
Abstract
The room-temperature study of Fe0.97Cr0.03 and Fe0.94Cr0.06 alloys was performed using the transmission Mössbauer spectroscopy (TMS). The TMS measurements allow to determine changes in chemical composition and content of iron oxides in the bulk of the studied samples during exposure to air at 570, 870 and 1070 K. The results indicate that the chemical composition of samples changes significantly, and it is strongly dependent on temperature at which the studied samples are exposed to air. The iron internal oxidation kinetics as well as the activation energies for the oxidation of iron atoms in studied alloys were calculated.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
1 Introduction
Today, the nuclear energy sector is looking for new structural materials which could be able to keep their design parameters for a long time during the exposure to factors such as corrosion, high temperature and high-energy neutron bombardment. These conditions will exist in new types of advanced fission reactors (generation IV) and fusion reactors. One of the most interesting and promising types of structural materials which could be used in those innovative nuclear facilities is high-chromium ferritic/martensitic steels [1]. Many studies of iron–chromium alloys, which are the base components of high-chromium ferritic/martensitic steels, show that a certain amount of chromium makes an iron alloy corrosion resistant. At ambient conditions, a thin and transparent film of chromium oxide rapidly forms on the open surface of the alloy preventing further oxidation and blocking corrosion. Unfortunately, upon ageing the Fe–Cr alloys with high chromium content often undergo transformation to either a high-temperature σ phase or separation into Fe-rich (α) and Cr-rich (α’) phases. These processes lead to significant degradation of mechanical properties of certain steel grades.
The main aim of this work is to expand the knowledge about corrosion process in Fe–Cr alloys with low chromium content at elevated temperatures. High-temperature corrosion is an important phenomenon which must be taken into account during designing of modern fission reactors such as the very-high-temperature reactor (VHTR) or the supercritical water reactor (SCWR). At the same time, for the Fe–Cr alloys with low chromium content the effect connected to separation into Fe-rich (α) and Cr-rich (α’) phases should be negligible. In this work, the Fe0.97Cr0.03 and Fe0.94Cr0.06 alloys which were exposed to air at 570, 870 and 1070 K were investigated by the transmission Mössbauer spectroscopy (TMS). These measurements allow to compare the oxidation process in two Fe–Cr alloys with different chromium contents under the same oxidation conditions. On the basis of obtained TMS data, it is possible to determine the changes in chemical composition and content of iron oxides in studied alloys during exposure to air as well as calculate the iron internal oxidation kinetics and the activation energies for the internal oxidation of iron atoms. These parameters are crucial for better understanding a corrosion process of Fe–Cr alloys during exposure to air.
2 Experimental details
The samples of Fe0.97Cr0.03 and Fe0.94Cr0.06 alloys were prepared in an arc furnace. Appropriate amounts of the 99.98% pure iron and 99.995% pure chromium were melted into a water-cooled copper crucible in argon atmosphere and quickly cooled to room temperature. Solidified alloys were remelted two times to ensure homogeneity. The weight losses during the melting process were below 0.2% of the original weight, so the compositions of the obtained ingots were close to nominal ones. Resulting ingots were cold-rolled to the final thickness of about 0.035 mm. In the next step, Fe0.97Cr0.03 and Fe0.94Cr0.06 foils were annealed in the vacuum at 1270 K for 2 h. To obtain homogeneous and defect-free samples [2], after annealing process, the foils were slowly cooled to room temperature during 6 h. The annealed samples were exposed to air at 570, 870 and 1070 K for several hours. Each step of samples processing was followed by TMS measurements which were taken at room temperature by means of a constant-acceleration POLON spectrometer of standard design, using a 3.7-GBq 57Co-in-Rh standard source with a full width at half maximum (FWHM) of 0.22 mm/s.
Each measured spectrum was fitted with a transmission integral for a sum of different six-line patterns corresponding to various isomer shifts IS, quadrupole splitting/shift QS as well as hyperfine fields B at 57Fe nuclei generated by different chemical states of 57Fe Mössbauer probes. The single-phase Fe–Cr alloy with bcc structure could be described by additive model which assumes that the influence of Cr atoms on B as well as the corresponding isomer shift IS on a subspectrum is additive and independent of the atom positions in the first two coordination shells of the nuclear probe [3]. In other words, it was accepted that for each subspectrum, the quantities B and IS are linear functions of the number n of Cr atoms located in the first two coordination shells of 57Fe. The functions can be written as follows:
where ΔB and ΔIS stand for the changes in B and IS with one Cr atom in the first two coordination shells of the Mössbauer probe. At the same time, it was assumed that the quadrupole splitting QS in a cubic lattice is equal to zero. All additional components which appeared in measured spectra were attributed to new compounds which were formed during exposure to air. Some of the obtained fits are presented in Figs. 1, 2, 3, 4, 5 and 6.

