factors regulate gene activity at several levels in both the nucleus and the cytoplasm. Epigenomics is a recently developing field of research aimed at defining rules of gene expression regulation/function through the action of a plethora of specific enzymes and binding proteins named erasers, readers and writers [32]. Along with guidance from long non-coding RNAs (lncRNAs), the effect results in hundreds of post-translational modifications to DNA or histone tails, affecting wholesale chromatin conformation at the nucleosomal level, affecting the accessibility of genomic elements to RNA polymerase and other transcription factors. The major epigenetic regulatory mechanisms discussed below include DNA methylation and histone modifications (Fig. 1). RNA-based epigenetic control represents an additional layer of gene regulation at the level of transcription or post-transcriptionally. This review will not address the roles of ncRNAs in heart failure, as it has been extensively covered elsewhere [35].

Key players in DNA-based epigenetic mechanisms of gene regulation including DNA methylation, histone modifications and ncRNA-based regulation (created by Biorender). HMT histone methyltransferase, HAT histone acetyltransferase, HDMT histone demethylase, HDAC histone deacetylase, DNMT DNA methyltransferases, TET ten-eleven translocation methylcytosine dioxygenases, TDG thymine DNA glycosylase
DNA methylation refers to the addition of a methyl group to 5′ position of cytosine residues (5mC) throughout the genome. DNA methylation is written by the family of enzymes known as DNA methyltransferases (DNMT1, DNMT2, and DNMT3) [108]. While DNA methylation can be dynamically regulated in response to various stimuli, it is generally stably maintained in many cases, particularly in differentiated cells where it plays a crucial role in determining cell identity and fate. DNA demethylation is limited to only a few specific loci in differentiated cells, and occurs through specific mechanisms that involve the activity of TET (ten-eleven translocation) family of enzymes in conjunction with other factors [108]. However, the process of systematic TET-determined DNA demethylation is known to occur during early embryonic development, particularly after zygote formation, where it plays a critical role in erasing epigenetic marks and enabling the establishment of new gene expression patterns [108]. Additionally, DNA methylation may be passively lost through cellular replication cycles due to a basal inefficacy of the maintenance DNMT1, and this phenomenon has been utilized as a marker of biological aging of different tissues in mammals [47].
Histone modifications include post-translational chemical modifications, which occur predominantly at histone N-terminal tails and primarily involve the amino acids lysine and arginine [7, 61]. Acetylation status of histones is related to the activity of two families of enzymes, known as histone acetyltransferases (HATs) and histone deacetylases (HDACs) that add and remove one or more acetyl groups mainly at lysine residues on the histone tails [61]. Histone methylation/demethylation is catalysed by histone methyltransferase/ demethylases and can occur on both lysine and arginine residues of histone tails. In contrast to histone acetylation, which unequivocally leads to transcription activation, histone methylation can increase or decrease transcriptional activation depending on the location of the targeted amino acid residues in the histone tail and the number of methyl groups added [39].
Epigenomics in heart failure
Complex diseases, in particular CVD, are characterized by cellular biochemical and morphological remodelling. Changes in cell identity such as dedifferentiation are permitted by wholesale reprogramming of the epigenome and the transcriptome. Targeting the key epigenetic erasers, writers or readers responsible would affect disease-associated gene regulation on a global scale, ideally reversing a number of pathophysiological pathways rather than just a single protein or pathway.
DNA demethylating agents and histone deacetylase inhibitors are already on the market and FDA-approved for the treatment of certain forms of cancer, and further are being developed [19, 37, 83, 96]. Pharmacological advances in the generation of highly specific and effective inhibitors and activators of epigenetic mediators are needed along with targeting strategies for epigenetic drugs to move closer to clinical reality.
DNA methylation has been reported as important in cardiovascular development and disease. However, despite their biological relevance, the impact of DNA methylating and de-methylating enzymes, only a few pieces of evidence accumulated that these epigenetic enzymes may play an important role in CVD [38, 80, 103, 113] Table 1]. Interestingly, recent reports outlined a new layer of regulation of TET function. It has been recently found that dysmetabolic conditions, such as diabetes, may alter DNA demethylation in association with TET relocation outside the nucleus, a phenomenon dependent on AMPK activity [124]. Whether TET extra-nuclear compartmentalization occurs in the heart of diabetic patients and whether this phenomenon has a role in this context is currently unknown.
Whilst DNA methylations are important epigenetic marks, covalent modifications on histone tails work in concert to affect chromatin structure and gene expression regulating progression of CVD. Members of the HDAC Class IIa family—HDAC5 and HDAC9—protect against hypertrophic remodelling. The genetic disruption of specific genes in mice results in heightened vulnerability to cardiac hypertrophy, as well as a weakened response to stimuli that contribute to the development of hypertrophy, such as calcineurin activation and increased pressure. The binding of HDAC5 and HDAC9 leads to inhibition of Mef2c, a transcription factor that upregulates pro-hypertrophy genes [132]. When exposed to a pro-hypertrophic stimulus, two stress-responsive kinases, CaMK and PKD, act upon HDAC5 and HDAC9 causing HDAC4-dependent phosphorylation. Phosphorylated HDACs then associate with the chaperone protein 14-3-3 and relocate from the nucleus to the cytoplasm. This release of HDAC5 and HDAC9 from Mef2c leads to their interaction with p300, and consequently transcription activation [58]. This mechanism could also be relevant for the cardiotoxic effects of doxorubicin. Namely, doxorubicin impairs cardiac function by modulating the activity or expression of important Ca2+ handling proteins, leading to altered calcium function, and activates CaMKIIδ resulting in hyperphosphorylated PLN-T17 and RYR2-S2814, which increases the open probability of the RYR2 and leads to a significantly augmented diastolic sarcoplasmic reticulum Ca2 + leak [44]. Weather this mechanism can directly influence HDACs and to what extent that play role in the cardiotoxicity remains to be determined.
Furthermore, a complex mechanism has recently been deduced by which prevention of O-GlcNAcylation (post-translational addition of β-linked N-acetylglucosamine) to (Ser)-642 of HDAC4 stopped proteolytic cleavage of HDAC4 to create a short N-terminal fragment of HDAC4 [62]. Reducing the production of the NT HDAC4 fragment protected against cardiac hypertrophy in the diabetic heart. What’s more, preventing the production of NT-HDAC4 attenuated the pro-hypertrophic Ca2 + /calmodulin-dependent protein kinase II-mediated phosphorylation at HDAC4 Ser-632.
Thus far, less is known about the role of altered histone methylation marks in cardiac hypertrophy and disease than that of the aforementioned histone tail acetylation marks. Thienpont et al., identified a mechanism by which binding of miR-217 to a non-canonical binding site in the untranslated region of euchromatic histone lysine methyltransferases (EHMTs) in CMs in response to pathological stimuli caused a reduction in H3K9me2 in hypertrophy-associated genes such as Nppa, Nppb and Myh7 [107]. Preventing the miR-217-induced loss of EHMT and H3K9me2 genetically or using antisense oligo therapy attenuated the cardiac hypertrophy in vitro or in vivo models.
Assembly of the repressive chromatin complex BRG1–G9a/GLP–DNMT3 at the adult myosin heavy chain (Myh6) gene promoter, with increased H3K9 and CpG methylation at the locus, has been associated with gene repression in pressure overloaded stressed mouse hearts [43]. Chromatin structure is determined by nucleosome positioning, histone modifications, and DNA methylation. How chromatin modifications are altered under pathological conditions (in a coordinated manner) remains elusive. In human hypertrophic hearts, the BRG1-G9a/GLP-DNMT3 complex is also activated; its level correlates with H3K9/CpG methylation, Myh6 repression, and cardiomyopathy.
RNA editing and the epitranscriptome in heart failure
RNA editing represents a set of different enzymatically mediated modifications of ribonucleotides within coding and non-coding RNA molecules. Some of the most prevalent types of modifications includes methylation of different nucleotides and conversion of one nucleotide to another. Although the first modified nucleoside—5-ribosyluracil, or pseudouridine—was discovered in the 1950s, studies have only recently been aimed at understanding the role of RNA modifications in the epigenetic control of pathophysiological responses in the cardiovascular system. Indeed, the chemical modification of mRNA, tRNA, and rRNA was once deemed to be a static phenomenon involved largely in fine-tuning the structure of those macromolecules. However, ribonucleic acid bases in coding and noncoding RNA can be modified in a dynamic, reversible manner that is suggestive of a regulatory code atop the primary sequence. In analogy with epigenetics, the effector proteins that lead to epitranscriptomic modifications are also referred to as writers, erasers and readers [129]. The writers introduce certain types of modification in RNA, while the erasers recognize these modifications and catalyse their removal. However, the downstream effects of these structural changes depend on the readers [129], the proteins that specifically interact with modified bases thus affecting RNA behaviour and resulting in defined downstream effects [129].
Epigenetic modifications of RNA are modifications to the structure of the RNA molecule that include both methylation and hydroxymethylation of the cytosine base, as well as modifications of the adenine base. As for DNA, RNA methylation is the addition of a methyl group to the cytosine base, which changes the structure of the RNA molecule and can have an effect on gene expression [53]. Hydroxymethylation of the cytosine base is similar to methylation, but the hydroxymethyl group is added instead of the methyl group. This modification is especially abundant in cardiac tissue [29, 31]. Modification of the adenine base can also occur and can be a form of epigenetic modification. This involves the addition of a methyl or hydroxymethyl group to the adenine base, which can also affect gene expression [24, 82, 117, 118]. Epigenetic modifications of RNA are important for gene expression and have been studied extensively in recent years [73].
