
Abstract
Real-time polymerase chain reaction (PCR) is commonly used for a sensitive and specific quantification of messenger RNA (mRNA). The levels of mRNA are frequently compared between two or more experimental groups. However, such comparisons require normalization procedures, and reference genes are frequently used for this purpose. We discuss pitfalls in normalization and specifically in the choice of reference genes. Reference genes, which prove suitable for some experimental conditions, are not necessarily similarly appropriate for others. Therefore,a proper validation of the suitability of a given reference gene or sets thereof is required for each experimental setting. Several computer programmes are available to aid such validation.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Gene expression analysis at the messenger RNA (mRNA) level has become increasingly important in biological research. Originally, it was mainly used to determine whether differences protein expression could be explained at the transcriptional level. However, it meanwhile has much wider applications. This includes measurement of mRNA in situations where quantification of the protein is difficult or cumbersome, e.g. the measurement of atrial natriuretic peptide mRNA as a marker of the hypertrophic phenotype of cardiomyocytes, but important findings continue to require confirmation at the protein level. Most recently, mRNA expression analysis is being used to provide insight into complex regulatory networks and to identify genes relevant to new biological processes or implicated in diseases. Although differences in measured mRNA levels can result from differential mRNA degradation, they are usually interpreted to reflect differential transcription. In this issue of Basic Research in Cardiology Brattelid et al. [1] highlight the problem of normalization using reference genes when measuring the regulation of gene expression at the mRNA level.
Normalization is a widely used approach when comparing the levels of lipids, proteins or mRNA in two or more groups of samples. Actually, it is being used so widely and routinely that the underlying assumptions are often ignored which can lead to dubious results and conclusions. For example, after myocardial infarction the amount of fibroblasts and extracellular matrix proteins is increased relative to cardiomyocytes in the myocardium. Comparing the diseased with the normal tissue might lead to false results and wrong conclusions when normalizing for the amount of tissue or for total protein content because the differences in cell type and amount of extracellular matrix proteins are not taken into account. Therefore, it is always important to be aware of the assumptions underlying normalization and of the implications of such, often untested assumptions for data interpretation.
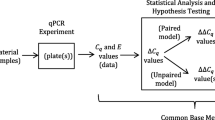
Apart from general considerations regarding the normalization of data in biology, special considerations apply to the normalization of quantitative real-time polymerase chain reaction (PCR) data [4]. While normalization for sample size, e.g. tissue volume or weight, can also be applied to real-time PCR data, this is only a first but on its own insufficient step as it can be difficult to ensure that different samples contain the same cellular material (see example above). Measuring total RNA or genomic DNA gives a good idea of the amount of cellular material. Normalization against genomic DNA is rarely used since it is difficult to coextract with RNA and it may vary in copy number per cell. Total RNA measurement is a good second step to control for experimental error, but it requires a good method of assessing quality and quantity. The measurement can be effected when RNA quality is suboptimal [2]. The drawback of normalization against total RNA is that it does not control for errors introduced at the reverse transcription step of PCR reactions. In addition, it primarily measures ribosomal RNA (rRNA) whereas real-time PCR aims to determine mRNA expression. Furthermore, normalization for total RNA assumes that the rRNA:mRNA ratio is the same in all groups, which might not always be the case [9]. Finally, rRNA is not present in purified mRNA and the high abundance of rRNA compared to mRNA makes it difficult to subtract the baseline value in real-time PCR analysis. Thus, markers of rRNA such as 18S or 28S rRNA might also be suboptimal as normalization factors in many settings.
Therefore, the most frequently used approach in real-time PCR is to normalize the expression level of a specific mRNA of interest against that of one or more reference genes [4]. This is based on the assumption that expression of the mRNA of interest is described relative to the overall transcriptional activity of a cell. Reference genes are often referred to as housekeeping genes assuming that those genes are expressed at a constant level in various tissues at all stages of development and are unaffected by the experimental treatment. The most commonly used reference genes are β-actin, glyceraldehyde-3-phosphate dehydrogenase (GAPDH), hypoxanthine phosphoribosyltransferase (HPRT), and β-2-microglobulin. Normalization to a reference gene is a simple method and frequently used because it can control many variables. An advantage of reference genes as compared to total or rRNA is that the reference gene is subject to the same conditions as the mRNA of interest. Both the reference gene and the gene of interest are measured using real-time PCR which simplifies the normalization, particularly if the number of amplification cycles during the PCR reaction is similar for the reference gene and the gene of interest. The reference gene might also be used as a control for different amounts of RNA used in the reverse transcription step, but this requires validation. Most classical reference genes (see above) have been introduced for normalization purposes when mRNA expression was assessed by methods such as Northern blotting and conventional endpoint reverse transcriptase PCR because of their high expression levels in all cells. For these techniques their use as positive control was acceptable, because only qualitative or semiquantitative changes were measured.
Real-time PCR provides high sensitivity and accurate mRNA quantification over a wide dynamic range of detection. This requires re-evaluation of the usefulness of previously used reference genes, but many studies continued to use the above mentioned classical reference genes for normalization without any validation of their suitability in real-time PCR. This potentially calls for trouble, as it has recently been found that the mRNA expression level of several reference genes is much less stable than one would hope. Several studies have reported that the expression levels of the classical reference genes can vary extensively and are thus unsuitable for the normalization. Moreover, several recent studies report that such variation may by due to active regulation of classical reference genes under certain experimental conditions (see: http://normalisation.gene-quantification.info).
