
Abstract
The aim of the study was to investigate whether pre-conditioning with CpG-oligodeoxynucleotides (CpG-ODN) may change cardiac ischemia/reperfusion (I/R)-dependent inflammation and modulates infarct size and cardiac performance. WT and TLR9-deficient mice were pre-treated with 1668-, 1612- and H154-thioate or D-Gal as control. Priming with 1668-thioate significantly induced inflammatory mediators in the serum and a concomitant increase of immune cells in the blood and spleen of WT mice. Furthermore, it induced myocardial pattern recognition receptors and pro-inflammatory cytokines peaking 2 h after priming and a continuous increase of IL-10. 16 h after pre-conditioning, myocardial ischemia was induced for 1 h. Infarct size determined after 24 h of I/R was reduced by 75 % due to pre-conditioning with 1668-thioate but not in the other groups. During reperfusion, cytokine expression in 1668-thioate primed mice increased further with IL-10 exceeding the other mediators by far. These changes were observed neither in animals pre-treated with 1612- or H154-thioate nor in TLR9-deficient mice. The 1668-thioate-dependent increase of IL-10 was further supported by results of a micro-array analysis 3 h after begin of reperfusion. Block of IL-10 signaling increased I/R size and prevented influence of priming. In the group pre-treated with 1668-thioate, cardiac function was preserved 24 h, 14 days and 28 days after I/R, whereas animals without pre-conditioning exhibited impaired heart function 24 h and 14 days after I/R. The excessive 1668-thioate-dependent IL-10 up-regulation during pre-conditioning and after I/R seems to be the key factor for reducing infarct size and improving cardiac function. This is in agreement with the finding that IL-10 block prevents cardioprotection by pre-conditioning.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Myocardial infarction (MI) is the result of an acute occlusion of a coronary artery due to plaque rupture and subsequent thrombosis [27]. The prognosis of patients suffering from MI is mainly determined by the infarct size (IS) [14], which in turn depends on the inflammatory response of the myocardium. It has been reported that preceding angina pectoris (AP) reduces the inflammatory response and thus attenuates subsequent ischemia/reperfusion (I/R) injury. This phenomenon termed ischemic pre-conditioning (IPC) has been confirmed in numerous animal models (for review see [12, 19]). Pre-conditioning (PC) can also be achieved by antecedent application of physical, e.g. hyper- or hypothermia or chemical stimuli, e.g. anesthetics and virulence factors [7, 21, 35]. PC of the heart has been reported to reduce IS to improve left ventricular function and to protect against lethal arrhythmia [17]. The actual knowledge of cardioprotection has been summarized in [3, 18].
The inflammatory response induced by I/R is mainly mediated by the innate immune system [11]. As part of innate immunity, pattern recognition receptors (PRRs), in particular Toll-like receptors (TLRs), have been linked to myocardial I/R [11]. TLRs are primarily expressed in immune cells, such as macrophages and dendritic cells. These immune cells, but also other cell types such as myocardial cells, are responsible for TLR signaling in the heart [25]. Especially, TLR2- and TLR4 deficiency as well as pre-conditioning with their respective ligands Pam3CSK4 and lipopolysacharide (LPS) has been shown to reduce myocardial infarct size [29, 39]. TLR stimulation as PC initially induces a transient up-regulation of inflammatory mediators, which in turn may modulate the subsequent inflammatory response and desensitize the heart to I/R [11]. On the other hand, I/R by itself is supposed to release endogenous ligands that bind as damage associated molecular patterns (DAMPs) to TLR2 and TLR4 [8, 47]. Finally, TLR deficiency as well as PC via TLR2, -4 and -9 stimulation converges in a diminished inflammatory response leading to reduced IS [28, 29, 45]. With respect to potential clinical application, TLR2 and TLR4 ligands, e.g. LPS, seem rather unsuitable for the use in humans due to serious side effects. However, synthetic TLR9 ligands such as CpG-oligodeoxynucleotides (CpG-ODNs) also induce a strong innate immune response with less harmful side effects for the human organism [24]. Thus, CpG-ODNs are currently applied in clinical trials as immunostimulatory agents as well as vaccine adjuvants [4, 32]. The immunostimulatory potential of CpG-ODNs depends on the CG-motif as well as on the degree of methylation, e.g. 1668-thioate is a stimulatory ODN. In addition, suppressive ODNs such as H154-thioate have the potential to specifically inhibit the immune activation and do not induce an inflammatory response [10, 41]. It has already been shown that priming with CpG-ODNs is able to protect the heart against the harmful consequences of hypertension as well as ischemia [7, 28, 44]. Pathway analysis of the TLR9-dependent innate immune response revealed an up-regulation of interferon-γ and cellular growth signaling during the priming phase as well as an up-regulation of pro- and anti-inflammatory cytokines [44]. Interestingly, it has recently been shown that IL-10 is able to attenuate I/R injury in the murine heart [5]. This can be taken as a sign that the activation of anti-inflammatory pathways is able to interfere with I/R injury.
Therefore, we tested the hypothesis whether the systemic inflammation caused by CpG-ODN pre-conditioning may change the I/R-dependent cardiac inflammation and whether this in turn is correlated to variations of infarct size and cardiac performance. As changes in inflammation due to I/R have to be separated from surgery-induced trauma, all experiments were performed in a closed-chest model as best approach to the clinical situation.
Materials and methods
All mice were handled in accordance with the Guide for Use and Care of Laboratory Animals (NIH publication No. 85-23, revised 1996). Protocols were approved by the local authority (LANUV, Recklinghausen, Germany).
Surgical techniques, experimental groups and protocols
10 to 12-week-old male C57BL/6 wild-type (WT; Charles River Laboratories) and TLR9-deficient (TLR9-D) mice (kindly provided by Professor Shizuo Akira, Osaka University, Osaka, Japan; back-crossed to a C57BL/6 background) were included. To prevent an inflammatory reaction due to surgical trauma, a chronic closed-chest model of myocardial I/R with left parasternal incision was applied. Surgical procedures and assessment of IS are described and depicted in Kim et al. [22] and in the supplement. After 5–7 days of recovery, all mice were treated with an intraperitoneal (i.p.) dose of d-GalactosamineN (d-GalN, 0.2 mg/g, Roth, Karlsruhe, Germany) to slow down the hepatic degradation of CpG-ODNs (Fig. 1) [38]. In control experiments, d-GalN alone did not induce an inflammatory response (data not shown). Animals were randomized to intraperitoneally receive a stimulatory CpG-ODN (1668-thioate; 5′-TCC-ATG-ACG-TTC-CTG-ATG-CT; TibMolBiol, Berlin Germany; 0.25 nmol/g), a non-CG containing ODN (1612-thioate; 5′-GCT-AGA-TGT-TAG-CGT5; TibMolBiol; 0.25 nmol/g), a suppressive ODN (H154-thioate; 5′-CCT-CAA-GCT-TGA-GGG-G-3; TibMolBiol, Berlin Germany; 0.25 nmol/g) or d-GalN as control. In pre-experiments, the dosage of CpG-ODN 1668-thioate was tested (0.05, 0.25, 0.5 and 1.0 nmol/g). Mortality during I/R experiments was around 15 %. This was not influenced by priming with 0.05 and 0.25 nmol/g but the higher doses increased mortality. 16 h after ODN-priming myocardial infarction was induced for 1 h, followed by 24 h, 14 or 28 days of reperfusion (for time course see Fig. 1). 16 h were chosen because we knew from earlier experiments that pre-conditioning with LPS leads to a reduction of infarct size after this time span [39] and that priming with CpG-ODN 1668-thioate applied under the same conditions (interval and concentration) as used here was able to attenuate cardiac hypertrophy induced by transverse aortic constriction [44].