Specifically, there are a number of RNA epigenetic modifications of which the most common include N6-methyladenosine (m6A), N5-methylcytosine (m5C), N1-methyl Adenine (m1A), N6,2′-O-dimethyladenosine (m6Am), N7-methyl guanosine (m7G) and the Cytosine hydroxylation [22, 121].
For the scope of this article we will focus on m6A that is considered to be the most prevalent epitranscriptomic modification, found on ~ 25% of transcripts at the genome-wide level (Fig. 2). It is most frequently located near the stop codons, the 5´ and 3´-UTR, and within long internal exons affecting the RNA stability, translation, and secondary structure formation [23]. M6A is produced in the reaction catalysed by the multi-subunit writer complex that adds a methyl group to the N6 position of adenosine [129]. This complex includes methyltransferase 3 (METTL3), which plays a catalytic role, methyltransferase 14 (METTL14) that acts as a RNA-binding domain, and the cofactors Wilms tumour‑associated protein‑1 (WATAP) and KIAA1429 [114, 116]. As a reversible modification, m6A can be demethylated by erasers such as fat mass and obesity-associated protein (FTO) and α-ketoglutarate-dependent dioxygenase alkB homolog 5 (ALKBH5) [54, 89]. The biological effects of m6A modifications go beyond their influence on RNA structure and stability. As mentioned above, these effects are mediated through the interaction with readers such as the YT521-B homology domain (YTHDF2) and the insulin-like growth factor-2 mRNA-binding protein (IGF2BP) that recognize and bind to specific m6A modifications [48, 138].

The most important players in m6A RNA modifications and possible downstream effects. writers, erasers and readers (created by Biorender). METTL3 methyltransferase 3, METTL14 methyltransferase 14, WATAP Wilms tumour‑associated protein‑1, FTO fat mass and obesity-associated protein, ALKBH5 α-ketoglutarate-dependent dioxygenase alkB homolog 5, YTHDF2 YT521-B homology domain, IGF2BP insulin-like growth factor-2 mRNA-binding protein
So far, several studies have demonstrated a critical role for m6A modifications in heart function. Namely, cardiac hypertrophy is associated with a significant increase in m6A methylation of mRNA isolated from cardiomyocytes (CMs), with enrichment in specific functional clusters of genes, such as those regulating kinases and intracellular signalling pathways [26]. In addition, through experiments taking advantage of METTL3 overexpression, it appears that METTL3 drives compensated hypertrophy in vivo. Moreover, following ischemia/reperfusion, upregulation of METTL3 has been implicated in the control of genes related to autophagy [91, 98, 99]. M6A hypermethylation of mRNA encoding for myosin regulatory light chain 2 is observed in failing hearts, and leads to a decrease in protein levels [59]. Coherently, inhibition of METTL3 blunts CM hypertrophic growth whereas increased expression of METTL3 is sufficient to stimulate its development [25]. In the intact animal, METTL3 overexpression induces adaptive growth of the heart without untoward functional effects whereas cardiac-specific knockout leads to heart failure upon stress or aging. These findings are indicative of a role for RNA methylation in maintaining cardiac homeostasis [33]. Additionally, FTO is downregulated in the failing heart, a phenomenon associated with increased deposition of m6A on CM mRNA and worsened contractile function [81, 94]. Indeed, FTO selectively demethylates transcripts encoding proteins involved in muscle contraction, sarcomere organization, and cardiac hypertrophy, preventing their degradation. On the other hand, overexpression of FTO is associated with reduced cardiac fibrosis and increased angiogenesis in mice with myocardial infarction, suggestive of a possible therapeutic value in targeting this mechanism [81].
M6A modification can also influence the functions of ncRNAs. For example, during miRNA biogenesis, pri-miRNAs are modified by METTL3-depending mechanism [2, 3]. These m6A modifications are recognized by “reader” heterogeneous nuclear ribonucleoproteins A2/B1, which in turn, stimulate the initiation of DICER-mediated processing through the recruitment of DGCR8 [111]. It seems that m6A modifications block A:G non-canonical base-paring, affecting the strength of miRNAs interaction with target mRNA [38]. In addition, two lncRNAs associated with MI, X‑inactive specific transcript (XIST) and Metastasis associated lung adenocarcinoma transcript 1 (MALAT1) are highly prone to m6A modification. XIST-mediated transcriptional repression of X-linked genes is dependent on m6A modification and binding of m6A “reader” RNA binding motif protein 15 (RBM15) [92]. In the case of MALAT1, m6A modifications induce conformational changes and increased binding by a number of RNA binding proteins (RBPs) [101]. Adenosine deaminase acting on RNA (ADAR1) also seems to play important role in heart function and development. In neonatal mice CMs, oxidative stress induced simultaneously ADAR1 and protein kinase PKR expression leading to increased apoptosis and inflammation [120]. However, overexpression of ADAR1 inhibited PKR activation, which is suggested to be a self-preservation mechanism in neonatal CMs that limits excessive apoptosis and inflammation. In the study by Moore et al., cardiac knockout of ADAR1 is associated with embryonic lethality leading to the conclusion that ADAR1 is essential for embryonic CM survival and proliferation [84]. In the same lines, Azzouzi et al. have induced an ADAR1 deletion in adult CMs through the use of a tamoxifen-inducible Cre recombinase under the control of the cardiac-specific α-myosin heavy chain promoter (αMHC) [27]. The study shows that targeted ADAR1 deletion leads to a significant increase in ADAR1‑null mice lethality, accompanied by induction of stress markers, overall reduced expression of miRNAs, severe ventricular remodelling and quick and spontaneous cardiac dysfunction, mediated through miR-199a-5p and unfolded protein response. In addition to coding RNAs, A-to-I editing plays important role in the function of miRNAs. So far, 2711 potential pri-miR editing sites have been described within approximately 80% of all human pri-miRs, implicating that A-to-I editing can have a tremendous impact on miRNA target specificity [67]. Increased A-to-I-editing of vasoactive miR-487b-3p has been found in ischemic muscle tissues undergoing neovascularization after induction of hind limb ischemia [110]. The edited mature miR-487b-3p has a unique targetome and promotes angiogenesis, in contrast to the canonical miR-487b-3p. It seems that vasoactive microRNA editing is a widespread phenomenon that enhances neovascularization in response to ischemia [119]. Other RNA modifications such as m1A, m3C, m5C, m7G, and their roles in the pathophysiology of hear diseases have been highly understudied.
Epigenetic and epitranscriptomic crosstalk
Interactions between the “epi” worlds of DNA and RNA have not been extensively studied. However, lately, new evidence is starting to reveal the importance of the interplay between epigenetic and epitranscriptomic mechanisms in the regulation of gene expression.
M6A writing complex can influence histone modification status and chromatin state indirectly by inducing m6A modifications to mRNAs transcribed from the genes that encode for epigenetic modifiers, affecting their stability and expression levels [11]. M6A hypomethylation of EZH2 and CBP/p300 mRNA through METTL3 and METTL14 knock-out in neural stem cells leads to a decrease of H3K27me3 (marker of transcriptional repression) and an increase of H3K27ac (marker of transcriptional activation) compromising the ability of the cells to proliferate [17, 119]. In mouse embryonic stem cells, METTL3 silencing leads to the elevation of two histone marks associated with active transcription (H3K4me3 and H3K27ac), resulting in increased chromatin accessibility and chromatin-associated regulatory RNAs (carRNAs), including promoter-associated RNAs, enhancer RNAs, and repeat RNAs [72]. Furthermore, Li et al. have studied how epigenetic signals respond to m6A methylation in human Flp-In HEK293 cells [68]. Knockdown of METTL3 results in a significant increase in H3K9me2 and minor upregulation of H3K27ac and H3K27me3 [49]. Besides indirect effects on epigenetic control, m6A writing complex can directly bind to chromatin remodelling factors and histone modifications. Direct binding of METTL14 to H3K36me3 brings the whole m6A writing machinery in near proximity of RNA polymerase II thus enabling the addition of m6A to nascent RNAs during transcription [115]. On the other hand, epigenetic mechanisms can also play important roles in the regulation of epitranscriptomic mechanisms. Histone acetyltransferase p300 increased H3K27 acetylation of METTL3 promoter, leading to increased METTL3 transcription and m6A hypermethylation [133]. Furthermore, decreased methylation of METTL3 promoter results in METTL3 downregulation and reduced m6A levels on pri-miR-25, disrupting its maturation [18]. Similarly, the demethylation of H3K4me3 in the METTL14 promoter by the lysine-specific histone demethylase 5C promoted the decreasing the transcription of METTL14 [127].
M6A readers are also able to take an active part in epigenetic-epitranscriptomic interplays. In the nucleus, m6A-marked intracisternal A particle (IAP) transcripts interact with YTHDC1, which recruits METTL3 and in turn promotes the association of METTL3 with chromatin [105]. In addition, METTL3 interacts with histone 3 lysine 9 (H3K9) tri-methyltransferase SETDB1 and its cofactor TRIM28, important for their localisation to IAPs [105]. YTHDC1 binds m6A-modified carRNAs leading to their degradation and decreased chromatin accessibility [72]. YTHDC1 promotes the demethylation of H3K9me2 through interaction with lysine demethylase 3B (KDM3B), ultimately leading to increased gene expression [68]. In the cytoplasm, another m6A reader, YTHDF2, destabilizes m6A methylated lysine demethylase 6B (KDM6B) mRNA leading to an increase in H3K27me3 levels [123]. On the other hand, miRNa-145 binds to the 3’-UTR of the YTHDF2 mRNA, resulting in its downregulation. Another interesting level of interplay is observed in smooth muscle cells under high-glucose conditions. Namely, circYTHDC2 targets the 3’-UTR of the TET2 mRNA leading to its degradation and in turn influence methylation of the DNA [128].