Room-temperature TMS spectra for the Fe0.97Cr0.03 sample exposed to air at 570 K for several hours, fitted with a sum of various number of six-line patterns corresponding to different chemical states of iron atoms

Room-temperature TMS spectra for the Fe0.97Cr0.03 sample exposed to air at 870 K for several hours, fitted with a sum of five six-line patterns corresponding to different chemical states of iron atoms

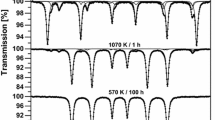
Room-temperature TMS spectra for the Fe0.97Cr0.03 sample exposed to air at 1070 K for several hours, fitted with a sum of various number of six-line patterns corresponding to different chemical states of iron atoms

Room-temperature TMS spectra for the Fe0.94Cr0.06 sample exposed to air at 570 K for several hours, fitted with a sum of various number of six-line patterns corresponding to different chemical states of iron atoms

Room-temperature TMS spectra for the Fe0.94Cr0.06 sample exposed to air at 870 K for several hours, fitted with a sum of six six-line patterns corresponding to different chemical states of iron atoms

Room-temperature TMS spectra for the Fe0.94Cr0.06 sample exposed to air at 1070 K for several hours, fitted with a sum of various number of six-line patterns corresponding to different chemical states of iron atoms
3 Results and discussion
As one can see in Figs. 1, 2, 3, 4, 5 and 6, the measured TMS spectra for Fe0.97Cr0.03 and Fe0.94Cr0.06 alloys change significantly with time of exposure to air. For samples exposed to air at 570 K for time shorter than 100 h, the TMS spectra analysis reveal existence of components (sextets) which could be assigned only to single-phase Fe–Cr alloy with bcc structure [3, 4]. For longer exposure time as well as for alloys exposed at 870 and 1070 K, the additional magnetically split components were observed. These subspectra were accounted for by adding to the model of the spectrum three sextets. One sextet with B ≈ 51.7 T, IS ≈ 0.37 mm/s, QS ≈ −0.2 mm/s could be a result of the α-Fe2O3 presence in the sample [5]. The last two sextets with B ≈ 48.9 T and 46.1 T, IS ≈ 0.24 mm/s and 0.68 mm/s, QS <0.1 mm/s correspond to the two different sites of Fe atoms in Fe3O4 compound [6]. Here, it is worth noting that the changes in relative intensities of two Fe3O4 sextets were observed. This fact suggests that stoichiometric and non-stoichiometric Fe3O4 were formed [7, 8]. At the same time, in all measured TMS spectra, there are no other components which could be connected with FeO, FeOOH or iron carbides [9–11]. The obtained main hyperfine parameters derived from the TMS spectra for studied samples are listed in Tables 1, 2. The above findings are important evidence that in Fe0.97Cr0.03 and Fe0.94Cr0.06 alloys exposed to air at elevated temperatures, only two types of compounds are formed: α-Fe2O3 and Fe3O4. Moreover, the further analysis of the TMS spectra parameters being the effective thicknesses T A (x) related to components of each spectrum can give detailed information about a chemical composition of studied samples. That is possible because the effective thicknesses are determined by the fractions c(x) of absorbing atoms corresponding to the components under consideration, and the proper relationship can be expressed as follows:
where σ 0 is the maximal cross section for nuclear γ resonance absorption, t A denotes the thickness of the absorber, N stands for the total number of resonant absorbing atoms 57Fe per unit volume, and f is the Lamb–Mössbauer factor. Assuming that the relative ratio of the Lamb–Mössbauer factors f α-Fe:f α-Fe2O3:f Fe3O4 is equal to 1:1.08:1.05 [12], the fraction c(x) can be easily calculated using the T A (x) values as in such case:
The computed c(x) values were used to find parameters c(Fe), c(Fe2O3) and c(Fe3O4) which are the total intensities of spectrum components related to the existence of 57Fe nuclear probes in α-Fe, α-Fe2O3 and Fe3O4, respectively. These parameters are listed in Tables 3, 4. Finally, to obtain a chemical composition of studied samples, it is necessary to take into account that the relative quantities of iron atoms in observed compounds are different and equal to 1:2:3 for α-Fe, α-Fe2O3 and Fe3O4, respectively. The chemical composition estimated under above-mentioned assumptions for two exposure temperatures (870 and 1070 K) is presented in Fig. 7. As one can expect, a few atomic percent of Cr impurity dissolved in iron matrix has a significant influence on oxidation process of iron atoms in studied materials. The comparison of obtained data with results presented in previous work which concerns oxidation of pure iron [13] suggests that the internal oxidation of iron atoms in Fe–Cr alloys slows with increasing Cr content.

