
Abstract
Amino sugars are increasingly used as indicators for the accumulation of microbial residues in soil and plant material. A reverse-phase high-performance liquid chromatography method was improved for the simultaneous determination of muramic acid, mannosamine, glucosamine and galactosamine in soil and plant hydrolysates via ortho-phthaldialdehyde (OPA) pre-column derivatisation and fluorescence detection. The retention time was reduced, and the separation of muramic acid and mannosamine was optimised by modifying the mobile phase. The effects of excitation wavelength, OPA reaction time, tetrahydrofuran concentration and pH value of the mobile phase on the amino sugar separation were tested. Quantification limits were in the range of 0.13 to 0.90 μg ml−1. No interferences exist from amino acids or other primary amines, occurring in soil and plant hydrolysates.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Amino sugars make a significant contribution of 5% to 12% to soil organic N (Stevenson 1982) and roughly 3% to soil organic C (Joergensen and Meyer 1990). Up to 26 amino sugars have been found in microorganisms (Sharon 1965), whereas four of them have been quantified in soil. These amino sugars are glucosamine, galactosamine, muramic acid and mannosamine (Amelung et al. 2008). It has repeatedly been shown that amino sugars are useful indicators for the accumulation of different types of microbial residues in soil (Amelung et al. 2002; Amelung 2003; Liang et al. 2006, 2007a, b). Fungal cell walls are the major source of glucosamine in soils (Appuhn and Joergensen 2006). Muramic acid occurs exclusively in bacterial cell walls, especially in the murein skeleton of Gram-positive species (Millar and Casida 1970; Kenne and Lindburg 1983). Also, bacteria contain glucosamine in their peptidoglycan cell wall, but only the glucosamine that occurs in excess to muramic acid may be attributed to fungal sources (Chantiny et al. 1997; Guggenberger et al. 1999; Amelung 2001). Appuhn and Joergensen (2006) determined average conversion factors of 9 to convert fungal glucosamine to fungal C and 45 to convert muramic acid to bacterial C.
Galactosamine contributes roughly one third to the total sum of amino sugars observed in soil and is also nearly exclusively of microbial origin (Engelking et al. 2007). Galactosamine contributed on average 4% to the total amino sugar concentration in cultured bacteria and 15% in cultured fungi (Engelking et al. 2007). However, the function of galactosamine within bacterial and fungal cells and consequently the processes behind galactosamine formation during decomposition processes are still unknown. However, galactosamine in soil is still attributed to be mainly of bacterial origin (Amelung et al. 2008), which has been sometimes supported by correlation analysis (Rottmann et al. 2010). Virtually nothing is known about the origin of mannosamine (Amelung 2001; Amelung et al. 2008), although Amelung et al. (1999) and Guggenberger et al. (1999) found significant amounts in different soils. Mannosamine has been found in fungal melanin (Coelho et al. 1997) and as component of sialic acids of Aspergillus fumigatus on their conidial surface (Wasylnka et al. 2001). Mannosamine, i.e. N-acetyl d-mannosamine containing sialic acids are present as protective capsular components of bacteria, invading mammals (Ferrero and Aparicio 2010). However, small amounts of mannosamine may be also present as common linkage units between peptidoglycan and other bacterial cell wall components such as glycerol teichoic acid (Yoneyama et al. 1982).
Several methods for the determination of amino sugars have been published. Highly specific gas chromatographic analyses require difficult off-line derivatisation steps, e.g. derivatisation of the hydrolysis products in volatile aldononitrile acetates (Zhang and Amelung 1996). High-performance liquid chromatography (HPLC) methods need either derivatisation steps (Diaz et al. 1996; Ekblad and Näsholm 1996; Appuhn et al. 2004) or special equipment for anion exchange chromatography combined with pulsed amperometric detection (Kaiser and Benner 2000; Benner and Kaiser 2003). This means that HPLC methods are generally less time-consuming as they employ no or automated on-line derivatisation.