Lastly, m6A erasers that decrease m6A levels in RNAs can ultimately influence histone marks via several mechanisms. ALKBH5 decreases m6A levels on lncRNA TRERNA1, then in turn act as a scaffold and recruit EZH2 leading to the increase in H3K27me3 modification of cyclin-dependent kinases inhibitor p21 promoter and the decrease of its expression [100]. In addition, ALKBH5 knockdown leads to the increase of H3K9me3 levels in mouse embryonic stem cells via unknown mechanisms [127].
The presented data strongly suggest that the cross-talk between epigenetic and epitranscriptomic mechanisms represents an important level of cellular regulation that should be more extensively explored, particularly in the context of heart disease. So far, we do not have clear information how these mechanisms could be important in the function of the different cardiac cell types. Thus, it seems important to drive the field in this direction and reveal how these complex interplays can contribute to our better understanding of the pathological process behind cardiac dysfunction. Mechanisms of epigenetics-epitranscriptomics cross-talk are summarized in Fig. 3.

Mechanisms of epigenetics-epitranscriptomics cross-talk (created by Biorender). METTL3 methyltransferase 3, METTL14 methyltransferase 14, YTHDC1 YTH domain-containing protein 1, EZH2 enhancer of zeste homolog 2
Integration of epigenomics data through machine learning algorithms
To untangle the complex biology of cellular mechanisms, and differences in multiple cells, tissues and organs caused by hear failure, multi-omics measurements are often necessary. Multi-omics analyses adopt a holistic view toward the universal detection of genes (genomics), epigenetic modification (epigenomics), RNA expression (transcriptomics), protein translation (proteomics) and metabolite production (metabolomics) that can characterize eventually specific sets of biological samples in a non‐targeted and non‐biased manner. The rationale of these approaches is that a complex system can be understood more thoroughly if considered as a whole. By integrating epigenomics with other regulatory layers, we integrate data in a broader context that improves our understanding of the complex pathophysiological responses at the origin and progression of cardiac dysfunction.
There are many computational approaches for the analysis of multi-omics data, reviewed in [4, 10, 57]. Several integration strategies have been proposed to ensure that the main methodological challenge of multi-omics integration is addressed, namely ensuring that the information contained in each molecular “view” is used in a complementary rather than conflicting way, and does not overshadow the information present in other omics. Network-based, model-based and machine-learning based approaches have been proposed to solve this issue, with successful results [45, 66, 109]. As with experimental data, the accessibility of models to the community is of paramount importance. The model developers should be encouraged to use established frameworks such as the Systems Biology Markup Language (SBML) to facilitate dissemination [50].
While the use of machine learning methods has generated much interest, it is often limited by a lack of appropriate training data. Furthermore, several other challenges remain, e.g. the presence of redundant information across the omics, and an effective strategy for interpreting the machine learning models while integrating the omics data. Despite these challenges, using bioinformatics and systems biology tools, researchers can make informed decisions, and develop models that automatically learn and improve from feedback. In this machine learning framework, both “supervised” and “unsupervised” methods can be used. In supervised learning, one aims to predict target functions associated with a sample (e.g. risk score associated with a patient). This can be achieved through classifiers, when the goal is to predict the class to which a new sample belongs (e.g. high-risk or low-risk patients), or regressors, when the goal is to predict a numerical quantity (e.g. an exact risk score for each patient). Conversely, in unsupervised learning, one aims at predicting latent associations or patterns among the variables. In this context, clustering techniques can reveal patterns in the data, while dimensionality reduction techniques (e.g. principal component analysis, multi-dimensional scaling) can elucidate the key contribution to the variation observed in CVD data and to the interactions with other diseases like type 2 diabetes [8]. Merging biological knowledge with in silico models has been shown to contribute towards biologically informed and interpretable machine learning methods, even with regularised regression techniques [78].
Supervised machine learning is a powerful technique when the response “labels” are available for each of the samples used to improve the model. With such labels, a model can learn both from data and from feedback on the predictions it makes. One of the first examples of machine learning in the context of CVD was a semi-supervised class discovery method that was developed to predict clinical parameters and risk factors [28]. Recently, data from a large cohort of 378,256 patients was fed to four machine-learning algorithms to predict the first cardiovascular event over 10 years, with the best performance achieved by neural networks [122]. Machine and deep learning tools can be also used to discover patterns from large and heterogeneous data like those obtained in epigenomics studies, thus deepening and enhancing our understanding of interplay between different layers of epigenomic landscape. Using deep learning, Zhao et al. developed a model for the prediction of heart failure with preserved ejection fraction that was based on the methylation profile of 25 DNA loci and 5 clinical features (age, diuretics use, body mass index, albuminuria, and serum creatinine) [136]. The developed method outperformed models including only clinical features or DNA methylation data. In another study, the use of machine learning approaches enabled the prenatal detection of foetal congenital heart defects based on methylation profiles in circulating cell-free DNA from maternal blood [6]. In the study by Fernández-Pérez et al., different machine learning models (elastic net regression, K-Nearest neighbours, random forest, and support vector machine models), and one deep learning approximation (multilayer perceptron model) were used to explore the influence of different clinical and lifestyle factors on global DNA methylation profiles in patients with cerebrovascular disease [30]. The best models were elastic net regression and multilayer perceptron model, although with modest capability, explaining around 35–40% of variability in methylation profiles. By using cross-combination of three machine learning methods, including best subset regression, regularization techniques, and random forest algorithm, Ma et al. discovered a signature of five genes that acts as m7G regulators that can distinguish patients with heart failure from healthy subjects [76]. As previously mentioned, added value of machine learning approaches is their ability to ingrate data that come from different levels of cellular complexity, so-called multi-omics data. Zhou et al. used a multi-omics approach to explore molecular signatures in a mouse model of heart failure [137]. This study included machine learning-based integrative analysis of scRNA-seq, scATAC-seq, bulk ATAC-seq and miRNA-seq data that provided mechanists insights in the complexity of pressure overload-induced heart failure.
When applying bioinformatics or systems biology tools, the quality of pre-processing and data control is the key to avoiding learning and propagating noise in the machine learning model. Effective feature engineering or feature selection, and attempting different strategies for data integration, are also paramount to extracting cross-omics information from the different omics variables. Different omics layers may require different omics-specific data extraction strategies. After extracting latent information from each omics, a final integration method is often required to collate the latent multi-omics information, discarding any partial overlap (e.g. between genomics, epigenomics, transcriptomics, proteomics and metabolomics). This multi-step approach (also called transformation-based or intermediate-stage omics data integration) has often proven successful when dealing with multi-omics data [21], but further research is needed to ensure that the information overlap between the omics can be reduced, and only the unique information that each contributing omics can be extracted before combining all parameters.
To better exploit predictive algorithms and avoid using them as “black boxes”, incorporating mechanistic information seems a promising direction for the research field. In this regard, information derived from cell modelling can be incorporated into machine learning pipelines, for instance at the stage of feature selection. The availability of genome sequences, combined with biochemical information, has enabled the reconstruction of genome-scale models of metabolism. These include all known metabolic reactions in the cell, and can also have different coverage and scope, e.g. integrating metabolism with regulation, signalling, protein interactions and further cellular routes [88]. For instance, CardioNet was the first reconstruction of the metabolic network of the human cardiomyocyte [56]. Deep learning, and in particular its multi-modal versions, have been proposed to learn from these combined multi-omic datasets, including those generated by metabolic models [130]. Simulation of these metabolic models can be incorporated into the machine learning methods, therefore providing biomarkers and useful information that make the machine learning predictions more interpretable (see Fig. 4).

A single profile (e.g. a patient) can be measured on different omic spaces (e.g. transcriptomic data and metabolic activity). The distance between two patients can therefore also be measured in different spaces. The distance according to the transcriptomic profile of the two patients is likely to differ from the distance between the same two patients, but measured according to metabolism. Methods for integration of multiple omics define a multi-omic distance, e.g. by combining the distances from the individual omic profiles, therefore providing a systems-level view of the omic profiles. This strategy mitigates the bias that a single-omic analysis may introduce
Conclusions and perspectives
Major and rapid advances in next-generation sequencing technologies to study the transcriptome and epigenome have greatly enhanced our understanding of the molecular basis of disease in cells and tissues of the cardiovascular system. A large number of groups have now published bulk or single-cell sequencing data from human heart samples and pre-clinical models of cardiovascular disease. The number of publications in PubMed describing “cardiovascular disease,” “RNA-sequencing,” and “epigenomics” has increased from a total of 21 to 322 in the last decade. These data sets have undoubtedly expanded our knowledge and understanding of disease mechanisms in cardiovascular disease and leads to promising trials of epigenetic drugs (e.g. NCT05350969) [9, 103, 104].