For example, variation in supposedly stable reference genes was shown in studies examining their expression in serum-stimulated fibroblasts [8]. In that study the expression levels of β-actin and GAPDH were increased whereas that of β-2 microglobulin was unaffected by serum when normalizing for amount of mRNA. Comparing 13 reference genes in tumour samples from different tissues HPRT was identified to be the best reference gene for normalization whereas β-2 microglobulin was unsuitable [5]. Another study demonstrates the active regulation of HPRT, β-actin and two other reference genes (hydroxymethylbilane synthase and peptidylprolyle isomerase B) during lymphocyte activation [7].
If inappropriate reference genes are used for normalization, the experimental results obtained can differ greatly from those using a validated reference gene. An example is the study of mRNA levels of IL-4 in whole blood from healthy volunteers and patients with tuberculosis (TB) before and after 6 months anti-TB treatment, in which normalization was performed with both GAPDH and a reference gene specifically validated for that condition, HuPO [3]. The increase of IL-4 expression in TB patients when normalized to HuPO disappeared using GAPDH for normalization, whereas the non-significant decrease in IL-4 after anti-TB treatment normalized for HuPO turned into a significant increase when GAPDH was used for normalization. Consequently, both false positive and false negative results may be obtained when using an inappropriate reference gene. Importantly, a suitable reference gene in some circumstances is not necessarily similarly appropriate in others. Therefore, it is important to validate whether the chosen reference genes can be used for accurate gene expression analysis in a specific experimental condition.
Validation of a reference gene requires removal of any non-specific variation in expression.This can be done using a recently introduced programme called geNorm (freely available at http://medgen.ugent.be/~jvdesomp/genorm/) that mathematically identifies the most suitable reference gene for a given experimental condition [10]. Its calculations use the geometric mean of the expression of the candidate genes and by elimination of the least stable control genes the two most stable control genes are identified. Other statistical programmes (Best-Keeper, Norm Finder) have also been developed to determine the most appropriate reference gene for a given experimental condition. They use different algorithms to analyse the variation in the expression of reference genes, and that may result in different recommendations for the most suitable reference gene. Independent of the calculation method being used, pairs of reference genes appear to yield more robust data than any single reference genes in many cases. Thus, all three programmes identified the same combination of two reference genes as being most suitable for normalization [6].
In conclusion, real-time PCR is a powerful technique for the quantitative analysis of mRNA levels but reliable results can only be obtained when using appropriate normalization procedures. In this regard, reference genes appear superior to total RNA or rRNA for normalization purposes. However, the reference gene of choice may differ between experimental conditions and, therefore, validation of the reference gene being used is required for any given setting. Because of the inherent variation in the expression of reference genes the use of multiple reference genes rather than one reference gene is recommended to ensure reliable normalization of real-time PCR [10]. Several statistical programmes can help to determine the most appropriate reference gene or set of genes.
References
Brattelid T, Tveit K, Birkeland JAK, Sjaastad I, Qvigstad E, Krobert KA, Hussain RI, Skomedal T, Osnes J, Levy FO (2007) Expression of mRNA encoding G protein- coupled receptors involved in congestive heart failure: a quantitative RTPCR study and the question of normalisation. Basic Res Cardiol (DOI 10.1007/s00395-007-0648-1)
Bustin SA, Nolan T (2004) Pitfalls of quantitative real-time reverse-transcription polymerase chain reaction. J Biomol Tech 15:155–166
Dheda K, Huggett J, Chang JS, Kim LU, Bustin SA, Johnson MA, Rook GAW, Zumla A (2005) The implications of using an inappropriate reference gene for real-time reverse transcription PCR data normalization. Anal Biochem 344:141–143
Huggett J, Dheda K, Bustin SA, Zumla A (2005) Real-time RT-PCR normalisation; strategies and considerations. Genes Immun 6:279–284
de Kok JB, Roelofs RW, Giesendorf BA, Pennings JL, Waas ET, Feuth T, Swinkels DW, Span PN (2005) Normalization of gene expression measurements in tumour tissues: comparison of 13 endogenous control genes. Lab Invest 85:154–159
Robinson TL, Sutherland IA, Sutherland J (2007) Validation of candidate bovine reference genes for use with real-time PCR. Vet Immunol Immunopathol 115:160–165
Røge R, Thorsen J, Tørring C, Øzbay A, Møller BK, Carstens J (2006) Commonly used reference genes are actively regulated in in vitro stimulated lymphocytes. Scand J Immunol 65:202–209
Schmittgen TD, Zakrajsek BA (2000) Effect of experimental treatment on housekeeping gene expression: validation by real-time, quantitative RT-PCR. J Biochem Biophys Methods 46:69–81
Solanas M, Moral R, Escrich E (2001) Unsuitability of using ribosomal RNA as loading control for Northern blot analyses related to the imbalance between messenger and ribosomal RNA content in rat mammary tumors. Anal Biochem 288:99–102
Vandesompele J, De Preter K, Pattyn F, Poppe B,Van Roy N, De Paepe A, Speleman F (2002) Accurate normalization of real-time quantitative RT-PCR data by geometric averaging of multiple internal control genes. Genome Biol http://genomebiology.com/2002/3/7/ research/0034
Author information
Authors and Affiliations
Corresponding author
Additional information
This Invited Editorial refers to the Original Contribution “Expression of mRNA encoding G protein-coupled receptors involved in congestive heart failure” — “A quantitative RT-PCR study and the question of normalisation” by T. Brattelid et al. which can be found under http://dx.doi.org/10.1007/10.1007/s00395-007-0648-1
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License ( https://creativecommons.org/licenses/by-nc/2.0 ), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
Hendriks-Balk, M.C., Michel, M.C. & Alewijnse, .E. Pitfalls in the normalization of real-time polymerase chain reaction data. Basic Res Cardiol 102, 195–197 (2007). https://doi.org/10.1007/s00395-007-0649-0
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00395-007-0649-0