Appuhn et al. (2004) were the first to describe a method to determine the four amino sugars simultaneously. However, this methodological approach was hampered by two drawbacks: (1) The method was reliably working only on the Agilent 1100 HPLC equipment and (2) mannosamine was insufficiently separated in most soil hydrolysates, which resulted in erroneous high values usually omitted in further publications (Appuhn and Joergensen 2006; Engelking et al. 2007). The objective of the present paper was improving the method of Appuhn et al. (2004) to give reliable results for all four amino sugars in soil and plant hydrolysates using different HPLC equipments.
Materials and methods
Soil and plant samples
Method optimisation was implemented using six different soil samples (0–10 cm) taken from four arable and two forest sites in Germany (Hessia and Lower Saxony) differing in physical, chemical, and microbial properties (Table 1) and five different plant litter samples (Table 2). Soil physical, chemical, and biological properties of the soil samples and total C and N of litter samples were determined as described by Probst et al. (2008).
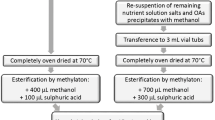
The amino sugar extraction was based on the method described by Appuhn et al. (2004) with minor modifications. Sieved (<2 mm) and air-dried soil (400 mg) or oven-dried (40°C) and steel ball-milled (Retsch, Haan, Germany) plant material (700 mg) was mixed with 10 ml of 6 M HCl. After 6 (soil) or 3 h (plant material) hydrolysis at 105°C, the samples were filtered over glass filters (Whatman GF/A). For the determination of the recovery rate, 0.3 ml of a 150 μmol l−1 standard solution were added to 0.3 ml of a quartz sand hydrolysate. A 0.3-ml aliquot was evaporated to dryness at 40–45°C to remove HCl, re-dissolved in water, evaporated a second time, and re-dissolved in 1 ml water. To test the effect of sample pH on the amino sugar amounts, another set of samples was prepared using 1 ml phosphate buffer solution pH 7 (containing 40.8 mmol l−1 Na2HPO4 × 2H2O and 25.9 mmol l−1 KH2PO4) to re-dissolve samples. After centrifugation at 5,000×g, the supernatant was frozen and stored at −18°C until analysis.
Solutions
All solutions were prepared with Milli-Q water produced via a Direct-Q 3 system (Millipore, Billerica, MA, USA). All other reagents were of high purity (≥95%). The buffer solution (pH 11) was prepared by dissolving 50 g H3BO3 in 900 ml water, adjusted to pH 11 with KOH (47% solution) and diluted to 1 l with water. This solution was stable for up to 12 months at 4°C. The reducing solution was prepared by adding 2.5 ml 2-mercaptoethanol to 100 ml buffer solution. This solution was stable for up to 6 months at 4°C in the dark. The derivatisation reagent was prepared by dissolving 25 mg ortho-phthaldialdehyde (OPA) in 2 ml methanol, mixed with 2 ml of reducing solution, and diluting to 44 ml with buffer solution. The reagent was stable for up to 7 days at 4°C in the dark.
For the standard stock solutions of the four amino sugars, standards (Sigma Aldrich, St. Louis, MO, USA) were dissolved in water to a concentration of 1,000 μmol l−1 (mannosamine, galactosamine, glucosamine) and 100 μmol l−1 (muramic acid), respectively, and stored at –18°C. Standard working solutions were prepared by diluting four aliquots of the stock solutions to a concentration range between 210 and 5 μmol l−1 (mannosamine, galactosamine, glucosamine) and 21 and 0.5 μmol l−1 (muramic acid), respectively. The working standard solutions were stable for over 12 months at −18°C.
For evaluating the interference of common amino acids and related compounds which are abundant in soil and plant hydrolysates, we measured two different amino acids standards (Sigma Aldrich, St. Louis, MO, USA). Amino acid standard solution 1 contained the following components at a concentration of 25 and 12 μmol l−1 for l-cysteine, respectively: l-alanine, ammonium chloride, l-arginine, l-aspartic acid, l-cystine, l-glutamic acid, glycine, l-histidine, l-isoleucine, l-leucine, l-lysine, l-methionine, l-phenylalanine, l-proline, l-serine, l-threonine, l-thyrosin and l-valine. Amino acid standard solution 2 contained the following components at a concentration of 50 μmol l−1: γ-amino-n-butyric acid, ammonium chloride, l-anserine, l-arginine, l-carnosine, creatinine, ethanolamine, l-histidine, l-homocystine, δ-dl-hydroxylysine, l-lysine, 1-methyl-l-histidine, 3-methyl-l-histidine, l-ornithine and l-tryptophan.