One major challenge associated to the widespread generation of big datasets is the unification of methodologies used to analyse data to be able to compare and integrate results generated by different research teams. This requires formulation of standardization of tools and analytical and appropriate education and training of individual researchers as well as minimal requirements for methodological reporting and data storage. Currently, none of these measures are in place, leading to poor reproducibility of data and massive underutilization of publicly available data, often generated from precious human specimens. The lack of consensus in sample storage, next-generation sequencing library preparation, data storage and analysis as well as thorough reporting of all the aforementioned is being recognized as holding back clinically important and relevant findings in the cardiovascular community [94].
Another aspect to consider is that alterations in chromatin, DNA modifications and RNA expression may be more consequences of disease rather than being the actual promoting factors for disease initiation and progression. Epigenetic and transcriptomic changes can be therefore adaptive in response to the pathological stressors. These mechanisms could be harnessed to protect the heart and vasculature in a disease context or even to prevent the detrimental effects of exposure to risk factors. Nevertheless, some modifications are disease-driving. Big data, including RNA-sequencing and epigenomics data, can provide an in-depth analysis of the state of cells or tissues at a given point in time or stage of disease progression.
A novel useful approach in studying epigenetic modifications can be through the use of gene editing tools based on CRISPR/Cas9 system. Modified Cas9 named dCas9 can be fused with genetic modifiers and used to introduce locus-specific modifications such as DNA methylation, histone methylation and acetylation [13]. Another application of this technology is live cell chromatin imaging, which allows to visualize specific locations in the genome in real time. This can be achieved by labelling dCas9 with fluorescent tags, which enables them to see the genomic loci in a single-, dual-, or multicolor way [13]. In addition, the use of Cas13 enzymes that bind to RNA and not DNA, opens new doors for site-specific epitranscriptomic research [13].
In order to distinguish between these various forms of molecular alteration and to identify relevant targets to guide drug development, it is essential to combine big data acquisition with functional cellular and pre-clinical studies using appropriate models. This validation requires a multi-disciplinary approach to translational cardiovascular disease, bringing together clinical and basic scientists, bioinformaticians and data scientists with molecular biologists, biochemists and medicinal chemists and experience animal technicians. These specialities are seldom found within a single research group. In this way, working in an interdisciplinary, collaborative manner, bringing together different expertise, such as that provided by the EU-CardioRNA COST Action, optimizes the opportunity to make breakthroughs with more sensitive and disease-specific diagnostic tools and treatments.
References
Acosta JN, Falcone GJ, Rajpurkar P, Topol EJ (2022) Multimodal biomedical AI. Nat Med 28:1773–1784. https://doi.org/10.1038/s41591-022-01981-2
Alarcón CR, Goodarzi H, Lee H, Liu X, Tavazoie S, Tavazoie SF (2015) HNRNPA2B1 is a mediator of m6A-dependent nuclear RNA processing events. Cell 162:1299–1308. https://doi.org/10.1016/j.cell.2015.08.011
Alarcón CR, Lee H, Goodarzi H, Halberg N, Tavazoie SF (2015) N 6-methyladenosine marks primary microRNAs for processing. Nature 519:482–485. https://doi.org/10.1038/nature14281
Arneson D, Shu L, Tsai B, Barrere-Cain R, Sun C, Yang X (2017) Multidimensional integrative genomics approaches to dissecting cardiovascular disease. Front Cardiovasc Med 4:8. https://doi.org/10.3389/fcvm.2017.00008
Awad S, Al-Haffar KM, Marashly Q, Quijada P, Kunhi M, Al-Yacoub N, Wade FS, Mohammed SF, Al-Dayel F, Sutherland G, Assiri A (2015) Control of histone H3 phosphorylation by CaMKII δ in response to haemodynamic cardiac stress. J Patho 235:606–618. https://doi.org/10.1002/path.4489
Bahado-Singh R, Friedman P, Talbot C, Aydas B, Southekal S, Mishra NK, Vishweswaraiah S (2023) Cell-free DNA in maternal blood and artificial intelligence: accurate prenatal detection of fetal congenital heart defects. Am J Obstet Gynecol 228:76-e1. https://doi.org/10.1016/j.ajog.2022.07.062
Bannister AJ, Kouzarides T (2011) Regulation of chromatin by histone modifications. Cell Res 21:381–395. https://doi.org/10.1038/cr.2011.22
Barna B, Matharoo K, Bhanwer AS (2014) A multifactorial dimensionality reduction model for gene polymorphisms and environmental interaction analysis for the detection of susceptibility for type 2 diabetic and cardiovascular diseases. Mol Cytogenet 7:1–2. https://doi.org/10.1186/1755-8166-7-s1-p116
Batkai S, Genschel C, Viereck J, Rump S, Bär C, Borchert T, Traxler D, Riesenhuber M, Spannbauer A, Lukovic D, Zlabinger K, Thum T (2021) CDR132L improves systolic and diastolic function in a large animal model of chronic heart failure. Eur Heart J 42:192–201. https://doi.org/10.1093/eurheartj/ehaa791
Bersanelli M, Mosca E, Remondini D, Giampieri E, Sala C, Castellani G, Milanesi L (2016) Methods for the integration of multi-omics data: mathematical aspects. BMC Bioinformatics 17:167–177. https://doi.org/10.1186/s12859-015-0857-9
Bove G, Amin S, Babaei M, Benedetti R, Nebbioso A, Altucci L, Del Gaudio N (2022) Interplay between m6A epitranscriptome and epigenome in cancer: current knowledge and therapeutic perspectives. Int J Cancer. https://doi.org/10.1002/ijc.34378
Bragazzi NL, Zhong W, Shu J, Abu Much A, Lotan D, Grupper A, Dai H (2021) Burden of heart failure and underlying causes in 195 countries and territories from 1990 to 2017. Eur J Prev Cardiol 28:1682–1690. https://doi.org/10.1093/eurjpc/zwaa147
Brandes RP, Dueck A, Engelhardt S, Kaulich M, Kupatt C, De Angelis MT, Wurst W (2021) DGK and DZHK position paper on genome editing: basic science applications and future perspective. Basic Res Cardiol 116:1–20. https://doi.org/10.1007/s00395-020-00839-3
Chapski DJ, Cabaj M, Morselli M, Mason RJ, Soehalim E, Ren S, Pellegrini M, Wang Y, Vondriska TM, Rosa-Garrido M (2021) Early adaptive chromatin remodeling events precede pathologic phenotypes and are reinforced in the failing heart. J Mol Cell Cardiol 160:73–86. https://doi.org/10.1016/j.yjmcc.2021.07.002
Chelladurai P, Boucherat O, Stenmark K, Kracht M, Seeger W, Bauer UM, Bonnet S, Pullamsetti SSSS (2021) Targeting histone acetylation in pulmonary hypertension and right ventricular hypertrophy. Br J Pharmacol 178:54–71. https://doi.org/10.1111/bph.14932
Chen H, Orozco LD, Wang J, Rau CD, Rubbi L, Ren S, Wang Y, Pellegrini M, Lusis AJ, Vondriska TM (2016) DNA methylation indicates susceptibility to isoproterenol-induced cardiac pathology and is associated with chromatin states. Circ Res 118:786–797. https://doi.org/10.1161/circresaha.115.305298
Chen J, Zhang YC, Huang C, Shen H, Sun B, Cheng X, Zhang YJ, Yang YG, Shu Q, Yang Y, Li X (2019) m6A regulates neurogenesis and neuronal development by modulating histone methyltransferase Ezh2. Genom Proteom Bionf 17:154–168. https://doi.org/10.1016/j.gpb.2018.12.007
Chen X, Xu M, Xu X, Zeng K, Liu X, Pan B, Li C, Sun L, Qin J, Xu T, He B, Wang S (2020) METTL14-mediated N6-methyladenosine modification of SOX4 mRNA inhibits tumor metastasis in colorectal cancer. Mol Cancer 19:1–16. https://doi.org/10.1186/s12943-020-01220-7
Chua GN, Wassarman KL, Sun H, Alp JA, Jarczyk EI, Kuzio NJ, Bennett MJ, Malachowsky BG, Kruse M, Kennedy AJ (2019) Cytosine-based TET enzyme inhibitors. ACS Med Chem Lett 10:180–185. https://doi.org/10.1021/acsmedchemlett.8b00474