Time course of experimental interventions and assessments. Non-linear scale for better identification of various interventions
Two further groups (with and without ODN-priming) of mice were treated i.p. with an anti-mouse monoclonal IL-10R1 antibody (5 μg diluted in 200 μl PBS; Biolegend, San Diego, CA, USA) directly after ODN-priming and 30 min prior to ischemia to block the influence of this anti-inflammatory cytokine during preconditioning and I/R [5].
Troponin T-measurement
350 μl blood was collected from the right carotid artery. The blood samples were analyzed with a Cardiac Reader® (Roche, Grenznach, Germany).
Multiplex cytokine assay
Blood was collected 2, 4, 6 or 16 h after priming and 30 min or 3 h after reperfusion. Blood was centrifuged at 3,400g for 10 min and the supernatant (plasma) was stored at −20 °C. After thawing, samples were analyzed immediately at 37 °C. Levels of TNF-α, IL-1β, IL-6, IL-10, IL-12, and IFNγ (Mouse Cytokine multi-Plex for Luminex™ laser, BioSource Europe) were determined using the microsphere array technique (Luminex 100 system, Luminex Corporation, Austin, TX, USA) as previously described [25].
Flow cytometry analysis of immune cells in the spleen and blood
Single cell suspensions of the spleens were generated by digestion with 1 mg/ml collagenase IV (Sigma, Taufkirchen, Germany) and 50 U/ml DNaseI (Sigma) in PBS with 3 % FCS for 30 min at 37 °C under mild stirring. Blood was collected by cardiac puncture in the presence of 0.5 M EDTA. One volume of blood was mixed with 20 volumes of Red Cell Lysis Buffer (145 mM NH4Cl, 100 μM EDTA, 12 mM NaHCO3). Spleen and blood cells were incubated with clone 2.4G2 culture supernatant to block Fc receptors. The following labeled antibodies from Pharmingen (BD Biosciences, San Diego, CA, USA) or eBiosciences (San Diego, CA, USA) were used at 1:200 dilution for staining of 1 × 106 cells: Gr1 PerCP-Cy5.5 (RB6-8C5), CD11c FITC (HL-3), CD11b APC (M1/70) and F4/80 PE (BM8). Dead cells were excluded with Hoechst 33342 stain [42, 43]. Sorting was performed on a FACSDiva cell sorter (BD Biosciences).
RNA-extraction and Taqman® RT-qPCR
The mRNA expression levels of TLR2 and -9 and as well as TNF-α, IL-1β, IL-6, and IL-10 were determined using Taqman® Realtime quantitative PCR (RT-qPCR; Applied Biosystems, Darmstadt, Germany). Total RNA was isolated with a standard protocol (Qiagen, Hilden, Germany). First-strand cDNA was synthesized using the High Capacity cDNA transcription kit (Applied Biosystems, Darmstadt, Germany) with random hexameric primers as described in the manufacturer’s protocol. RT-qPCR was performed and analyzed with 1/10 diluted cDNA according to the manufacturer’s instructions on an ABI Prism 7900 Sequence Detection System and SDS 2.2 Software (Applied Biosystems). Target gene expression was normalized to an internal control (glyceraldehyde-3-phosphate dehydrogenase, GAPDH). Relative RT-qPCR was performed using TaqMan Gene expression Master Mix (part 4369016; Applied Biosystems) with the following primers: GAPDH (Mm99999915_g1), TLR2 (Mm01213945_g1), TLR9 (Mm00446193_m1), IL-1β (Mm99999061_g1), IL-6 (Mm01210732_g1), IL-10 (Mm00439616_m1) and TNF-α (Mm00443258_m1). All murine primers were measured using FAM TAMRA chemistry and the relative standard curve method. At the end of the RT-qPCR cycle, dissociation curve analysis was performed to ascertain the amplification of a single PCR product.
Gene-expression analysis
Microarray analysis was used to monitor gene expression in the heart 30 min and 3 h after begin of reperfusion.
Microarray hybridization
Cy3 labeled reference and Cy5 labeled sample cDNAs (10 μl each) were combined, denaturated by heating for 2 min at 98 °C and mixed with 36 μl of hybridization solution at 42 °C (Ambion, Austin, TX, USA). Murine microarrays (NimbleGen, Roche, Basel, Switzerland) were overlaid with this solution and hybridized for 18 h at 42 °C using an actively mixing MAUI hybridization system (BioMicro Systems, Salt Lake City, UT, USA). Post hybridization the arrays were washed in 1× SSC/0.05 % SDS and 0.1× SSC, centrifuged to remove remaining liquid with unbound cDNA, and dried. Intensity values were generated using an array scanner (NimbleGen). Data were up-loaded to the mAdb database (Microarray Database, a collaboration of CIT/BIMAS and NCI/CCR at the NIH; http://nciarray.nci.nih.gov/) and formatted via the export function for use with BRB ArrayTools (Biometric Research Branch, NCI, Frederick, MY, USA).
Analysis of gene expression
Data from three independent experiments/time points and three untreated controls were used for statistical analyses. Expression analyses were performed using BRB ArrayTools. Data were background corrected, flagged values were removed, spots in which both signals were <100 were filtered out, ratios were log base 2 transformed and lowess intensity-dependent normalization was used to adjust for differences in labeling intensities of the Cy3 and Cy5 dyes [46]. Analysis was restricted to genes present on >50 % of the arrays after filtering. The gene expression profile of all treatment groups was compared to that of the control groups. A p value cutoff of 0.0001 was used to identify genes, whose expressions were significantly up-regulated after CpG-ODN stimulation when compared to controls. Data were evaluated using Ingenuity Pathway Analysis (IPA, Ingenuity Systems Inc., Redwood City, CA, USA). IPA maps each gene within a global molecular network, pathways and functional groups developed from information contained in the Ingenuity Pathways Knowledge Base (see: https://analysis.ingenuity.com/pa/info/help/Ingenuity_Network_Algorithm_Whitepaper_FINAL(2) [6]).
Cardiac pressure–volume measurements
Measurements of cardiac function were performed with a 1.4 french pressure conductance catheter (SPR-839, Millar Instruments, Houston, TX, USA) 24 h, 14 and 28 days after reperfusion. Recordings were performed under reduced isoflurane anesthesia (1 vol%) and controlled body temperature. For detailed description, see [44].
Statistics
Data are presented as mean ± SEM. Statistical analysis was performed by Student’s unpaired t test used to compare means between groups. Groups larger than two were analyzed with one-way ANOVA with Newman–Keuls post hoc test using GraphPad PRISM5 (La Jolla, CA, USA). p < 0.05 was considered statistically significant. For microarray analysis, genes that were differentially expressed in the treatment groups were identified using a random-variance t test with a significance level of p < 0.0001.