Chromatographic conditions
Chromatographic separations were performed on a Phenomenex (Aschaffenburg, Germany) Hyperclone C18 (ODS) column (125 mm length × 4 mm diameter, 5 μm particle size, 12 nm pore size), protected by a Phenomenex C18 security guard cartridge (4 mm length × 2 mm diameter). The column was placed in a column oven set at 35°C. The HPLC system consisted of a Dionex (Germering, Germany) P 580 gradient pump, a Dionex Ultimate WPS-3000TSL analytical autosampler with in-line split-loop injection and thermostat and a Dionex RF 2000 fluorescence detector set at 445 nm emission and 330 nm excitation wavelength with medium sensitivity.
An autosampler designed for automated pre-column derivatisation was used because of the need of a high injection precision and an effective external needle wash for eliminating carryover. Another autosampler tested (Dionex ASI 100) yielded poor reproducibility. Vials with OPA reagent and samples as well as vials for preparation were stored in the autosampler at 15°C. For derivatisation, 50 μl OPA reagent and 30 μl sample were mixed in a preparation vial, and after 120 s reaction time, 15 μl of the indole derivates were injected.
The mobile phase consisted of two eluents. Eluent A was a 97.8/0.7/1.5 (v/v/v) mixture of a water phase, methanol and tetrahydrofuran (THF). The water phase contained 52 mmol sodium citrate and 4 mmol sodium acetate, adjusted to pH 5.3 with HCl. Then, methanol and THF were added. Eluent B consisted of 50% water and 50% methanol (v/v). For degassing and sterilisation, both eluents were filtered over 0.2-μm-pore size hydrophilic propylene membrane filters. The mobile phase was delivered at a flow rate of 1.5 ml min−1. The amino sugar separation was performed isocratically, but a gradient was used for cleaning the column after every run. Every run was starting at an eluent A/B v/v composition of 93/7 for 19 min. A linear gradient was run to reach 80% B after 3 min and remaining isocratic for 3 min. A reverse gradient to 93/7 within 3 min was followed by 2 min isocratic run after which the column is preconditioned for the next sample. It is important to start the OPA derivatisation only at the beginning of every run and not during column preconditioning of the previous run. Otherwise, the equilibration time is not long enough for a good reproducibility.
Results
Method optimisation
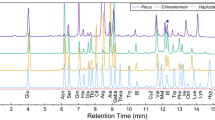
The chromatographic conditions used in the optimised method provided a good separation within approximately 19 min (Fig. 1). Retention times of examined amino sugars were: (1) muramic acid t R = 8.4 min, (2) mannosamine t R = 9.4 min, (3) galactosamine t R = 15.4 min and (4) glucosamine t R = 16.6 min. The components of the amino acid standard solution 1 occurred at 4.7, 10.5, 11.7, 12.6 and > 20.0 min, respectively (Fig. 2b). The retention times of the amino acid standard solution 2 were at 10.5, 11.8, 18.1 and >20 min, respectively (Fig. 2c). For testing the absence of any interference potential, an amino sugar standard (170 μmol l−1) was mixed with the amino acid standard 2 (500 μmol l−1) at the ratio of 5:1. All four amino sugar peaks were well separated from those of the amino acids (Fig. 2d).

Chromatograms of a a standard mixture consisting of 13 μmol l−1 muramic acid, 130 mannosamine, 130 galactosamine, 130 μmol l−1 glucosamine and of (b) a soil hydrolysate (arable 1)

Chromatograms of a an amino sugar standard mixture consisting of 17 μmol l−1 muramic acid, 170 mannosamine, 170 galactosamine, 170 μmol l−1 glucosamine and of b an amino acid standard 1, c an amino acid standard 2 and d an amino sugar standard spiked with an amino acid standard 2, components of amino acid standards (see Materials and methods)
Analysis of a standard mixture at different excitation wavelengths, different OPA reaction times and different pH values of the eluent revealed maxima for the area of all four amino sugar peaks at an excitation wavelength of 330 nm, an OPA reaction time of 120 s, an eluent pH of 5.3 (with the exception of mannosamine), respectively (Fig. 3). Increasing THF concentration in the eluent from 0.75% to 1.50% yielded a better resolution between muramic acid and mannosamine as well as to shorter retention times for all four amino sugars (Fig. 3d).