Costantino S, Paneni F (2022) The epigenome in atherosclerosis. Handb Exp Pharmacol 270:511–535. https://doi.org/10.1007/164_2020_422
Culley C, Vijayakumar S, Zampieri G, Angione C (2020) A mechanism-aware and multiomic machine-learning pipeline characterizes yeast cell growth. Proc Natl Acad Sci U S A 117:18869–18879. https://doi.org/10.1073/pnas.2002959117
Delatte B, Wang F, Ngoc LV, Collignon E, Bonvin E, Deplus R, Calonne E, Hassabi B, Putmans P, Awe S, Wetzel C (2016) Transcriptome-wide distribution and function of RNA hydroxymethylcytosine. Science 351:282–285. https://doi.org/10.1126/science.aac5253
Dominissini D, Moshitch-Moshkovitz S, Schwartz S, Salmon-Divon M, Ungar L, Osenberg S, Cesarkas K, Jacob-Hirsch J, Amariglio N, Kupiec M, Sorek R, Rechavi G (2012) Topology of the human and mouse m6A RNA methylomes revealed by m6A-seq. Nature 485:201–206. https://doi.org/10.1038/nature11112
Dominissini D, Moshitch-Moshkovitz S, Schwartz S, Salmon-Divon M, Ungar L, Osenberg S, Rechavi G (2012) Topology of the human and mouse m6A RNA methylomes revealed by m6A-seq. Nature 485:201–206. https://doi.org/10.1038/nature11112
Dorn LE, Lasman L, Chen J, Xu X, Hund TJ, Medvedovic M, Accornero F (2019) The N6-methyladenosine mRNA methylase METTL3 controls cardiac homeostasis and hypertrophy. Circulation 139:533–545. https://doi.org/10.1161/CIRCULATIONAHA.118.036146
Dorn LE, Lasman L, Chen J, Xu X, Hund TJ, Medvedovic M, Hanna JH, Van Berlo JH, Accornero F (2019) The N6-methyladenosine mRNA methylase METTL3 controls cardiac homeostasis and hypertrophy. Circulation 139:533–545. https://doi.org/10.1161/circulationaha.118.036146
El Azzouzi H, Vilaça AP, Feyen DA, Gommans WM, De Weger RA, Doevendans PA, Sluijter JP (2020) Cardiomyocyte specific deletion of ADAR1 causes severe cardiac dysfunction and increased lethality. Front Cardiovasc Med 7:30. https://doi.org/10.3389/fcvm.2020.00030
Elmarakeby HA, Hwang J, Arafeh R, Crowdis J, Gang S, Liu D, AlDubayan SH, Salari K, Kregel S, Richter C, Arnoff TE (2021) Biologically informed deep neural network for prostate cancer discovery. Nature 598:348–352. https://doi.org/10.1038/s41586-021-03922-4
Fang S, Li J, Xiao Y, Lee M, Guo L, Han W, Huang Y (2019) Tet inactivation disrupts YY1 binding and long-range chromatin interactions during embryonic heart development. Nat Commun 10:4297. https://doi.org/10.1038/s41467-019-12325-z
Fernández-Pérez I, Jiménez-Balado J, Lazcano U, Giralt-Steinhauer E, Rey Álvarez L, Cuadrado-Godia E, Jiménez-Conde J (2023) Machine learning approximations to predict epigenetic age acceleration in stroke patients. Int J Mol Sci 24:2759. https://doi.org/10.3390/ijms24032759
Fu L, Guerrero CR, Zhong N, Amato NJ, Liu Y, Liu S, Wang Y (2014) Tet-mediated formation of 5-hydroxymethylcytosine in RNA. J Am Chem Soc 136:11582–11585. https://doi.org/10.1021/ja505305z
Gillette TG, Hill JA (2015) Readers, writers, and erasers: chromatin as the whiteboard of heart disease. Circ Res 116:1245–1253. https://doi.org/10.1161/circresaha.116.303630
Gilsbach R, Schwaderer M, Preissl S, Grüning BA, Kranzhöfer D, Schneider P, Nührenberg TG, Mulero-Navarro S, Weichenhan D, Braun C, Dreßen M, Hein L (2018) Distinct epigenetic programs regulate cardiac myocyte development and disease in the human heart in vivo. Nat Commun 9:391. https://doi.org/10.1038/s41467-017-02762-z
Glezeva N, Moran B, Collier P, Moravec CS, Phelan D, Donnellan E, Russell-Hallinan A, O’Connor DP, Gallagher WM, Gallagher J, McDonald K, Watson CJ (2019) Targeted DNA methylation profiling of human cardiac tissue reveals novel epigenetic traits and gene deregulation across different heart failure patient subtypes. Circ Heart Fail 12:e005765. https://doi.org/10.1161/circheartfailure.118.005765
Gomes CPC, Schroen B, Kuster GM, Robinson EL, Ford K, Squire IB, EU-CardioRNAC. O. S. T. (2020) Action (CA17129). Regulatory RNAs in heart failure. Circulation 141:313–328. https://doi.org/10.1161/CIRCULATIONAHA.119.042474
Gonzalez-Jaramillo V, Portilla-Fernandez E, Glisic M, Voortman T, Bramer W, Chowdhury R, Roks AJ, Jan Danser AH, Muka T, Nano J, Franco OH (2019) The role of DNA methylation and histone modifications in blood pressure: a systematic review. J Hum Hypertens 33:703–715. https://doi.org/10.1038/s41371-019-0218-7
Gorica E, Mohammed SA, Ambrosini S, Calderone V, Costantino S, Paneni F (2022) Epi-drugs in heart failure. Front Cardiovasc Med 9:923014. https://doi.org/10.3389/fcvm.2022.923014
Greco CM, Kunderfranco P, Rubino M, Larcher V, Carullo P, Anselmo A, Kurz K, Carell T, Angius A, Latronico MV, Papait R, Condorelli G (2016) DNA hydroxymethylation controls cardiomyocyte gene expression in development and hypertrophy. Nat Commun 7:12418. https://doi.org/10.1038/ncomms12418
Greer EL, Shi Y (2012) Histone methylation: a dynamic mark in health, disease and inheritance. Nat Rev Genet 13:343–357. https://doi.org/10.1038/nrg3173
Greißel A, Culmes M, Burgkart R, Zimmermann A, Eckstein HH, Zernecke A, Pelisek J (2016) Histone acetylation and methylation significantly change with severity of atherosclerosis in human carotid plaques. Cardiovasc Pathol 25:79–86. https://doi.org/10.1016/j.carpath.2015.11.001
Groenewegen A, Rutten FH, Mosterd A, Hoes AW (2020) Eur J Heart Fail 22:1342–1356. https://doi.org/10.1002/ejhf.1858
Haas J, Frese KS, Park YJ, Keller A, Vogel B, Lindroth AM, Weichenhan D, Franke J, Fischer S, Bauer A, Marquart S, Meder B (2013) Alterations in cardiac DNA methylation in human dilated cardiomyopathy. EMBO Mol Med 5:413–429. https://doi.org/10.1002/emmm.201201553
Han P, Li W, Yang J, Shang C, Lin CH, Cheng W, Hang CT, Cheng HL, Chen CH, Wong J, Chang XY, CP. (2016) Epigenetic response to environmental stress: assembly of BRG1–G9a/GLP–DNMT3 repressive chromatin complex on Myh6 promoter in pathologically stressed hearts. Biochim Biophys Acta 1863:1772–1781. https://doi.org/10.1016/j.bbamcr.2016.03.002
Haupt LP, Rebs S, Maurer W, Hübscher D, Tiburcy M, Pabel S, Streckfuss-Bömeke K (2022) Doxorubicin induces cardiotoxicity in a pluripotent stem cell model of aggressive B cell lymphoma cancer patients. Basic Res Cardiol 117:13. https://doi.org/10.1007/s00395-022-00918-7
Hira MT, Razzaque MA, Angione C, Scrivens J, Sawan S, Sarker M (2021) Integrated multi-omics analysis of ovarian cancer using variational autoencoders. Sci rep 11:6265. https://doi.org/10.1038/s41598-021-85285-4
Hohl M, Wagner M, Reil JC, Müller SA, Tauchnitz M, Zimmer AM, Lehmann LH, Thiel G, Böhm M, Backs J, Maack C (2013) HDAC4 controls histone methylation in response to elevated cardiac load. J Clin Invest 123:1359–1370. https://doi.org/10.1172/jci61084
Horvath S (2013) DNA methylation age of human tissues and cell types. Genome Biol 14:1–20. https://doi.org/10.1186/gb-2013-14-10-r115
Huang H, Weng H, Sun W, Qin X, Shi H, Wu H, Zhao BS, Mesquita A, Liu C, Yuan CL, Hu YC, Chen J (2018) Recognition of RNA N 6-methyladenosine by IGF2BP proteins enhances mRNA stability and translation. Nat Cell Biol 20:285–295. https://doi.org/10.1038/s41556-018-0045-z
Huang H, Weng H, Zhou K, Wu T, Zhao BS, Sun M, Chen Z, Deng X, Xiao G, Auer F, Klemm L, Chen J (2019) Histone H3 trimethylation at lysine 36 guides m6A RNA modification co-transcriptionally. Nature 567:414–419. https://doi.org/10.1038/s41586-019-1016-7
Hucka M, Finney A, Sauro HM, Bolouri H, Doyle JC, Kitano H, Arkin AP, Bornstein BJ, Bray D, Cornish-Bowden A, Cuellar AA, Wang J (2003) The systems biology markup language (SBML): a medium for representation and exchange of biochemical network models. Bioinformatics 19:524–531. https://doi.org/10.1093/bioinformatics/btg015
Hulikova A, Park KC, Loonat AA, Gunadasa-Rohling M, Curtis MK, Chung YJ, Swietach P (2022) Alkaline nucleoplasm facilitates contractile gene expression in the mammalian heart. Basic Res Cardiol 117:17. https://doi.org/10.1007/s00395-022-00924-9