Results
Pre-conditioning with CpG-ODNs
Inflammatory mediators in the serum
1668-thioate significantly increased protein content in the serum of all determined inflammatory mediators during the priming phase, whereas stimulation with H154- and 1612-thioate did not change protein content of any of the investigated mediators. 1668-thioate up-regulated protein content of TNF-α (Fig. 2a), IL-1β (Fig. 2b), IL-6 (Fig. 2c), IL-12 (Fig. 2f) transiently with TNF-α and IL-6 reaching a maximum level after 2 h, IL-12 after 4 h and IL-1β after 6 h. IL-10 rose continuously reaching a plateau at 6 h (Fig. 2d). IFNγ production at least in part depends on IL-12, which rises 6 h after priming reaching the level of significance at 16 h (Fig. 2e). Pro-inflammatory mediators (IL-1β, IL-6, IL-12) had nearly returned to baseline level just before ischemia/reperfusion with the exception of TNF-α and IFNγ. In contrast, the anti-inflammatory mediator IL-10 had reached a high level at the start of cardiac injury. To prove TLR9-dependency of 1668-thioate stimulation, inflammatory parameters were also recorded in the serum of TLR9-D mice. None of the investigated mediators could be induced in TLR9-D animals (Supplement Figure 1).

Cytokine (a–e TNF-α, IL-1β, IL-6, IL-10, IL-12) and IFNγ (f) protein in blood serum measured at 2, 4, 6 and 16 h after priming with 1668-thioate, H154-thioate and 1612-thioate. All investigated mediators were up-regulated during the priming phase by 1668-thioate stimulation, whereas the other CpG-ODNs did not affect protein levels in the serum (n = 5/group, *p < 0.05 *significant group)
Inflammatory cells in serum and spleen
To further characterize the systemic inflammation caused by pre-conditioning, inflammatory cells were analyzed by flow cytometry of serum and spleen 6 and 16 h after stimulation (Fig. 3a, b). The number of polymorphous nucleic cells (PMN) in the serum significantly rose fivefold within the first 6 h and stayed high until 16 h (Fig. 3a). Macrophages (MPs) in the serum were divided into three groups Gr1low, Gr1interm and Gr1high. Gr1low macrophages were not influenced by priming, while the number of Gr1interm and Gr1high macrophages increased significantly by 3- and 30-fold, respectively, within the first 6 h remaining high until 16 h (Fig. 3a). In the spleen, the number of macrophages rose significantly by a factor of 1.5 between 6 and 16 h after stimulation (Fig. 3b). Splenic conventional dendritic cells (cDCs) proved to be insensitive to stimulation with 1668-thioate. Red pulp macrophages (RPM) rose twofold according to the same time pattern as MPs; however, their general number was low. Splenic PMNs became up-regulated within the first 6 h after stimulation and stayed elevated until 16 h (Fig. 3b). Taken together, PMNs as well as MPs in serum and spleen were significantly elevated at the onset of ischemia due to priming with 1668-thioate.

Flow cytometry of blood (a) and spleen (b) cells 6 and 16 h after priming with 1668-thioate and after d-GalN application. a Representative dot plots show the distribution of blood polymorph nuclear cells (PMN) as well as Gr1low, Gr1intermediate and Gr1high macrophages (MP) and bar graphs depict the quantitative evaluation of cell types 6 and 16 h after priming. b Representative dot plots and part of the gating tree used for detection of splenic MP, dendritic cells (cDC), red pulp macrophages (RPM) and PMN. Quantitative evaluation of MP and RPM is demonstrated in bar graphs 6 and 16 h after 1668-thioate stimulation (n = 4/group; *p < 0.05)
PRRs and cytokines in the myocardium
In line with the analysis of the blood serum, cytokine mRNA expression was analyzed in cardiac tissue; in addition, the expression of TLR2 and TLR9 was monitored (Fig. 4a–f). 1668-thioate injection resulted in a significant increase of all parameters during the priming phase with TLR2 (Fig. 4a), TNF-α (Fig. 4c), IL-1β (Fig. 4d) and IL-6 (Fig. 4e) peaking already after 2 h. TLR9 (Fig. 4b) expression reached the level of significance not until 16 h after 1668-thioate application. Similar to blood serum, IL-10 (Fig. 4f) continuously rose during the priming phase reaching a 200-fold increase after 16 h. Neither 1612-thioate nor H154-thioate was able to increase the mRNA expression of any inflammatory mediator. None of the genes of interest was influenced by CpG-ODN stimulation in TLR9-D mice at any time point (Supplement Fig. 2). Taken together, TLR2, TLR9, TNF-α and IL-10 were still elevated above control at the onset of cardiac ischemia.

RT-qPCR of mRNA expression of PRRs (a, b) and cytokines (c–f) in the heart measured at 2, 4, 6 and 16 h after priming with 1668-thioate, H154-thioate and 1612-thioate. Stimulation with 1668-thioate differentially up-regulated both PRRs and all cytokines during the priming phase (n = 8/group, *p < 0.05; *significant group)
Ischemia–reperfusion-dependent changes
Infarct markers and inflammatory mediators in the serum
In clinical practice, troponin T concentration in the serum is taken as a marker of myocardial injury. Therefore, troponin T concentration in murine serum was measured 24 h after reperfusion (Fig. 5a, b). The average troponin T concentration in 1668-thioate pre-conditioned mice was about three times smaller than in control animals pointing towards a decreased myocardial IS. To investigate whether troponin T may be employed as a marker of infarct size in small mammals such as mice, the troponin T concentration was plotted against infarct size (Fig. 5b). The correlation analysis revealed a linear slope of 2.081 ± 1.170E−008 with a correlation-coefficient of r = 0.67.

Recordings of different markers in the serum after ischemia/reperfusion. a Troponin T was measured 24 h after reperfusion. Priming with 1668-thioate significantly reduced serum troponin T. b Plot of serum troponin T (values taken from A) versus infarct size. (a, b slope: 2.081 ± 1.170E−008; correlation-coefficient: r = 0,67; n = 11/10, *p < 0.05). c–h Cytokine (c–g TNF-α, IL-1β, IL-6, IL-10, IL-12) and interferon-γ (h) protein in blood serum measured at 30 min and 3 h after reperfusion (n = 8/group, *p < 0.05; two * indicate that two groups differ significantly from the other marked groups)
I/R injury caused a local inflammation in the myocardium, which may lead to a transient systemic inflammation. Therefore, inflammatory markers were monitored in the serum 30 min and 3 h after re-opening of the LAD (Fig. 5c–h). Interestingly, TNF-α (Fig. 5c), IL-1β (Fig. 5d), IL-6 (Fig. 5e), IL-10 (Fig. 5f), Il-12 (Fig. 5g), and IFNγ (Fig. 5h) were not significantly increased at both time points in the d-GalN group. However, all parameters except IL-6 were significantly elevated in the 1668-thioate primed mice. Surprisingly, TNF-α was also increased in the 1612-thioate pre-treated animals, whereas H154-thioate and I/R did not cause significant changes.
Infarct size, PRRs and cytokines in the left ventricle
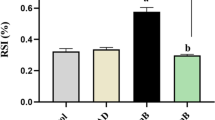
Infarct size in d-GalN or H-154-thioate and 1612-thioate pre-treated animals was around 35–40 % of AAR 24 h after reperfusion (Fig. 6a), whereas infarct size of 1668-thioate pre-treated animals stayed below 10 % of AAR, i.e. infarct size of 1668-thioate primed mice was less than a third of that of control animals (Fig. 6a). This is in agreement with the troponin T concentration in the serum (Fig. 5a). To demonstrate the TLR9-dependency of reduced infarct size, TLR9-D animals had also been pre-treated with 1668-thioate. TLR9-deficiency itself resulted in a significantly blunted infarct size of about 23 % of AAR, which could not be further reduced by 1668-thioate pre-treatment in these mice (Fig. 6a).