Optimisation of HPLC parameters: a sensitivity of the amino sugar determination as a function of OPA reaction time; b optimisation of the excitation wavelength; c changes in area as a function of eluent pH value; d change in resolution between muramic acid (MurN) and mannosamine (ManN) peaks and in retention time of MurN, ManN, galactosamine (GalN) and glucosamine (GlcN) as a function of tetrahydrofuran (THF) concentration in eluent (error bars represent ±SD)
The calibration curves (Fig. 4) were linear in the range from 5 to 210 μmol l−1 (mannosamine, galactosamine, glucosamine) and from 0.5 to 21 μmol l−1 (muramic acid), respectively, with good correlation coefficients and standard deviations (Table 3). The coefficient of variation was roughly 2% for intraday and 5% for interday precision, respectively (Table 3). This reflects the reproducibility and the precision of the method excluding sample preparation. The accuracy (expressed as recovery) for the four analytes was determined by spiked quartz sand hydrolysate with the standard mixture solution. The results of the recovery of mannosamine, galactosamine and muramic acid ranged from 105% to 115%. The recovery rate for glucosamine was 81%. The limit of quantification (LOQ) depended on the amino sugar and varied from 0.5 to 5 μmol l−1 for the standard mixture solution (Table 3).

Calibration curves of reference amino sugars (error bars represent ±SD)
The content of the different amino sugars increased in the order mannosamine < muramic acid < galactosamine < glucosamine (Table 4). In three arable soils, mannosamine was below the LOQ value. The glucosamine content in the soil samples ranged from 980 to 2,730 μg g−1 soil and contributed between 55% and 62% to the total amino sugar content. Galactosamine and muramic acid contributed on average 35% and 5%, respectively, mannosamine, if present, only 2%. The glucosamine content in the plant litter material ranged from 36 (maize leaves) to 1,550 μg g−1 dry weight (amaranth straw) and contributed 58 (maize leaves) to 93% (wheat straw) to the total amino sugar content (Table 5). Muramic acid was below the LOQ values in green pea and maize leaves. Mannosamine was below LOQ in maize leaves and wheat straw, but contributed roughly 20% to the total amino sugar content in pea leaves. Soil samples dissolved in water had significantly higher amino sugar contents than those dissolved in buffer solution (Table 4). In contrast, the sample solvent had no effect on the amino sugar amount in plant material (Table 5).
Discussion
Optimisation of the mobile phase
Studies by Appuhn et al. (2004) and Zelles (1988) described a mobile phase with 0.75% THF. However, Zelles (1988) had no satisfactory separation and Appuhn et al. (2004) noted long retention times (32.4 min for glucosamine). In our study, we tested increased THF concentrations and found an optimum concentration of 1.5% (Fig. 1). This yielded in shorter retention times for all amino sugars and a slightly better resolution between muramic acid and mannosamine in comparison to Appuhn et al. (2004). Both, Hodgin (1979) and Jarrett et al. (1986) had already studied the impact of THF on the amino acid separation by reversed phase chromatography. In our study, we could observe similar complex effects caused by THF. On the one hand, an increasing THF concentration caused muramic acid to change its elution position with mannosamine. On the other hand, increasing THF concentration caused better separation and shorter retention times (Fig. 3d). Roggendorf and Spatz (1981) found that THF accelerated compounds containing ether groups. This is probably the explanation for a better resolution between muramic acid and mannosamine and why muramic acid eluted faster than mannosamine with increased THF concentration, as muramic acid contains two ether groups and mannosamine only one. However, increasing THF concentration beyond 1.6%, we could not achieve reproducible peak separation anymore. For example, an occasional retention time shift for up to 2 min was observed, which caused an overlay of matrix- and amino sugar peaks. For this reason, we decided to use a THF concentration of 1.5%.