Imam MU, Ismail M (2017) The impact of traditional food and lifestyle behavior on epigenetic burden of chronic disease. Glob Chall 1:1700043. https://doi.org/10.1002/gch2.201700043
Jacob R, Zander S, Gutschner T (2017) The dark side of the epitranscriptome: chemical modifications in long non-coding RNAs. Int J Mol Sci 18:2387. https://doi.org/10.3390/ijms18112387
Jia G, Fu YE, Zhao XU, Dai Q, Zheng G, Yang Y, Yi C, Lindahl T, Pan T, Yang YG, He C (2011) N 6-methyladenosine in nuclear RNA is a major substrate of the obesity-associated FTO. Nat Chem Biol 7:885–88. https://doi.org/10.1038/nchembio.687
Jiang DS, Yi X, Li R, Su YS, Wang J, Chen ML, Liu LG, Hu M, Cheng C, Zheng P, Zhu XH, Wei X (2017) The histone methyltransferase mixed lineage leukemia (MLL) 3 may play a potential role in clinical dilated cardiomyopathy. Mol Med 23:196–203. https://doi.org/10.2119/molmed.2017.00012
Karlstädt A, Fliegner D, Kararigas G, Ruderisch HS, Regitz-Zagrosek V, Holzhütter HG (2012) CardioNet: a human metabolic network suited for the study of cardiomyocyte metabolism. BMC Syst Biol 6:1–20. https://doi.org/10.1186/1752-0509-6-114
Kim M, Tagkopoulos I (2018) Data integration and predictive modeling methods for multi-omics datasets. Mol omics 14:8–25. https://doi.org/10.1039/c7mo00051k
Klose RJ, Zhang Y (2007) Regulation of histone methylation by demethylimination and demethylation. Nat Rev Mol Cell Biol 8:307–318. https://doi.org/10.1038/nrm2143
Kmietczyk V, Riechert E, Kalinski L, Boileau E, Malovrh E, Malone B, Gorska A, Hofmann C, Varma E, Jürgensen L, Kamuf-Schenk V, Völkers M (2019) Tm6A-mRNA methylation regulates cardiac gene expression and cellular growth. Life Sci Alliance 2:e201800233. https://doi.org/10.26508/lsa.201800233
Koczor CA, Lee EK, Torres RA, Boyd A, Vega JD, Uppal K, Yuan F, Fields EJ, Samarel AM, Lewis W (2013) Detection of differentially methylated gene promoters in failing and nonfailing human left ventricle myocardium using computation analysis. Physiol Genomics 45:597–605. https://doi.org/10.1152/physiolgenomics.00013.2013
Kouzarides T (2007) Chromatin modifications and their function. Cell 128:693–705. https://doi.org/10.1016/j.cell.2007.02.005
Kronlage M, Dewenter M, Grosso J, Fleming T, Oehl U, Lehmann LH, Falcão-Pires I, Leite-Moreira AF, Volk N, Gröne HJ, Müller OJ, Backs J (2019) O-GlcNAcylation of histone deacetylase 4 protects the diabetic heart from failure. Circulation 140:580–594. https://doi.org/10.1161/circulationaha.117.031942
Kurozumi A, Nakano K, Yamagata K, Okada Y, Nakayamada S, Tanaka Y (2019) IL-6 and sIL-6R induces STAT3-dependent differentiation of human VSMCs into osteoblast-like cells through JMJD2B-mediated histone demethylation of RUNX2. Bone 124:53–61. https://doi.org/10.1016/j.bone.2019.04.006
Laugier L, Frade AF, Ferreira FM, Baron MA, Teixeira PC, Cabantous S, Ferreira LR, Louis L, Rigaud VO, Gaiotto FA, Bacal F (2017) Whole-genome cardiac DNA methylation fingerprint and gene expression analysis provide new insights in the pathogenesis of chronic Chagas disease cardiomyopathy. Clin Infect Dis 65:1103–1111. https://doi.org/10.1093/cid/cix506
Lee J, Hyeon DY, Hwang D (2020) Single-cell multiomics: technologies and data analysis methods. Exp Mol Med 52:1428–1442. https://doi.org/10.1038/s12276-020-0420-2
Leon-Mimila P, Wang J, Huertas-Vazquez A (2019) Relevance of multi-omics studies in cardiovascular diseases. Front Cardiovasc Med 6:91. https://doi.org/10.3389/fcvm.2019.00091
Li L, Song Y, Shi X, Liu J, Xiong S, Chen W, Fu Q, Huang Z, Gu N, Zhang R (2018) The landscape of miRNA editing in animals and its impact on miRNA biogenesis and targeting. Genome Res 28:132–143. https://doi.org/10.1101/gr.224386.117
Li Y, Xia L, Tan K, Ye X, Zuo Z, Li M, Xiao R, Wang Z, Liu X, Deng M, Cui J, Xia L (2020) N 6-Methyladenosine co-transcriptionally directs the demethylation of histone H3K9me2. Nat Genet 52:870–877. https://doi.org/10.1038/s41588-020-0677-3
Liao X, Kennel PJ, Liu B, Nash TR, Zhuang R, Godier-Furnemont AF, Xue C, Lu R, Colombo PC, Uriel N, Reilly MP, Topkara VK (2022) Genome-wide DNA methylation profiling of the failing human heart with mechanical unloading identifies LINC00881 as an essential regulator of calcium handling in the cardiomyocyte. bioRxiv 2022-03. https://doi.org/10.1101/2022.03.01.482163
Lin X, Li F, Xu F, Cui RR, Xiong D, Zhong JY, Zhu T, Shan SK, Wu F, Xie XB, Yuan LXB, LQ. (2019) Aberration methylation of miR-34b was involved in regulating vascular calcification by targeting Notch1. Aging (Albany NY) 11:3182. https://doi.org/10.18632/aging.101973
Lin X, Xu F, Cui RR, Xiong D, Zhong JY, Zhu T, Li F, Wu F, Xie XB, Mao MZ, Liao XB, Yuan LQ (2018) Arterial calcification is regulated via an miR-204/DNMT3a regulatory circuit both in vitro and in female mice. Endocrinology 159:2905–2916. https://doi.org/10.1210/en.2018-00320
Liu J, Dou X, Chen C, Chen C, Liu C, Xu MM, Zhao S, Shen B, Gao Y, Han D, He C (2020) N 6-methyladenosine of chromosome-associated regulatory RNA regulates chromatin state and transcription. Science 367:580–586. https://doi.org/10.1126/science.aay6018
Liu N, Pan T (2015) RNA epigenetics. Transl Res 165:28–35. https://doi.org/10.1016/j.trsl.2014.04.003
Liu R, Jin Y, Tang WH, Qin L, Zhang X, Tellides G, Hwa J, Yu J, Martin KA (2013) Ten-eleven translocation-2 (TET2) is a master regulator of smooth muscle cell plasticity. Circulation 128:2047–2057. https://doi.org/10.1161/circulationaha.113.002887
Luo X, Hu Y, Shen J, Liu X, Wang T, Li L, Li J (2022) Integrative analysis of DNA methylation and gene expression reveals key molecular signatures in acute myocardial infarction. Clin Epigenet 14:1–13. https://doi.org/10.1186/s13148-022-01267-x
Ma C, Tu D, Xu Q, Wu Y, Song X, Guo Z, Zhao X (2023) Identification of m7G regulator-mediated RNA methylation modification patterns and related immune microenvironment regulation characteristics in heart failure. Clinic Epigenetics 15:1–21. https://doi.org/10.1186/s13148-023-01439-3
Madsen A, Höppner G, Krause J, Hirt MN, Laufer SD, Schweizer M, Tan WL, Mosqueira D, Anene-Nzelu CG, Lim I, Foo RS, Stenzig J (2020) An important role for DNMT3A-mediated DNA methylation in cardiomyocyte metabolism and contractility. Circulation 142:1562–1578. https://doi.org/10.1161/circulationaha.119.044444
Magazzù G, Zampieri G, Angione C (2021) Multimodal regularized linear models with flux balance analysis for mechanistic integration of omics data. Bioinformatics 37:3546–3552. https://doi.org/10.1093/bioinformatics/btab324
Maleszewska M, Gjaltema RA, Krenning G, Harmsen MC (2015) Enhancer of zeste homolog-2 (EZH2) methyltransferase regulates transgelin/smooth muscle-22α expression in endothelial cells in response to interleukin-1β and transforming growth factor-β2. Cell Signal 27:1589–1596. https://doi.org/10.1016/j.cellsig.2015.04.008
Martinez SR, Gay MS, Zhang L (2015) Epigenetic mechanisms in heart development and disease. Drug Discov today 20:799–811. https://doi.org/10.1016/j.drudis.2014.12.018
Mathiyalagan P, Adamiak M, Mayourian J, Sassi Y, Liang Y, Agarwal N, Jha D, Zhang S, Kohlbrenner E, Chepurko E, Chen J, Sahoo S (2019) FTO-dependent N6-methyladenosine regulates cardiac function during remodeling and repair. Circulation 139:518–532. https://doi.org/10.1161/circulationaha.118.033794
Meyer KD, Patil DP, Zhou J, Zinoviev A, Skabkin MA, Elemento O, Jaffrey SR (2015) 5′ UTR m6A promotes cap-independent translation. Cell 163:999–1010. https://doi.org/10.1016/j.cell.2015.10.012
Montalvo-Casimiro M, González-Barrios R, Meraz-Rodriguez MA, Juárez-González VT, Arriaga-Canon C, Herrera LA (2020) Epidrug repurposing: discovering new faces of old acquaintances in cancer therapy. Front Oncol 10:605386. https://doi.org/10.3389/fonc.2020.605386