Recordings of infarct size (IS) and PRRs as well as cytokines from the heart after ischemia/reperfusion. a IS in percent of area at risk (AAR) evaluated by TTC staining after 24 h of reperfusion. Priming with 1668-thioate significantly reduced IS. Blocking with IL-10 antibody effectively blocked protection by 1668-thioate priming. IS in TLR9-D mice was significantly lower than in WT mice. Priming of WT animals with 1668-thioate reduced IS to a higher degree than TLR9-deficiency. Priming of TLR9-D mice with 1668-thioate was ineffective (n = 8/group, *p < 0.05 vs. WT 1668; # p < 0.05 vs. WT control). RT-qPCR of mRNA expression of PRRs (b, c) and cytokines (d–g) recorded at 30 min and 3 h of reperfusion. Priming with 1668-thioate resulted in a significant increase of TLR9, TNF-α, IL-6 and IL-10 mRNA expression at both time points (n = 8/group, *p < 0.05; *significant group)
Messenger RNA expression of markers of cardiac inflammation was monitored 30 min and 3 h after reperfusion (Fig. 6b–g). In accordance with findings in blood serum, I/R alone did not result in an increased expression of these markers in the myocardium. However, TLR9 (Fig. 6c), TNF-α (Fig. 6d), IL-6 (Fig. 6f) and IL-10 (Fig. 6g) were clearly and significantly up-regulated 30 min and 3 h after I/R in the 1668-thioate primed group. Interestingly, extremely high values were reached in case of IL-6 and IL-10. To control whether these high levels of IL-10 are involved in the beneficial influence of ODN-priming, we blocked the IL-10 receptor, IL-10R1, with a specific antibody in WT animals with and without priming. Blockade of the IL-10R1 led to a significant increase of infarct size. In the mice without priming, the infarct size amounted to 57 % of AAR after IL-10R1 blockade compared to 35 % with intact IL-10 signaling (Fig. 6a). In case of 1668-thioate priming, IL-10R1 inhibition resulted in an infarct size of 61 % of AAR compared to 8 % without antibody.
Micro-array analysis of the myocardium
For a more detailed analysis, micro-array investigations were performed on myocardial tissue 4 h after pre-conditioning as well as 30 min and 3 h after reperfusion. All treatment groups were normalized to untreated, healthy mice. During pre-conditioning phase, mice pre-treated with 1668-thioate were compared to sham mice whereas after reperfusion four groups (d-GalN alone, sham and 1668-, 1612-thioate pre-treatment) were investigated. 4 h after priming, the IL-10 pathway expression revealed a clear, albeit less prominent up-regulation than the IFN pathway (IL-10 pathway: p < 2.1E−3 vs. IFN signaling pathway: p < 1.2E−11). We also evaluated the expression of NF-κB pathway inhibitors TNFAIP3, NFKBIA, TRIM30 and TNIP1, most of which were up-regulated (TNFAIP3: 3.0; NFkBIA: 3.7; TRIM30: 5.04; TNIP1: 0.0).
During reperfusion, sham operation did not induce gene expression above a random distribution at the time of reperfusion. I/R itself (d-GalN) increased expression in a relatively small number of genes (30 min: 10 genes, 3 h: 76 genes, respectively), which is in accordance with the RT-qPCR results on inflammatory mediators. Pre-treatment with 1612-thioate only moderately increased gene expression in the heart after I/R (30 min: 50 genes; 3 h: 131 genes). On the other hand, 1668-thioate pre-treatment resulted in an up-regulation of 635 genes 30 min after I/R, increasing further towards 3 h with 697 up-regulated genes.
Ingenuity pathway analysis (IPA) was used to identify the pattern of regulatory interactions underlying 1668-thioate-dependent gene activation after I/R. 1668-thioate priming in conjunction with I/R up-regulated specific functional groups (Table 1). The analysis of functional groups of activated genes revealed that “cellular movement” (p < 4.1E−07) and “inflammatory response” (p < 4.68E−07) were markedly induced. Regarding up-regulated pro-inflammatory pathways, the “pattern recognition receptor pathway” (p < 3.4E−10), “interferon pathway” (p < 1.1E−8) and the “IL-6 pathway” (p < 2.4E−7) were most prominent (Table 2). Therefore, up-regulated genes of the interferon pathway are displayed in detail in Fig. 7b. Among the elevated decisive genes of the interferon pathway is the transcription factor STAT1, which in turn induces the STAT1-dependent protein IRF1 thereby inhibiting NFκBp65. At the same time, the anti-inflammatory IL-10 pathway (Fig. 7a) was significantly up-regulated in the microarray analysis (p < E−8, Table 2). This was in concordance with RT-qPCR analysis that revealed a massive up-regulation in IL-10 mRNA (Fig. 6g).

Results of ingenuity pathway analysis in the heart 1668-thioate primed WT mice 3 h after reperfusion. a IL-10-dependent suppressive target genes were significantly elevated (p < E−4; gray color). b Significant mRNA up-regulation of constituents involved in the IFNγ pathway (p < E−4; red color). A minimum of three animals per group were independently analyzed (stringency cut-off: p < 0.0001 above controls, up-regulated mRNAs are depicted in gray or red color)
Analysis of cardiac function
Functional parameters characterizing the cardiovascular system were measured 24 h, 14 days and 28 days after I/R with a pressure–volume catheter (Fig. 8a–h). An obvious pattern of response was that all recorded parameters [heart rate (HR), stroke volume (SV), cardiac output (CO), ejection fraction (EF), stroke work (SW), end-systolic volume (ESV), velocity of pressure increase (dP/dt max) and of pressure decrease (dP/dt min)] were impaired 24 h after I/R in the animals without pre-conditioning. Priming with 1668-thioate prevented at least partially this deterioration of cardiac function. This general pattern was preserved at least until 14 days. 28 days after I/R, the cardiac function of animals without pre-conditioning had largely recovered to the level of the 1668-thioate primed mice.

Hemodynamic parameters, a heart rate (HR), b stroke volume (SV), c cardiac output (CO), d ejection fraction (EF), e stroke work (SW), f end-systolic volume (ESV), g velocity of pressure increase (dP/dt max), h velocity of pressure decrease (dP/dt min) after 24 h, 14 days and 28 days of reperfusion (n = 10, *p < 0.05). After 24 h of reperfusion, I/R leads to a significant impairment of all measured parameters except HR, which was prevented by priming with 1668-thioate. After 14 days, the effect of 1668-thioate priming was still maintained
Discussion
The objective of this study was to characterize local and systemic inflammatory responses to CpG-ODN pre-conditioning before and after cardiac I/R injury and thus to elucidate how the CpG-ODN 1668-thioate can protect the heart from I/R injury and preserve cardiac function. The most important signaling pathways were visualized via micro-array technique of regulated genes. Finally, the functional consequences of priming with 1668-thioate were monitored by measuring hemodynamic characteristics in vivo.