Lindroth and Mopper (1979) showed that a decreasing pH value of the mobile phase yields better separation between the amino acids. However, they tested only a pH value range between 5.9 and 7.9. To examine whether a lower mobile phase pH shows a better separation, we tested a pH value range from 4 to 6 in the mobile phase. We obtained no better separation at low pH, but we did observe that pH values below 5 reduced the fluorescence response (Fig. 3c) as already reported by Jarrett et al. (1986). Our results confirm the pH value chosen by Zelles (1988) and Appuhn et al. (2004).
Excitation wavelength optimisation
The optimal excitation wavelength is a compromise between increasing the fluorescence intensity of amino sugar peaks and decreasing fluorescence intensity of other primary amines of the matrix. For instance, at a wavelength of 280 nm, a high fluorescence signal for all primary amines is observed, unfortunately also for interfering peaks like ammonia (Jarrett et al. 1986). Several studies about amino sugar determination (Mimura and Delmas 1983; Zelles 1988; Appuhn et al. 2004) use an excitation wavelength of 340 nm except for Rönkkö et al. (1994), who suggested an excitation wavelength of 310 nm. But opposed to these studies, other authors like Lindroth and Mopper (1979) and Jarrett et al. (1986) recommended an excitation wavelength of 330 nm for amino acid methods. Jarrett et al. (1986) confirmed this adjustment by testing several excitation wavelengths for amino acid determination. We tested excitation wavelengths in a range between 300 and 340 nm to find the optimal excitation wavelength for amino sugar determination (Fig. 3a). With an optimum excitation wavelength of 330 nm, our result is consistent with that reported by Jarrett et al. (1986).
Ortho-phthaldialdehyde (OPA) reaction time
To obtain a high fluorescence response, we tested different reaction times for the OPA derivatisation. The fluorescence intensity of the muramic acid–isoindole-derivate was relatively constant whereas the other three amino sugars derivates decompose after 120 s reaction time (Fig. 3b). Lindroth and Mopper (1979) suggested that electron donating groups (in our case the carboxyl-group of muramic acid) have a stabilising effect on the isoindole group. This assumption is supported by Roth (1971), who showed in his study notably high fluorescence intensity for amino acids with two carboxyl-groups (aspartic acid, glutamic acid). Moreover, it would explain the stability and the ten times higher fluorescence response of the muramic acid derivates. We presume that the low fluorescence response of mannosamine is closely related to the conformation of mannosamine. The primary amino group of this molecule is located in an axial conformation, whereas the other three amino sugars contain the amino group in an equatorial conformation. Steric effects hinder the OPA reaction in mannosamine, which proceeds more easily in an equatorial conformation.
Validation parameters
In general, our validation parameters (Table 3) of this HPLC method are similar with validation parameters reported earlier (Zelles 1988; Appuhn et al. 2004). Zhang and Amelung (1996) described a reliable and sensitive amino sugar determination via gas chromatography. Their LOQ ranged from 10 μg ml−1 (muramic acid) to 20 μg ml−1 (other three amino sugars). In contrast to that, we reached a higher sensitivity with LOQ of 0.5 μmol l−1, which is equal to 0.13 μg ml−1 for muramic acid and 5.0 μmol l−1, which is equal to 0.90 μg ml−1 for the other three amino sugars. Furthermore, with gas chromatographic methods, a time-consuming off-line derivatisation procedure is needed for obtaining volatile components.
Our LOQ is similar to that of other HPLC methods described by Kaiser and Benner (2000), Diaz et al. (1996) or Ekblad and Näsholm (1996). Kaiser and Benner (2000) used a pulsed amperometric detector whereas Diaz et al. (1996) applied a fluorescence detector with 6-aminoquinolyl-N-hydroxysuccinmydyl-carbamate as derivatisation reagent. Ekblad and Näsholm (1996) used also fluorometric detection and 9-fluorenylmethylchloroformate for derivatisation. Diaz et al. (1996) determined only glucosamine and galactosamine. The peaks were well separated but each amino sugar yielded two peaks. Ekblad and Näsholm (1996) examined only glucosamine, which eluted in three peaks. Kaiser and Benner (2000) determined all four amino sugars with two different methods based on anion exchange chromatography.