Moore JB IV, Sadri G, Fischer AG, Weirick T, Militello G, Wysoczynski M, Gumpert AM, Braun T, Uchida S (2020) The A-to-I RNA editing enzyme Adar1 is essential for normal embryonic cardiac growth and development. Circ Res 127:550–552. https://doi.org/10.1161/circresaha.120.316932
Movassagh M, Choy MK, Goddard M, Bennett MR, Down TA, Foo RS (2010) Differential DNA methylation correlates with differential expression of angiogenic factors in human heart failure. PLoS ONE 5:e8564. https://doi.org/10.1371/journal.pone.0008564
Movassagh M, Choy MK, Knowles DA, Cordeddu L, Haider S, Down T, Siggens L, Vujic A, Simeoni I, Penkett C, Goddard M, Foo RS (2011) Distinct epigenomic features in end-stage failing human hearts. Circulation 124:2411–2422. https://doi.org/10.1161/circulationaha.111.040071
Nührenberg TG, Hammann N, Schnick T, Preißl S, Witten A, Stoll M, Gilsbach R, Neumann FJ, Hein L (2015) Cardiac myocyte de novo DNA methyltransferases 3a/3b are dispensable for cardiac function and remodeling after chronic pressure overload in mice. PLoS ONE 10:e0131019. https://doi.org/10.1371/journal.pone.0131019
O’Brien EJ, Monk JM, Palsson BO (2015) Using genome-scale models to predict biological capabilities. Cell 161:971–987. https://doi.org/10.1016/j.cell.2015.05.019
Ougland R, Zhang CM, Liiv A, Johansen RF, Seeberg E, Hou YM, Remme J, Falnes PØ (2004) AlkB restores the biological function of mRNA and tRNA inactivated by chemical methylation. Mol cell 16:107–116. https://doi.org/10.1016/j.molcel.2004.09.002
Papait R, Serio S, Pagiatakis C, Rusconi F, Carullo P, Mazzola M, Salvarani N, Miragoli M, Condorelli G (2017) Histone methyltransferase G9a is required for cardiomyocyte homeostasis and hypertrophy. Circulation 136:1233–1246. https://doi.org/10.1161/circulationaha.117.028561
Pastore N, Brady OA, Diab HI, Martina JA, Sun L, Huynh T, Lim JA, Zare H, Raben N, Ballabio A, Puertollano R (2016) TFEB and TFE3 cooperate in the regulation of the innate immune response in activated macrophages. Autophagy 12:1240–1258. https://doi.org/10.1080/15548627.2016.1179405
Patil DP, Chen CK, Pickering BF, Chow A, Jackson C, Guttman M, Jaffrey SR (2016) m6A RNA methylation promotes XIST-mediated transcriptional repression. Nature 537:369–373. https://doi.org/10.1038/nature19342
Pepin ME, Ha CM, Crossman DK, Litovsky SH, Varambally S, Barchue JP, Pamboukian SV, Diakos NA, Drakos SG, Pogwizd SM, Wende AR (2019) Genome-wide DNA methylation encodes cardiac transcriptional reprogramming in human ischemic heart failure. Lab Invest 99:371–386. https://doi.org/10.1038/s41374-018-0104-x
Robinson EL, Baker AH, Brittan M, McCracken I, Condorelli G, Emanueli C, Srivastava PK, Gaetano C, Thum T, Vanhaverbeke M, Angione C, EU-CardioRNA COST Action CA17129 (2022) Dissecting the transcriptome in cardiovascular disease. Cardiovasc Res 118:1004–1019. https://doi.org/10.1093/cvr/cvab117
Robinson EL, Drawnel FM, Mehdi S, Archer CR, Liu W, Okkenhaug H, Alkass K, Aronsen JM, Nagaraju CK, Sjaastad I, Sipido KR, Roderick HL (2022) MSK-mediated phosphorylation of histone H3 Ser28 couples MAPK signalling with early gene induction and cardiac hypertrophy. Cells 11:604. https://doi.org/10.3390/cells11040604
Rodríguez-Paredes M, Esteller M (2011) Cancer epigenetics reaches mainstream oncology. Nat Med 17:330–339. https://doi.org/10.1038/nm.2305
Savarese G, Becher PM, Lund LH, Seferovic P, Rosano GM, Coats AJ (2022) Global burden of heart failure: a comprehensive and updated review of epidemiology. Cardiovasc Res 118:3272–3287. https://doi.org/10.1093/cvr/cvac013
Song H, Feng X, Zhang H, Luo Y, Huang J, Lin M, Jin J, Ding X, Wu S, Huang H, Yu T (2019) METTL3 and ALKBH5 oppositely regulate m6A modification of TFEB mRNA, which dictates the fate of hypoxia/reoxygenation-treated cardiomyocytes. Autophagy 15:1419–1437. https://doi.org/10.1080/15548627.2019.1586246
Song H, Pu J, Wang L, Wu L, Xiao J, Liu Q, Chen J, Zhang M, Liu Y, Ni M, Mo J (2015) ATG16L1 phosphorylation is oppositely regulated by CSNK2/casein kinase 2 and PPP1/protein phosphatase 1 which determines the fate of cardiomyocytes during hypoxia/reoxygenation. Autophagy 11:1308–1325. https://doi.org/10.1080/15548627.2015.1060386
Song W, Fei F, Qiao F, Weng Z, Yang Y, Cao B, Yue J, Xu J, Zheng M, Li J (2022) ALKBH5-mediated N6-methyladenosine modification of TRERNA1 promotes DLBCL proliferation via p21 downregulation. Cell Death Discov 8:25. https://doi.org/10.1038/s41420-022-00819-7
Spitale RC, Flynn RA, Zhang QC, Crisalli P, Lee B, Jung JW, Kuchelmeister HY, Batista PJ, Torre EA, Kool ET, Chang HY (2015) Structural imprints in vivo decode RNA regulatory mechanisms. Nature 519:486–490. https://doi.org/10.1038/nature14263
Stahlschmidt SR, Ulfenborg B, Synnergren J (2022) Multimodal deep learning for biomedical data fusion: a review. Brief Bioinform 23:bbab569. https://doi.org/10.1093/bib/bbab569
Stenzig J, Schneeberger Y, Löser A, Peters BS, Schaefer A, Zhao RR, Ng SL, Höppner G, Geertz B, Hirt MN, Tan W (2018) Pharmacological inhibition of DNA methylation attenuates pressure overload-induced cardiac hypertrophy in rats. J Mol Cell Cardiol 120:53–63. https://doi.org/10.1016/j.yjmcc.2018.05.012
Stratton MS, Bagchi RA, Felisbino MB, Hirsch RA, Smith HE, Riching AS, Enyart BY, Koch KA, Cavasin MA, Alexanian M, Song K, McKinsey TA (2019) Dynamic chromatin targeting of BRD4 stimulates cardiac fibroblast activation. Circ Res 125:662–677. https://doi.org/10.1161/circresaha.119.315125
Tan B, Zhou K, Liu W, Prince E, Qing Y, Li Y, Han L, Qin X, Su R, Pokharel SP, Yang L, Chen J (2022) RNA N6-methyladenosine reader YTHDC1 is essential for TGF-beta-mediated metastasis of triple negative breast cancer. Theranostics 12:5727. https://doi.org/10.7150/thno.71872
Tang H, Zeng Z, Shang C, Li Q, Liu J (2021) Epigenetic regulation in pathology of atherosclerosis: a novel perspective. Front Genet 12:2643. https://doi.org/10.3389/fgene.2021.810689
Thienpont B, Aronsen JM, Robinson EL, Okkenhaug H, Loche E, Ferrini A, Brien P, Alkass K, Tomasso A, Agrawal A, Bergmann O, Roderick HL (2017) The H3K9 dimethyltransferases EHMT1/2 protect against pathological cardiac hypertrophy. J Clin Invest 127:335–348. https://doi.org/10.1172/jci88353
Traube FR, Carell T (2017) The chemistries and consequences of DNA and RNA methylation and demethylation. RNA Biol 14:1099–1107. https://doi.org/10.1080/15476286.2017.1318241
Vahabi N, Michailidis G (2022) Unsupervised multi-omics data integration methods: a comprehensive review. Front Gen. https://doi.org/10.3389/fgene.2022.854752
van der Kwast RV, van Ingen E, Parma L, Peters HA, Quax PH, Nossent AY (2018) Adenosine-to-inosine editing of microRNA-487b alters target gene selection after ischemia and promotes neovascularization. Circ Res 122:444–456. https://doi.org/10.1161/circresaha.117.312345
van der Kwast RV, Quax PH, Nossent AY (2019) An emerging role for isomiRs and the microRNA epitranscriptome in neovascularization. Cells 9:61. https://doi.org/10.3390/cells9010061
Virolainen SJ, VonHandorf A, Viel KC, Weirauch MT, Kottyan LC (2022) Gene–environment interactions and their impact on human health. Genes Immun. https://doi.org/10.1038/s41435-022-00192-6
Vujic A, Robinson EL, Ito M, Haider S, Ackers-Johnson M, See K, Methner C, Figg N, Brien P, Roderick HL, Skepper J (2015) Experimental heart failure modelled by the cardiomyocyte-specific loss of an epigenome modifier, DNMT3B. J Mol Cell Cardiol 82:174–183. https://doi.org/10.1016/j.yjmcc.2015.03.007
Wang P, Doxtader KA, Nam Y (2016) Structural basis for cooperative function of Mettl3 and Mettl14 methyltransferases. Mol cell 63:306–317. https://doi.org/10.1016/j.molcel.2016.05.041