During pre-conditioning, pro-inflammatory mediators such as TNF-α, IL-1β and IL-6 increased in a time-dependent manner in the blood and in the heart. For the explanation of the beneficial effect of pre-conditioning, the level of pro-inflammatory mediators directly before the onset of ischemia seems to be most relevant. Interestingly, TNF-α was the only pro-inflammatory cytokine, which was still significantly elevated 16 h after priming. IFNγ was extremely high in the blood just before the ischemic insult. However, an early and prominent up-regulation of IFNγ can induce regulatory proteins like interferon regulatory factors (IRF). In this context, Stevens et al. demonstrated that protection from I/R brain injury after priming with CpG-ODNs requires IFR3 and IRF7 [40]. In our hands, IFNγ reached its maximum 16 h after priming, thereby following the preceding up-regulation of IL-12. This observation can be explained as IFNγ is produced in leukocytes like NK cells and T-cells on stimulation with IL-12 and IL-18 [20, 36]. In the blood and the heart, the anti-inflammatory mediator IL-10 was at its highest level 6 and 16 h after priming with 1668-thioate, i.e. at the onset of cardiac ischemia, anti-inflammatory mediators were well above control levels. Neither priming with 1612-thioate nor with H154-thioate induced any significant change in inflammatory mediators during the priming phase underlining the lack of inflammatory potency of these ODNs [23]. Priming-induced increases of cytokines were absent in the blood and hearts of TLR9-D mice proving their dependency on TLR9 signaling. Consequently, infarct size in TLR9-D animals was insensitive to priming with 1668-thioate.
In a study with CpG-ODN 1826 pre-conditioning, Cao et al. [7] interrupted the TLR9-signaling further downstream during priming, i.e. blocking PI3K/Akt and thus, also prevented the pre-conditioning influence of CpG-ODNs. This branch of the TLR9-signaling pathway seems to be common with the TLR2 signaling as the same inhibition strategy also prevented pre-conditioning via TLR2 [15].
Micro-array analysis during the priming phase supported the findings of RT-qPCR. For better comparison with earlier results of Mathur et al. [28], we also focused on NF-κB pathway inhibitors. Re-calculation of these data to fold-change revealed a somewhat higher up-regulation of the gene expression of TNFAIP3, NFKBIA, TRIM30 in our hands (Mathur vs. own data: TNFAIP3: 2.6 vs. 3.0; NFkBIA: 2.2 vs. 3.7; TRIM30: 3.8 vs. 5.04). Concurrently those investigations found that TNIP1 was not elevated. Hence, switching on NF-κB pathway inhibitors during pre-conditioning with CpG-ODNs seems to be a relatively stable phenomenon, which contributes to cardiac protection.
However, in our hands the most obvious effect of pre-conditioning was the up-regulation of IL-10 as demonstrated in the RT-qPCR. The micro-array analysis performed after pre-conditioning with 1668-thioate also detected a significant albeit less prominent up-regulation compared with the interferon pathway (IL-10 pathway: 2.1E−3 vs. IFN signaling pathway: 1.2E−11). However, the micro-array analysis may underscore the role of the IL-10 pathway during priming, as this analysis was performed 4 h after application of 1668-thioate, i.e. at a time point when IL-10 was still in the rising phase according to RT-qPCR.
16 h after priming with 1668-thioate, GR-1high/(F4/80)high monocytes were significantly elevated in the blood, whereas GR-1low/(F4/80)high reparative monocytes [13] had not been influenced by pre-conditioning. Possibly, the elevated inflammatory GR-1high/(F4/80)high monocytes were able to invade the infarct area faster than without pre-conditioning. In addition, a switch from GR-1high/(F4/80)high to reparative GR-1low/(F4/80)high may be promoted by the raised IL-10 concentration in the blood and in the heart [2].
24 h after I/R, significant differences in troponin T levels in the blood between control (d-GalN alone) and 1668-thioate primed animals were detected (Fig. 5a, b). This can be taken as an indication of differences in infarct size. Possibly, this difference in troponin T levels is present much earlier as Mathur et al. [28] found differences in troponin I already 6 h after reperfusion in a mouse model. The early appearance of differences in troponin prompted us to monitor the levels of inflammatory mediators in blood and heart 30 min and 3 h after I/R. Interestingly, I/R by itself did not enhance any systemic marker of inflammation in the blood at the investigated time points. However, animals primed with 1668-thioate exhibited increased levels of TNF-α, IL-1β, IL-10, IL-12, and IFNγ either at both or only at one time point after reperfusion (Fig. 5c–h). Pro-inflammatory cytokines were increased mainly at the early time point, whereas IL-10 seemed to become up-regulated delayed.
In the heart, mRNA expression of TNF-α, IL-1β, IL-6, and IL-10 did only partially reflect the situation in the blood. Again none of the cytokines was induced by I/R alone. TNF-α, IL-6 and IL-10 were increasingly expressed over time, whereas IL-1β did not reach the level of significance at any time point. Interestingly, 1668-thioate priming had raised IL-10 mRNA expression by a factor of 200 until the start of I/R. 30 min after reperfusion, IL-10 mRNA expression was already more than doubled compared to the start of I/R. At 3 h after reperfusion, the IL-10 mRNA had again increased two times, i.e. IL-10 was 1,000× higher than in untreated hearts at this time. Taken together, there were signs of an increased inflammation after I/R in the blood and the heart of mice pre-treated with 1668-thioate. IL-6, which has been reported to exhibit pro- as well as anti-inflammatory properties [34], was expressed relatively high in this group. However, the expression of the anti-inflammatory cytokine IL-10 in the heart after priming with 1668-thioate and I/R outbalanced all other investigated mediators by far. It should be noted that the source of the pro- as well as anti-inflammatory cytokines was not directly investigated here. Thus, cytokines may derive from either immune cells or sedentary cells of the heart.
Micro-array analysis performed in this study (Fig. 7, Tables 1, 2) revealed that I/R itself increased expression in a relatively small number of genes further supporting the results from the RT-qPCR analysis. In contrast, 1668-thioate pre-treatment in conjunction with cardiac I/R resulted in significant up-regulation of 635 genes 30 min after reperfusion and persisted towards 3 h. The massive, CpG-ODN priming-dependent up-regulation of gene expression after I/R can be taken as a sign of increased inflammation. However, the extremely high expression of IL-10 mRNA demonstrated by RT-qPCR is further supported by the pathway analysis as different downstream components of IL-10 signaling had been up-regulated consequently (Fig. 7a). Furthermore, some of the initially pro-inflammatory pathways finally lead to the induction of suppressive networks, which terminate the inflammatory processes, e.g. IFNγ up-regulation induces IRF as mentioned above. Interestingly, IL-10 has recently been shown to be the central mediator of remote ischemic pre-conditioning of the heart [5]. As remote pre-conditioning up-regulated IL-10 in the ischemic organ, this IL-10 was able to protect the heart against I/R injury. This finding was further confirmed by the fact that IL-10 deficient mice could not profit from remote ischemic pre-conditioning. In the present study, we found that priming with CpG-ODN massively up-regulated IL-10 in the blood as well as in the heart. This was seen during the priming phase and even more extensively after I/R. As Cai et al. demonstrated that application of IL-10 prior to I/R induced phosphorylation of Akt, the massive increase of IL-10 observed in our hands may be the clue to the Akt-phosphorylation, which was shown by Cao et al. [7] to be a reason for smaller infarct size after CpG pre-conditioning. Interestingly, IL-10 up-regulation seems also to attenuate cardiac remodeling after I/R injury, which may improve cardiac long-term performance [33].