Chemical composition of the Fe0.97Cr0.03 and Fe0.94Cr0.06 alloys exposed to air at 870 and 1070 K in function of exposure time
To analyse thermal oxidation of iron in Fe–Cr alloys, the Deal–Grove model was applied [14]. This model requires that the oxidation of Fe follows a mechanism whereby a thin layer of oxide is formed by place exchange after adsorption has occurred with further oxidation occurring due to an electric field mechanism. The electric field is formed by electrons tunnelling from the metal to the adsorbed oxygen and is able to support ion transport through the place exchange and formed thin oxide layer enabling the oxide layer to increase in thickness. Due to fact that TMS technique is not sensitive for thin oxide layer (below 100 nm), the parabolic regime (for thick films) was assumed:
where FeOX is a fraction of oxidised iron atoms in studied alloys at time t and k denotes the reaction rate constant which is equal to:
where E A is activation energy for iron oxidation, k B stands for Boltzmann constant, k 0 is pre-exponential factor, and T denotes temperature. Surprisingly, during analysis of the iron oxidation kinetic curves, it was found that parabolic regime is not valid and the logarithmic oxidation kinetics was much more plausible:
where k denotes a reaction rate constant and C is a constant. The parabolic and logarithmic iron oxidation kinetic curves fitted to experimental data obtained for the Fe0.97Cr0.03 and Fe0.94Cr0.06 alloys exposed to air at 870 and 1070 K are presented in Fig. 8. The knowledge of the reaction rate k, for two different temperatures, T 1 = 870 K and T 2 = 1070 K combined with Eq. (5), enabled determination of the activation energy EA, for the oxidation of iron atoms in the temperature interval of 870–1070 K, using the following equation:
This equation is valid assuming that the pre-exponent factor k 0 in Eq. (5) is temperature independent. The estimated reaction rate constants as well as activation energies of iron internal oxidation process for studied Fe–Cr alloys are presented in Table 5. The obtained EA values for Fe–Cr alloys are much higher than the activation energy for the oxidation reaction of pure Fe which was determined to be 32(6) kJ/mol [15]. This is in accord with the observation that the iron oxidation process in Fe–Cr alloys is much slower than the one in pure iron sample. Moreover, the higher EA value for Fe0.94Cr0.06 alloy also agrees with previous conclusion that the internal oxidation of iron atoms slows with increasing Cr content.