Effect of sample solvent
Dorresteijn et al. (1996) reported that the OPA derivatisation is a very complex reaction with three possible products: hydrolysis of OPA, stabilisation of OPA by 2-mercaptoethanol to yield an ortho-phthalaldehyde-2-mercaptoethanol complex and reaction of OPA with 2-mercaptoethanol and amino sugars yielding the isoindole-derivate needed for amino sugar analysis. This complexity makes it difficult to find the optimal conditions for the derivatisation. However, pH of the reaction mixture, the concentration of 2-mercaptoethanol and the reaction time are probably the most important variables to control. The effect of the reaction time is discussed above.
With regard to method accuracy, Dorresteijn et al. (1996) suggested Milli-Q water to be best suited. Appuhn et al. (2004) also used water as sample solvent in their study. However, since sample pH is between 2 and 3 in the sample extract, we examined if better accuracy for this method is possible with higher pH during OPA reaction. Sample extract pH was increased to values about 5 with phosphate buffer. In general, buffered samples showed higher standard deviations. Significant differences between water and buffer were only observed for the soil samples, particularly for the basalt-derived clayey soil forest 1, which contained high contents of aluminium and iron oxides. The hydrolysis products of these oxides presumably form as central ions complexes in the presence of ligands like phosphate. If these complexes bind amino sugars, they may not be completely available for OPA derivatisation anymore. With respect to the higher coefficients of variation and the lower amino sugar content using buffer as sample solvent, we recommend to use water as more reliable sample solvent.
Concentrations of amino sugars in soil and plant material samples
Our amino sugar contents in soil (Table 4) compare well with those reported in the literature from arable and forest soils (Zelles 1988; Guggenberger et al. 1999; Joergensen et al. 2010). Guggenberger et al. (1999) obtained in different arable soils a total amino sugar composition of 64% glucosamine, 31% galactosamine, 3% muramic acid, and 2% mannosamine. Amelung et al. (1999) found in different grassland soils 60% glucosamine, 33% galactosamine, 5% muramic acid and 2% mannosamine. Liang et al. (2007a) measured in different forest soils 67% glucosamine, 21% galactosamine, 8% muramic acid and 4% mannosamine. Ding et al. (2010) detected 55% glucosamine, 40% galactosamine and 5% muramic acid, but no mannosamine in soil samples amended with different amounts maize residues after 38 weeks of incubation. Also, Glaser et al. (2004) did not find mannosamine in all samples. Liang et al. (2006, 2007b) measured 72% glucosamine, 8% galactosamine, 13% muramic acid and 7% mannosamine in soybean straw and maize stalks. Amelung et al. (1999), Guggenberger et al. (1999), Liang et al. (2006, 2007a, b) and Ding et al. (2010) all used after conversion of the amino sugars to aldonitrile acetates the gas chromatographic method described in detail by Zhang and Amelung (1996). Benner and Kaiser (2003) measured 63% glucosamine, 17% galactosamine, 5% mannosamine and 16% muramic acid in marine particulate organic matter, with pulsed amperometric detection and ion exchange chromatography, a method without a derivatisation step.
It is not clear whether the absence of mannosamine in the low-biomass arable soils is due to the missing sensitivity as the majority of the data reported for mannosamine has been obtained in high biomass soils (Guggenberger et al. 1999; Turrión et al. 2002; Liang et al. 2007b). In our three soils with detectable mannosamine data, the concentration is in the range obtained by others (Amelung et al. 1999; Guggenberger et al. 1999). High mannosamine percentages, as in our pea leaves, were also, one time, observed in litter layer (Turrión et al. 2002).
Conclusions
Our method allows fast, quantitative and reproducible HPLC determination of the four most important amino sugars in soil and plant litter. The separation between muramic acid and mannosamine was optimised in combination with shorter retention times in comparison with previously published methods. No interferences exist from amino acids or other primary amines, occurring in soil and plant hydrolysates.