Pre-treatment with 1668-thioate in our study led to a significant reduction of IS of more than 75 % (Fig. 6a). The amount of IS reduction seems to be pronounced, but Ha et al. also obtained a distinct reduction of infarct size (65 %) due to LPS priming using a comparable I/R protocol [16]. Recently, Cao et al. [7] reported a smaller reduction of infarct size by about 35 % in an open-chest model after CpG-ODN priming. The higher degree of cardiac protection obtained in our hands may be attributed to differences in the applied CpG-ODNs (1668-thioate vs. 1826), the duration of priming and/or reperfusion (16 h/24 h vs. 1 h/4 h) as well as to our superior surgery model (closed-chest vs. open-chest model) [22, 26]. In a further study on myocardial I/R, where the effect of pre-conditioning with CpG-ODN 2395 was estimated by troponin I measurement [28], pre-conditioning induced lower troponin I levels in the blood indicating reduction of infarct size. Taken together, three study groups reported a cardio-protective influence of pre-conditioning with CpG-ODNs using different ODNs and diverse time-frames. Thus, the ODN-dependent cardio-protection seems to be a stable mechanism. Here, we show that this effect was TLR9-dependent as 1668-thioate priming did not affect infarct volume in TLR9-D animals. However, the TLR9-deficiency by itself reduced infarct size by about 35 % (Fig. 6a). This phenomenon reported here for the first time needs further studies to be explained. To confirm the above-mentioned hypothesis that IL-10 up-regulation is an essential step for the beneficial effect of ODN-priming we interrupted the IL-10-dependent signaling pathway by block of IL-10R1 with an appropriate antibody. This intervention caused a significant increase of infarct size, which may be attributed to block of physiological amounts of IL-10. In the case of IL-10R1 inhibition, the effect of previous ODN-priming on infarct size was completely prevented. Taken together, IL-10 seems to be the essential regulator, which transmits cardioprotection following ODN-priming. All cells in the heart, i.e. immune cells as well as sedentary cells like cardiomyocytes and cardiofibroblasts, may be influenced by IL-10. However, the cellular target of IL-10 was not elucidated in the present study but shall be pursued in further investigations.
After myocardial I/R, a decline in cardiac functional parameters is expected [48]. Mice suffering from I/R exhibited smaller SV, CO, EF, SW, dP/dt max and dP/dt min at 24 h and 14 days of reperfusion as well as an increased ESV at 24 h. This shows that the ischemic event elicited in our experiments is sufficient to cause the anticipated pathology. Interestingly, all mentioned parameters were superior to the I/R group if the animals had been primed with 1668-thioate. This beneficial influence of CpG-ODN pre-conditioning lasted until 14 days after I/R. Most of the measured characteristics remained nearly identical to sham animals in 1668-thioate pre-treated mice although they had suffered from 1 h of ischemia. Interestingly, other groups using different ODNs and different times for pre-conditioning found comparable improvements of cardiac function after I/R by application of ultrasound techniques [7, 28]. Only the present investigation demonstrates that during remodeling, the resting performance of the hearts in the I/R group recovered reaching the level of the 1668-thioate I/R group 28 days after reperfusion.
Taken together, three different CpG-ODNs (1668-thioate, 1826 [7] and 2395 [28]) applied in a different time-frame for priming (16 h, 1 h [7], 24 h [28]) and in different studies all led to successful pre-conditioning. Thus, it may be speculated that there is a common mechanism, by which CpG-ODNs induce effective organ protection. As priming with CpG-ODNs rapidly leads to a long-lasting systemic inflammation, the time-window for pharmacological pre-conditioning seems to be wide. The increase of IL-10 reported here for the first time may be regarded as the common mechanism for pre-conditioning in all three studies. In an investigation with ischemic pre-conditioning, IL-10 administration has been shown to protect the heart against I/R injury via PI3K/Akt signaling [5]. CpG-ODN-dependent cardioprotection could also be prevented by disruption or block of the PI3K/Akt signaling pathway [7]. Thus, the remaining link to be elucidated was whether the CpG-ODN-dependent activation of PI3K/Akt signaling is dependent on IL-10 up-regulation. The present study shows for the first time that priming with a synthetic CpG-ODN massively increases IL-10 expression in the blood and in the heart before and after I/R. In a reverse approach, IL-10 signaling was inhibited and consequently, infarct size rose and ODN-priming remained ineffective. Therefore, the mechanism, by which CpG-ODN is able to protect the heart, seems to be the induction of IL-10 expression and a consecutive activation of the PI3K/Akt pathway.
A transfer of these new findings from mice to humans may be hampered by differences in inflammatory responses between both species [37]. However, a correlation between the anti-inflammatory cytokine IL-10 and acute myocardial infarction in humans has been shown [9]. Furthermore, pre-conditioning as well as exercise seems to induce cardioprotective factors in humans, which are able to overcome the inter-species differences as they were able to decrease infarct size and cardiac function in isolated rat hearts [31]. Although pre-conditioning is not suited to reduce I/R injury in case of a sudden myocardial infarction, it may be applied prior to elective cardiac surgery. Here, application of remote ischemic pre-conditioning (RIPC) to humans before cardiac surgery has induced indications of cardioprotective effects [1]. Furthermore, RIPC is actually being evaluated in a multi-center clinical trial [30]. The cardioprotective influence of IL-10 demonstrated in this and other studies may also be transferred to the clinical setting by direct application of IL-10 prior to cardiac surgery.
References
Albrecht M, Zitta K, Bein B, Wennemuth G, Broch O, Renner J, Schuett T, Lauer F, Maahs D, Hummitzsch L, Cremer J, Zacharowski K, Meybohm P (2013) Remote ischemic preconditioning regulates HIF-1α levels, apoptosis and inflammation in heart tissue of cardiosurgical patients: a pilot experimental study. Basic Res Cardiol 108:314. doi:10.1007/s00395-012-0314-0
Arnold L, Henry A, Poron F, Baba-Amer Y, van Rooijen N, Plonquet A, Gherardi RK, Chazaud B (2007) Inflammatory monocytes recruited after skeletal muscle injury switch into antiinflammatory macrophages to support myogenesis. J Exp Med 204:1057–1069. doi:10.1084/jem.20070075