4 Conclusions
The results reported in this paper can be concluded as follows:
-
1.
The TMS measurements of Fe0.97Cr0.03 and Fe0.94Cr0.06 alloys allow to determine changes in chemical composition and the presence of newly formed compounds in studied materials during exposure to air at 570, 870 and 1070 K.
-
2.
The comparison of data obtained for Fe0.97Cr0.03 and Fe0.94Cr0.06 samples results in conclusion that the internal oxidation of iron atoms slows with increasing Cr content.
-
3.
It was found that parabolic regime for internal oxidation of iron atoms in diluted Fe–Cr alloys is not valid and the logarithmic oxidation kinetics was much more plausible.
-
4.
The estimation of the reaction rate k, for two different temperatures, T 1 = 870 K and T 2 = 1070 K, enabled determination of the activation energy E A for the internal oxidation of iron atoms in studied Fe–Cr alloys. E A = 0.46(2) eV for Fe0.97Cr0.03 and E A = 0.60(3) eV for Fe0.94Cr0.06.
References
I. Cook, Materials research for fusion energy. Nat. Mater. 5, 77–80 (2006)
R. Idczak, R. Konieczny, J. Chojcan, A study of defects in iron-based binary alloys by the Mössbauer and positron annihilation spectroscopies. J. Appl. Phys. 115, 103513 (2014)
I. Vincze, I.A. Campbell, Mössbauer measurements in iron base alloys with transition metals. J. Phys. F 3, 647–663 (1973)
Rafał Idczak, Karolina Idczak, Robert Konieczny, Oxidation and surface segregation of chromium in Fe–Cr alloys studied by Mössbauer and X-ray photoelectron spectroscopy. J. Nucl. Mater. 452, 141–146 (2014)
R. Zboril, M. Mashlan, D. Petridis, Iron(III) oxides from thermal processess synthesis, structural and magnetic properties, Mössbauer spectroscopy characterization, and applications. Chem. Mater. 24, 969–982 (2002)
B.J. Evans, S.S. Hafner, 57Fe hyperfine fields in magnetite (Fe3O4). J. Appl. Phys. 40, 1411–1413 (1969)
J.M. Daniels, A. Rosencwaig, Mössbauer spectroscopy of stoichiometric and non-stoichiometric magnetite. J. Phys. Chem. Solids 30, 156l–1571 (1969)
F.C. Voogt, T. Hibma, G.L. Zhang, M. Hoefman, L. Niesen, Growth and characterization of non-stoichiometric magnetite Fe3–δO4 thin films. Surf. Sci. 331–333, 1508–1514 (1995)
L.F. Checherskaya, V.P. Romanov, P.A. Tatsienko, Mössbauer effect in Wüstite. Phys. Stat. Sol. a 19, K177–K182 (1973)
F. Van der Woude, A.J. Dekker, Mössbauer Effect in α-FeOOH. Phys. Stat. Sol. 13, 181–193 (1966)
B. Kniep, A. Constantinescu, D. Niemeier, K.D. Becke, An in situ Mössbauer Study of the Formation of Cementite, Fe3C. Z. Anorg. Allg. Chem. 629, 1795–1804 (2003)
D.A. Channing, M.J. Graham, A study of iron oxidation processes by mössbauer spectroscopy. Corros. Sci. 12, 271–289 (1972)
K. Idczak, R. Idczak, R. Konieczny, An investigation of the corrosion of polycrystalline iron by XPS, TMS and CEMS. Phys. B 491, 37–45 (2016)
B.E. Deal, A.S. Grove, General relationship for the thermal oxidation of silicon. J. Appl. Phys. 36, 3770–3778 (1965)
A.P. Grosvenor, B.A. Kobe, N.S. McIntyre, Activation energies for the oxidation of iron by oxygen gas and water vapour. Surf. Sci. 574, 317–321 (2005)
Acknowledgements
This work was supported by the Polish Ministry of Science and Higher Education under the “Iuventus Plus” programme in the years 2015–2017, project number IP2014 015573.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Idczak, R. Internal oxidation process in diluted Fe–Cr alloys: a transmission Mössbauer spectroscopy study. Appl. Phys. A 122, 1009 (2016). https://doi.org/10.1007/s00339-016-0544-3
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s00339-016-0544-3