References
Amelung W (2001) Methods using amino sugars as markers for microbial residues in soil. In: Lal JM, Follett RF, Stewart BA (eds) Assessment methods for soil carbon. Lewis Publishers, Boca Raton, pp 233–272
Amelung W (2003) Nitrogen biomarkers and their fate in soil. J Plant Nutr Soil Sci 166:677–686
Amelung W, Zhang X, Flach KW, Zech W (1999) Amino sugars in native grassland soils along a climosequence in North America. Soil Sci Soc Am J 63:6–2
Amelung W, Lobe I, Du Preez CC (2002) Fate of microbial residues in sandy soils of the South African Highveld as influenced by prolonged arable cropping. Eur J Soil Sci 53:29–35
Amelung W, Brodowski S, Sandhage-Hofmann A, Bol R (2008) Combining biomarker with stable isotope analyses for assessing the transformation and turnover of soil organic matter. Adv Agron 100:155–250
Appuhn A, Joergensen RG (2006) Microbial colonisation of roots as a function of plant species. Soil Biol Biochem 38:040–051
Appuhn A, Joergensen RG, Scheller E, Wilke B (2004) The automated determination of glucosamine, galactosamine, muramic acid and mannosamine in soil and root hydrolysates by HPLC. J Plant Nutr Soil Sci 167:17–21
Benner R, Kaiser K (2003) Abundance of amino sugars and peptidoglycan in marine particulate and dissolved organic matter. Limnol Oceanogr 48:118–128
Chantiny MH, Angers DH, Prévost D, Vézina LP, Cahlifour FP (1997) Soil aggregation and fungal and bacterial biomass under annual and perennial cropping systems. Soil Sci Soc Am J 61:262–267
Coelho RRR, Sacramento DR, Linhares LF (1997) Amino sugars in fungal melanins and soil humic acids. Eur J Soil Sci 48:425–429
Diaz J, Lliberia JL, Comellas L, Broto-Puig F (1996) Amino acid and amino sugar determination by derivatization with 6-aminoquinolyl-N-hydroxysuccinimidyl carbamate followed by high-performance liquid chromatography and fluorescence detection. J Chromatogr A 719:171–179
Ding X, Zhang X, He H, Xie H (2010) Dynamics of soil amino sugar pools during decomposition processes of corn residues as affected by inorganic N addition. J Soils Sediments 10:758–766
Dorresteijn RC, Berwald LG, Zomer G, de Gooijer CD, Wieten G, Beuvery EC (1996) Determination of amino acids using o-phthalaldehyde-2-mercaptoethanol derivatization. Effect of reaction conditions. J Chromatogr A 724:159–167
Ekblad A, Näsholm T (1996) Determination of chitin in fungi and mycorrhizal roots by an improved HPLC analysis of glucosamine. Plant Soil 178:29–35
Engelking B, Flessa H, Joergensen RG (2007) Shifts in amino sugar and ergosterol contents after addition of sucrose and cellulose to soil. Soil Biol Biochem 39:2111–2118
Ferrero MÁ, Aparicio LR (2010) Biosynthesis and production of polysialic acids in bacteria. Appl Microbiol Biotechnol 86:1621–1635
Glaser B, Turrión MB, Alef K (2004) Amino sugars and muramic acid—biomarkers for soil microbial community structure analysis. Soil Biol Biochem 36:399–407
Guggenberger G, Frey SD, Six J, Paustian K, Elliott ET (1999) Bacterial and fungal cell wall residues in conventional and no-tillage agroecosystems. Soil Sci Soc Am J 63:1188–1198
Hodgin JC (1979) The separation of pre-column o-phthalaldehyde derivatized amino acids by high performance liquid chromatography. J Liquid Chromatog Rel Technol 2:1047–1059
Jarrett HW, Cooksy KD, Ellis B, Anderson JM (1986) The separation of o-phthalaldehyde derivates of amino acids by reversed-phase chromatography on octylsilica columns. Anal Biochem 153:189–198
Joergensen RG, Meyer B (1990) Chemical change in organic matter decomposing in and on a forest Rendzina under beech (Fagus sylvatica L.). J Soil Sci 41:17–27
Joergensen RG, Mäder P, Fließbach A (2010) Long-term effects of organic farming on fungal and bacterial residues in relation to microbial energy metabolism. Biol Fertil Soils 46:303–307
Kaiser K, Benner R (2000) Determination of amino sugars in environmental samples with high salt content by high-performance anion-exchange chromatography and pulsed amperometric detection. Anal Chem 72:2566–2572