Bell R, Beeuwkes R, Botker HE, Davidson S, Downey J, Garcia-Dorado D, Hausenloy DJ, Heusch G, Ibanez B, Kitakaze M, Lecour S, Mentzer R, Miura T, Opie L, Ovize M, Ruiz-Meana M, Schulz R, Shannon R, Walker M, Vinten-Johansen J, Yellon D (2012) Trials, tribulations and speculation! Report from the 7th Biennial Hatter Cardiovascular Institute Workshop. Basic Res Cardiol 107:300. doi:10.1007/s00395-012-0300-6
Bode C, Zhao G, Steinhagen F, Kinjo T, Klinman DM (2011) CpG DNA as a vaccine adjuvant. Expert Rev Vaccines 10:499–511. doi:10.1586/erv.10.174
Cai ZP, Parajuli N, Zheng X, Becker L (2012) Remote ischemic preconditioning confers late protection against myocardial ischemia-reperfusion injury in mice by upregulating interleukin-10. Basic Res Cardiol 107:277. doi:10.1007/s00395-012-0277-1
Calvano SE, Xiao W, Richards DR, Felciano RM, Baker HV, Cho RJ, Chen RO, Brownstein BH, Cobb JP, Tschoeke SK, Miller-Graziano C, Moldawer LL, Mindrinos MN, Davis RW, Tompkins RG, Lowry SF (2005) A network-based analysis of systemic inflammation in humans. Nature 437:1032–1037. doi:10.1038/nature03985
Cao Z, Ren D, Ha T, Liu L, Wang X, Kalbfleisch J, Gao X, Kao R, Williams D, Li C (2012) CpG-ODN, the TLR9 agonist, attenuates myocardial ischemia/reperfusion injury: involving activation of PI3 K/Akt signaling. Biochim Biophys Acta 1832:96–104. doi:10.1016/j.bbadis.2012.08.008
Chen CJ, Kono H, Golenbock D, Reed G, Akira S, Rock KL (2007) Identification of a key pathway required for the sterile inflammatory response triggered by dying cells. Nat Med 13:851–856. doi:10.1038/nm1603
Dominguez-Rodriguez A, Abreu-Gonzalez P, de la Rosa A, Vargas M, Ferrer J, Garcia M (2005) Role of endogenous interleukin-10 production and lipid peroxidation in patients with acute myocardial infarction treated with primary percutaneous transluminal coronary angioplasty, interleukin-10 and primary angioplasty. Intern J Cardiol 99:77–81. doi:10.1016/j.ijcard.2003.12.027
Ehrentraut SF, Dörr A, Ehrentraut H, Lohner R, Lee SH, Hoeft A, Baumgarten G, Knuefermann P, Boehm O, Meyer R (2012) Vascular dysfunction following polymicrobial sepsis: role of pattern recognition receptors. PLoS ONE 7:e44531. doi:10.1371/journal.pone.0044531
Eltzschig HK, Eckle T (2011) Ischemia and reperfusion—from mechanism to translation. Nat Med 17:1391–1401. doi:10.1038/nm.2507
Frangogiannis NG (2012) Regulation of the inflammatory response in cardiac repair. Circ Res 110:159–173. doi:10.1161/CIRCRESAHA.111.243162
Geissmann F, Manz MG, Jung S, Sieweke MH, Merad M, Ley K (2010) Development of monocytes, macrophages, and dendritic cells. Science 327:656–661. doi:10.1126/science.1178331
Gibbons RJ, Valeti US, Araoz PA, Jaffe AS (2004) The quantification of infarct size. J Am Coll Cardiol 44:1533–1542. doi:10.1016/j.jacc.2004.06.071
Ha T, Hu Y, Liu L, Lu C, McMullen JR, Kelley J, Kao RL, Williams DL, Gao X, Li C (2010) TLR2 ligands induce cardioprotection against ischaemia/reperfusion injury through a PI3K/Akt-dependent mechanism. Cardiovasc Res 87:694–703. doi:10.1093/cvr/cvq116
Ha T, Hua F, Liu X, Ma J, McMullen JR, Shioi T, Izumo S, Kelley J, Gao X, Browder W, Williams DL, Kao RL, Li C (2008) Lipopolysaccharide-induced myocardial protection against ischaemia/reperfusion injury is mediated through a PI3 K/Akt-dependent mechanism. Cardiovasc Res 78:546–553. doi:10.1093/cvr/cvn037
Hausenloy DJ, Yellon DM (2011) The therapeutic potential of ischemic conditioning: an update. Nat Rev Cardiol 8:619–629. doi:10.1038/nrcardio.2011.85
Heusch G (2013) Cardioprotection: chances and challenges of its translation to the clinic. Lancet 381:166–175. doi:10.1016/S0140-6736(12)60916-7
Heusch G, Kleinbongard P, Skyschally A, Levkau B, Schulz R, Erbel R (2012) The coronary circulation in cardioprotection: more than just one confounder. Cardiovasc Res 94:237–245. doi:10.1093/cvr/cvr271
Kannan Y, Yu J, Raices RM, Seshadri S, Wei M, Caligiuri MA, Wewers MD (2011) IκBζ augments IL-12- and IL-18-mediated IFNγ production in human NK cells. Blood 117:2855–2863. doi:10.1182/blood-2010-07-294702
Khaliulin I, Clarke SJ, Lin H, Parker J, Suleiman MS, Halestrap AP (2007) Temperature preconditioning of isolated rat hearts—a potent cardioprotective mechanism involving a reduction in oxidative stress and inhibition of the mitochondrial permeability transition pore. J Physiol 581:1147–1161. doi:10.1113/jphysiol.2007.130369
Kim SC, Boehm O, Meyer R, Hoeft A, Knufermann P, Baumgarten G (2012) A murine closed-chest model of myocardial ischemia and reperfusion. J Vis Exp 65:e3896. doi:10.3791/3896
Klaschik S, Tross D, Shirota H, Klinman DM (2010) Short- and long-term changes in gene expression mediated by the activation of TLR9. Mol Immunol 47:1317–1324. doi:10.1016/j.molimm.2009.11.014
Klinman DM, Klaschik S, Sato T, Tross D (2009) CpG oligonucleotides as adjuvants for vaccines targeting infectious diseases. Adv Drug Deliv Rev 61:248–255. doi:10.1016/j.addr.2008.12.012
Knuefermann P, Schwederski M, Velten M, Krings P, Ehrentraut H, Rüdiger M, Boehm O, Fink K, Dreiner U, Grohe C, Hoeft A, Baumgarten G, Koch A, Zacharowski K, Meyer R (2008) Bacterial DNA induces myocardial inflammation and reduces cardiomyocyte contractility: role of toll-like receptor 9. Cardiovasc Res 78:26–35. doi:10.1093/cvr/cvn011
Lips DJ, van der Nagel T, Steendijk P, Palmen M, Janssen BJ, van Dantzig JM, de Windt LJ, Doevendans PA (2004) Left ventricular pressure–volume measurements in mice: comparison of closed-chest versus open-chest approach. Basic Res Cardiol 99:351–359. doi:10.1007/s00395-004-0476-5
Matar F, Mroue J (2012) The management of thrombotic lesions in the cardiac catheterization laboratory. J Cardiovasc Transl Res 5:52–61. doi:10.1007/s12265-011-9327-6
Mathur S, Walley KR, Boyd JH (2011) The Toll-like receptor 9 ligand CPG-C attenuates acute inflammatory cardiac dysfunction. Shock 36:478–483. doi:10.1097/SHK.0b013e31822d6442
Mersmann J, Berkels R, Zacharowski P, Tran N, Koch A, Iekushi K, Dimmeler S, Granja TF, Boehm O, Claycomb WC, Zacharowski K (2010) Preconditioning by toll-like receptor 2 agonist Pam3CSK4 reduces CXCL1-dependent leukocyte recruitment in murine myocardial ischemia/reperfusion injury. Crit Care Med 38:903–909. doi:10.1097/CCM.0b013e3181ce50e6