Wang Q, Chen C, Ding Q, Zhao Y, Wang Z, Chen J, Jiang Z, Zhang Y, Xu G, Zhang J, Zhou J, Wang S (2020) METTL3-mediated m6A modification of HDGF mRNA promotes gastric cancer progression and has prognostic significance. Gut 69:1193–1205. https://doi.org/10.1136/gutjnl-2019-319639
Wang X, Feng J, Xue Y, Guan Z, Zhang D, Liu Z, Gong Z, Wang Q, Huang J, Tang C, Zou T, Yin P (2017) Structural basis of N6-adenosine methylation by the METTL3-METTL14 complex. Nature 542:260–260. https://doi.org/10.1038/nature21073
Wang X, Lu Z, Gomez A, Hon GC, Yue Y, Han D, He C (2014) N 6-methyladenosine-dependent regulation of messenger RNA stability. Nature 505:117–120. https://doi.org/10.1038/nature12730
Wang X, Zhao BS, Roundtree IA, Lu Z, Han D, Ma H, He C (2015) N6-methyladenosine modulates messenger RNA translation efficiency. Cell 161:1388–1399. https://doi.org/10.1016/j.cell.2015.05.014
Wang Y, Li Y, Yue M, Wang J, Kumar S, Wechsler-Reya RJ, Zhang Z, Ogawa Y, Kellis M, Duester G, Zhao JC (2018) N 6-methyladenosine RNA modification regulates embryonic neural stem cell self-renewal through histone modifications. Nat Neurosci 21:195–206. https://doi.org/10.1038/s41593-017-0057-1
Wang Y, Men M, Xie B, Shan J, Wang C, Liu J, Zheng H, Yang W, Xue S, Guo C (2016) Inhibition of PKR protects against H2O2-induced injury on neonatal cardiac myocytes by attenuating apoptosis and inflammation. Sci Rep 6:38753. https://doi.org/10.1038/srep38753
Wang Y, Zhu K, Dai R, Li R, Li M, Lv X, Yu Q (2022) Specific interleukin-1 inhibitors, specific interleukin-6 inhibitors, and GM-CSF blockades for COVID-19 (at the edge of sepsis): a systematic review. Front Pharmacol 12:804250. https://doi.org/10.3389/fphar.2021.804250
Weng SF, Reps J, Kai J, Garibaldi JM, Qureshi N (2017) Can machine-learning improve cardiovascular risk prediction using routine clinical data? PLoS ONE 12:e0174944. https://doi.org/10.1371/journal.pone.0174944
Wu C, Chen W, He J, Jin S, Liu Y, Yi Y, Gao Z, Yang J, Yang J, Cui J, Zhao W (2020) Interplay of m6A and H3K27 trimethylation restrains inflammation during bacterial infection. Sci Adv 6:eaba0647. https://doi.org/10.1126/sciadv.aba0647
Wu D, Hu D, Chen H, Shi G, Fetahu IS, Wu F, Rabidou K, Fang R, Tan L, Xu S, Liu H, Shi YG (2018) Glucose-regulated phosphorylation of TET2 by AMPK reveals a pathway linking diabetes to cancer. Nature 559:637–641. https://doi.org/10.1038/s41586-018-0350-5
Wu TT, Ma YW, Zhang X, Dong W, Gao S, Wang JZ, Zhang LF, Lu D (2020) Myocardial tissue-specific Dnmt1 knockout in rats protects against pathological injury induced by Adriamycin. Lab Invest 100:974–985. https://doi.org/10.1038/s41374-020-0402-y
Xia Y, Brewer A, Bell JT (2021) DNA methylation signatures of incident coronary heart disease: findings from epigenome-wide association studies. Clin Epigenet 13:1–16. https://doi.org/10.1186/s13148-021-01175-6
Xu W, Li J, He C, Wen J, Ma H, Rong B, Diao J, Wang L, Wang J, Wu F, Tan L (2021) METTL3 regulates heterochromatin in mouse embryonic stem cells. Nature 591:317–321. https://doi.org/10.1038/s41586-021-03210-1
Yuan J, Liu Y, Zhou L, Xue Y, Lu Z, Gan J (2021) YTHDC2-Mediated circYTHDC2 N6-methyladenosine modification promotes vascular smooth muscle cells dysfunction through inhibiting ten-eleven translocation 2. Front Cardiovasc Med 8:686293. https://doi.org/10.3389/fcvm.2021.686293
Zaccara S, Ries RJ, Jaffrey SR (2019) Reading, writing and erasing mRNA methylation. Nat Rev Mol Cell Biol 20:608–624. https://doi.org/10.1038/s41580-019-0168-5
Zampieri G, Vijayakumar S, Yaneske E, Angione C (2019) Machine and deep learning meet genome-scale metabolic modeling. PLoS Comput Biol 15:e1007084. https://doi.org/10.1371/journal.pcbi.1007084
Zarzour A, Kim HW, Weintraub NL (2019) Epigenetic regulation of vascular diseases. Arterioscler Thromb Vasc Biol 39:984–990. https://doi.org/10.1161/ATVBAHA.119.312193
Zhang CL, McKinsey TA, Chang S, Antos CL, Hill JA, Olson EN (2002) Class II histone deacetylases act as signal-responsive repressors of cardiac hypertrophy. Cell 110:479–488. https://doi.org/10.1016/s0092-8674(02)00861-9
Zhang J, Bai R, Li M, Ye H, Wu C, Wang C, Li S, Tan L, Mai D, Li G, Pan L (2019) Excessive miR-25-3p maturation via N 6-methyladenosine stimulated by cigarette smoke promotes pancreatic cancer progression. Nat Commun 10:1858. https://doi.org/10.1038/s41467-019-09712-x
Zhang QJ, Chen HZ, Wang L, Liu DP, Hill JA, Liu ZP (2011) The histone trimethyllysine demethylase JMJD2A promotes cardiac hypertrophy in response to hypertrophic stimuli in mice. J Clin Invest 121:447–2456. https://doi.org/10.1172/jci46277
Zhao L, You T, Lu Y, Lin S, Li F, Xu H (2021) Elevated EZH2 in ischemic heart disease epigenetically mediates suppression of NaV1. 5 expression. J Mol Cell Cardiol 153:95–103. https://doi.org/10.1016/j.yjmcc.2020.12.012
Zhao X, Sui Y, Ruan X, Wang X, He K, Dong W, Fang X (2022) A deep learning model for early risk prediction of heart failure with preserved ejection fraction by DNA methylation profiles combined with clinical features. Clin Epigenet 14:1–15. https://doi.org/10.1186/s13148-022-01232-8
Zhou X, Zhang S, Zhao Y, Wang W, Zhang H (2022) A multi-omics approach to identify molecular alterations in a mouse model of heart failure. Theranostics 12:1607. https://doi.org/10.7150/thno.68232
Zhu T, Roundtree IA, Wang P, Wang X, Wang L, Sun C, Tian Y, Li J, He C, Xu Y (2014) Crystal structure of the YTH domain of YTHDF2 reveals mechanism for recognition of N6-methyladenosine. Cell Res 24:1493–1496. https://doi.org/10.1038/cr.2014.152
Zurek M, Aavik E, Mallick R, Ylä-Herttuala S (2021) Epigenetic regulation of vascular smooth muscle cell phenotype switching in atherosclerotic artery remodeling: a mini-review. Front Genet 12:719456. https://doi.org/10.3389/fgene.2021.719456
Acknowledgements
This article is based upon work from EU-CardioRNA COST Action CA17129 (www.cardiorna.eu) and EU-AtheroNET COST Action CA21153 (https://www.cost.eu/actions/CA21153) funded by COST (European Cooperation in Science and Technology; www.cost.eu).
Funding
M.S. is funded the Ministry of Education, Science and Technological Development, Republic of Serbia through Grant Agreement with University of Belgrade-Faculty of Pharmacy No: 451–03-9/2021–14/200161, European Union (HORIZON-MSCA-2021-SE-01–01—MSCA Staff Exchanges 2021 CardioSCOPE 101086397, HORIZON-MSCA-2021-PF- MAACS 101064175). ELR is supported by an American Heart Association Postdoctoral fellowship (#829504) and a Colorado Clinical and Translational Sciences Institute (CCTSI) Pilot Grant award (#CO‐J‐22‐C.A. acknowledges a Network Development Award from The Alan Turing Institute, grant TNDC2-100022. CG is supported by Ricerca Corrente—Rete cardiologica IRCCS; Progetto di Rete Aging "Next Promising" Ricerca Corrente 2022; Regione Lombardia, "Progetto Immunhub". FM is supported by the Italian Ministry of Health (“Ricerca Corrente”, RF-2019–12368521, The Italian Cardiology Network IRCCS RCR-2022–23682288), by Telethon Foundation (#4462 GGP19035A), AFM-Telethon (# 23054), EU Horizon 2020 (COVIRNA, Grant #101016072) and cardioRNA COST action (CA17129). Y.D. is funded by the EU Horizon 2020 project COVIRNA (Grant # 101016072), the National Research Fund (grants # C14/BM/8225223, C17/BM/11613033, and COVID-19/2020–1/14719577/miRCOVID), CardioRNA COST action (CA17129), the Ministry of Higher Education and Research, and the Heart Foundation—Daniel Wagner of Luxembourg.
Author information
Authors and Affiliations
Consortia
Corresponding author
Ethics declarations
Conflict of interest
Y.D. owns patents related to diagnostic and therapeutic applications of RNAs.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Sopic, M., Robinson, E.L., Emanueli, C. et al. Integration of epigenetic regulatory mechanisms in heart failure. Basic Res Cardiol 118, 16 (2023). https://doi.org/10.1007/s00395-023-00986-3
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1007/s00395-023-00986-3