Kenne LK, Lindburg B (1983) Bacterial polysaccharides. In: Aspinall GO (ed) The polysaccharides. Academic, New York, pp 287–353
Liang C, Zhang X, Rubert KF, Balser TC (2006) Effect of plant materials on microbial transformation of amino sugars in three soil microcosms. Biol Fertil Soils 43:631–639
Liang C, Fujinuma R, Wei LP, Balser TC (2007a) Tree species-specific effects on soil microbial residues in an upper Michigan old-growth forest system. Forestry 80:65–72
Liang C, Zhang X, Balser TC (2007b) Net microbial amino sugar accumulation process in soil as influenced by different plant material inputs. Biol Fertil Soils 44:1–7
Lindroth P, Mopper K (1979) High performance liquid chromatography determination of subpicomole amounts of amino acids by precolumn fluorescence derivatization with o-phthaldialdehyde. Anal Chem 51:1667–1674
Millar WN, Casida LE (1970) Evidence for muramic acid in soil. Can J Microbiol 16:299–304
Mimura T, Delmas D (1983) Rapid and sensitive method for muramic acid determination by high performance liquid chromatography with precolumn fluorescence derivatization. J Chromatogr 280:91–98
Probst B, Schüler C, Joergensen RG (2008) Vineyard soils under organic and conventional management—Microbial biomass and activity indices and their relation to soil chemical properties. Biol Fertil Soils 44:443–450
Roggendorf E, Spatz R (1981) Systematic use of tetrahydrofuran in reversed-phase high-performance liquid chromatography. An example of the selectivity benefits of ternary mobile phases. J Chromatogr 204:263–268
Rönkkö R, Pennanen T, Smolander A, Kitunen V, Kortemaa H, Haahtela K (1994) Quantification of Frankia strains and other root-associated bacteria in pure cultures and in the rhizosphere of axenic seedlings by high-performance liquid chromatography-based muramic acid assy. Appl Environ Microbiol 60:3672–3678
Roth M (1971) Fluorescence reaction for amino acids. Anal Chem 43:880–882
Rottmann N, Siegfried K, Buerkert A, Joergensen RG (2010) Litter decomposition in fertilizer treatments of vegetable crops under irrigated subtropical conditions. Biol Fertil Soils. doi:10.1007/s00374-010-0501-9
Sharon N (1965) Distribution of amino sugars in microoganisms, plants and invertebrates. In: Balasz EA, Jeanlanx RW (eds) The amino sugars, part 2A. Distribution and biological role. Academic, New York, pp 1–45
Stevenson FJ (1982) Organic forms of soil nitrogen. In: Stevenson FJ (ed) Nitrogen in agricultural soils. American Society of Agronomy, Madison, pp 101–104
Turrión MB, Glaser B, Zech W (2002) Effects of deforestation on contents and distribution of amino sugars within particle-size fractions of mountain soils. Biol Fertil Soils 35:49–53
Wasylnka JA, Simmer MI, Moore MM (2001) Differences in sialic acid density in pathogenic and non-pathogenic Aspergillus species. Microbiol 147:869–877
Yoneyama T, Koike Y, Arakawa Y, Yokoyama HK, Sasaki Y, Kawamura T, Araki Y, Ito E, Takao S (1982) Distribution of mannosamine and mannosaminuronic acid among cell walls of Bacillus species. J Bacteriol 149:15–21
Zelles L (1988) The simultaneous determination of muramic acid and glucosamine in soil by high-performance liquid chromatography with precolumn fluorescence derivatization. Biol Fertil Soils 6:125–130
Zhang X, Amelung W (1996) Gas chromatographic determination of muramic acid, glucosamine, mannosamine, and galactosamine in soils. Soil Biol Biochem 28:1201–1206
Acknowledgments
We gratefully acknowledge Gabriele Dormann for her technical assistance. Caroline Indorf was funded by the German Research Foundation (DFG).
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License (https://creativecommons.org/licenses/by-nc/2.0), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
Indorf, C., Dyckmans, J., Khan, K.S. et al. Optimisation of amino sugar quantification by HPLC in soil and plant hydrolysates. Biol Fertil Soils 47, 387–396 (2011). https://doi.org/10.1007/s00374-011-0545-5
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00374-011-0545-5