Meybohm P, Zacharowski K, Cremer J, Roesner J, Kletzin F, Schaelte G, Felzen M, Strouhal U, Reyher C, Heringlake M, Schon J, Brandes I, Bauer M, Knuefermann P, Wittmann M, Hachenberg T, Schilling T, Smul T, Maisch S, Sander M, Moormann T, Boening A, Weigand MA, Laufenberg R, Werner C, Winterhalter M, Treschan T, Stehr SN, Reinhart K, Hasenclever D, Brosteanu O, Bein B, Group RIPH-SI (2012) Remote ischaemic preconditioning for heart surgery. The study design for a multi-center randomized double-blinded controlled clinical trial—the RIPHeart-Study. Eur Heart J 33:1423–1426. doi:10.1093/eurheartj/ehs131
Michelsen MM, Stottrup NB, Schmidt MR, Lofgren B, Jensen RV, Tropak M, St-Michel EJ, Redington AN, Botker HE (2012) Exercise-induced cardioprotection is mediated by a bloodborne, transferable factor. Basic Res Cardiol 107:260. doi:10.1007/s00395-012-0260-x
Murad YM, Clay TM (2009) CpG oligodeoxynucleotides as TLR9 agonists: therapeutic applications in cancer. BioDrugs 23:361–375. doi:10.2165/11316930-000000000-00000
Parajuli N, Yuan Y, Zheng X, Bedja D, Cai ZP (2012) Phosphatase PTEN is critically involved in post-myocardial infarction remodeling through the Akt/interleukin-10 signaling pathway. Basic Res Cardiol 107:248. doi:10.1007/s00395-012-0248-6
Pedersen BK, Febbraio MA (2008) Muscle as an endocrine organ: focus on muscle-derived interleukin-6. Physiol Rev 88:1379–1406. doi:10.1152/physrev.90100.2007
Riess ML, Stowe DF, Warltier DC (2004) Cardiac pharmacological preconditioning with volatile anesthetics: from bench to bedside? Am J Physiol 286:H1603–H1607. doi:10.1152/ajpheart.00963.2003
Schoenborn JR, Wilson CB (2007) Regulation of interferon-γ during innate and adaptive immune responses. Adv Immunol 96:41–101. doi:10.1016/S0065-2776(07)96002-2
Seok J, Warren HS, Cuenca AG, Mindrinos MN, Baker HV, Xu W, Richards DR, McDonald-Smith GP, Gao H, Hennessy L, Finnerty CC, Lopez CM, Honari S, Moore EE, Minei JP, Cuschieri J, Bankey PE, Johnson JL, Sperry J, Nathens AB, Billiar TR, West MA, Jeschke MG, Klein MB, Gamelli RL, Gibran NS, Brownstein BH, Miller-Graziano C, Calvano SE, Mason PH, Cobb JP, Rahme LG, Lowry SF, Maier RV, Moldawer LL, Herndon DN, Davis RW, Xiao W, Tompkins RG, Host Response Inflammation to Injury LSCRP (2013) Genomic responses in mouse models poorly mimic human inflammatory diseases. Proc Nat Act Sci USA 110:3507–3512. doi:10.1073/pnas.1222878110
Silverstein R (2004) D-galactosamine lethality model: scope and limitations. J Endotoxin Res 10:147–162. doi:10.1179/096805104225004879
Stapel H, Kim SC, Osterkamp S, Knuefermann P, Hoeft A, Meyer R, Grohe C, Baumgarten G (2006) Toll-like receptor 4 modulates myocardial ischaemia-reperfusion injury: role of matrix metalloproteinases. Eur J Heart Fail 8:665–672. doi:10.1016/j.ejheart.2006.03.005
Stevens SL, Leung PY, Vartanian KB, Gopalan B, Yang T, Simon RP, Stenzel-Poore MP (2011) Multiple preconditioning paradigms converge on interferon regulatory factor-dependent signaling to promote tolerance to ischemic brain injury. J Neurosci 31:8456–8463. doi:10.1523/JNEUROSCI.0821-11.2011
Stunz LL, Lenert P, Peckham D, Yi AK, Haxhinasto S, Chang M, Krieg AM, Ashman RF (2002) Inhibitory oligonucleotides specifically block effects of stimulatory CpG oligonucleotides in B cells. Eur J Immunol 32:1212–1222. doi:10.1002/1521-4141(200205)32:5<1212:AID-IMMU1212>3.0.CO;2-D
Tittel AP, Heuser C, Ohliger C, Knolle PA, Engel DR, Kurts C (2011) Kidney dendritic cells induce innate immunity against bacterial pyelonephritis. J Am Soc Nephrol 22:1435–1441. doi:10.1681/ASN.2010101072
Ueda Y, Kondo M, Kelsoe G (2005) Inflammation and the reciprocal production of granulocytes and lymphocytes in bone marrow. J Exp Med 201:1771–1780. doi:10.1084/jem.20041419
Velten M, Duerr GD, Pessies T, Schild J, Lohner R, Mersmann J, Dewald O, Zacharowski K, Klaschik S, Hilbert T, Hoeft A, Baumgarten G, Meyer R, Boehm O, Knuefermann P (2012) Priming with synthetic oligonucleotides attenuates pressure overload-induced inflammation and cardiac hypertrophy in mice. Cardiovasc Res 96:422–432. doi:10.1093/cvr/cvs280
Wang E, Feng Y, Zhang M, Zou L, Li Y, Buys ES, Huang P, Brouckaert P, Chao W (2011) Toll-like receptor 4 signaling confers cardiac protection against ischemic injury via inducible nitric oxide synthase- and soluble guanylate cyclase-dependent mechanisms. Anesthesiology 114:603–613. doi:10.1097/ALN.0b013e31820a4d5b
Yang YH, Dudoit S, Luu P, Lin DM, Peng V, Ngai J, Speed TP (2002) Normalization for cDNA microarray data: a robust composite method addressing single and multiple slide systematic variation. Nucleic Acids Res 30:e15. doi:10.1093/nar/30.4.e15
Yu M, Wang H, Ding A, Golenbock DT, Latz E, Czura CJ, Fenton MJ, Tracey KJ, Yang H (2006) HMGB1 signals through toll-like receptor (TLR) 4 and TLR2. Shock 26:174–179. doi:10.1097/01.shk.0000225404.51320.82
Zia A, Kern KB (2011) Management of postcardiac arrest myocardial dysfunction. Curr Opin Crit Care 17:241–246. doi:10.1097/MCC.0b013e3283447759
Acknowledgments
Financial support by the Else-Kröner-Fresenius Stiftung (Grant Nr. P46/08//A22/08), and BONFOR. We thank Susanne Schulz for excellent technical assistance and Dr. Catherine Widman for critically reading the manuscript.
Conflict of interest
The authors declare that they do not have any conflict of interest.
Author information
Authors and Affiliations
Corresponding author
Additional information
P. Markowski, O. Boehm, R. Meyer and P. Knuefermann contributed equally to this work.
Electronic supplementary material
Below is the link to the electronic supplementary material.
395_2013_376_MOESM2_ESM.docx
Supplement Figure 1 Cytokine (a-e: TNF-α, IL-1β, IL-6, IL-12) and interferon-γ (f) proteins in blood serum of WT and TLR9-D mice measured at 2, 4, 6 and 16 h after priming with 1668-thioate. All investigated mediators were up-regulated during the priming phase by 1668-thioate stimulation in WT but not in TLR9-D mice (n = 5/group, *: p < 0.05). Supplement Figure 2. RT-qPCR of mRNA expression of PRRs (a, b) and cytokines (c-f) in the heart of WT and TLR9-D mice measured at 2, 4, 6 and 16 h after priming with 1668-thioate. Stimulation with 1668-thioate differentially up-regulated TLR2 and -9 and the cytokines TNF-α, IL-1β, IL-6, and IL-10 during the priming phase in WT but not in TLR9-D mice (n = 8/group, *: p < 0.05) (DOCX 83 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution License which permits any use, distribution, and reproduction in any medium, provided the original author(s) and the source are credited.
About this article
Cite this article
Markowski, P., Boehm, O., Goelz, L. et al. Pre-conditioning with synthetic CpG-oligonucleotides attenuates myocardial ischemia/reperfusion injury via IL-10 up-regulation. Basic Res Cardiol 108, 376 (2013). https://doi.org/10.1007/s00395-013-0376-7
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1007/s00395-013-0376-7