
Abstract
Down syndrome (DS) is the most frequent genetic disorder leading to intellectual disabilities and is caused by three copies of human chromosome 21. Mouse models are widely used to better understand the physiopathology in DS or to test new therapeutic approaches. The older and the most widely used mouse models are the trisomic Ts65Dn and the Ts1Cje mice. They display deficits similar to those observed in DS people, such as those in behavior and cognition or in neuronal abnormalities. The Ts65Dn model is currently used for further therapeutic assessment of candidate drugs. In both models, the trisomy was induced by reciprocal chromosomal translocations that were not further characterized. Using a comparative genomic approach, we have been able to locate precisely the translocation breakpoint in these two models and we took advantage of this finding to derive a new and more efficient Ts65Dn genotyping strategy. Furthermore, we found that the translocations introduce additional aneuploidy in both models, with a monosomy of seven genes in the most telomeric part of mouse chromosome 12 in the Ts1Cje and a trisomy of 60 centromeric genes on mouse chromosome 17 in the Ts65Dn. Finally, we report here the overexpression of the newly found aneuploid genes in the Ts65Dn heart and we discuss their potential impact on the validity of the DS model.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Down syndrome (DS) remains the most frequent genetic cause of mental retardation in humans. The genetic basis, trisomy of human chromosome 21 (Hsa21), happens in 1 out of 700–800 births, with a sex ratio of 3 boys for 2 girls (Huret and Sinet 2000) and it is estimated that there are more than 217,800 cases a year throughout the world (Christianson et al. 2006). This is certainly an underestimation considering that this syndrome accounts for approximately 2% of miscarriages (Contestabile et al. 2010). The clinical picture of this syndrome is extremely complex, but the most disabling phenotypes are certainly the intellectual disabilities, with delayed learning and cognition, the appearance of Alzheimer-like disease while aging, the morphological anomalies, the locomotor deficit, and cardiac malformations.
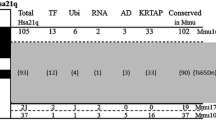
To improve our knowledge of DS, mouse models have been created in recent years. At the genomic level, the long arm of Hsa21 comprises 33.7 Mb of DNA, containing approximately 291 annotated genes in hg19.refGene (Pruitt et al. 2003). Among those genes, 181 have a counterpart in the mouse genome according to the Mammalian Orthology section of the Mouse Genome Informatics (The Jackson Laboratory, Bar Harbor, ME). These genes are located in three syntenic regions localized on different mouse chromosomes (Mmu for Mus musculus). The largest region is found on Mmu16, approximately 37 Mb, including 119 orthologous genes from Lipi to Zfp295. The next segment, including 20 orthologous genes from Umodl1 to Hsf2 bp, is found on Mmu17; and, finally, the most telomeric region, encompassing 42 orthologous genes between Cstb and Prmt2, is on Mmu10. The order and relative orientation of the genes are well preserved between the two species (Dierssen et al. 2009). Several DS murine models were developed over the years using controlled chromosomal engineering techniques (Brault et al. 2006, 2007; Li et al. 2007; Pereira et al. 2009; Yu et al. 2010a, b). Before that, the first models selected carried translocations of specific segments homologous to Hsa21. Briefly, the Ts(16C-tel)1Cje (abbreviated Ts1Cje) was isolated after a targeting experiment aimed at inactivating the Sod1 gene on Mmu16 (Sago et al. 1998). In this line the translocated segment starts upstream of a knockout allele of Sod1 with 67 functional genes and is fused to the telomeric end of Mmu12 (Huang et al. 1997; Pruitt et al. 2003). The Ts(1716)65Dn line (abbreviated Ts65Dn) was isolated after X-ray irradiation, carrying a segment with 122 genes homologous to Hsa21 starting upstream of Mrpl39 and finishing at the telomeric end of Mmu16, which is translocated to a small centromeric part of Mmu17 (Davisson et al. 1990).
With more than 190 references, Ts65Dn has been extensively studied as a DS mouse model. Notably, Ts65Dn mice show deficits in cognition, learning, and memory, plus additional anomalies related to alterations observed in DS people (Reeves et al. 1995). For example, alterations of Ts65Dn cognitive functions have been correlated with changes in the number of neurons (Moldrich et al. 2007; Roper et al. 2006), decrease in long-term potentiation (LTP), and increase in GABAergic inhibition (Belichenko et al. 2004, 2009; Costa and Grybko 2005; Kleschevnikov et al. 2004; Popov et al. 2011). Based on this observation, several treatments were successfully evaluated in Ts65Dn DS models to restore hippocampus-dependent learning (Braudeau et al. 2011; Costa et al. 2008; Fernandez et al. 2007; Moran et al. 2002; Rueda et al. 2008; Salehi et al. 2009).
However, the exact position of the translocation breakpoint between Mmu17 and Mmu16 is not precisely defined in the Ts65Dn. Accordingly, the list of genes located on the centromeric part of Mmu17 in the translocated chromosome was not precisely known. Thus, the effect of this additional trisomy of Mmu17 genes on DS-related phenotypes is difficult to evaluate. In addition, the genotyping is based mainly on genomic quantification, with modest discrimination in the high-throughput process (Liu et al. 2003). An alternative using polymorphic genetic markers was recently proposed (Lorenzi et al. 2010). Nevertheless, classical crossing-over introduces confusion in genotyping and limits the alternative method to a triage (AD and YH, personal communication), not improving the efficiency of Ts65Dn genotyping.
Here we report the characterization of the translocation breakpoints in both the Ts65Dn and the Ts1Cje line. We also propose a new genotyping protocol using classical PCR to facilitate the use of the Ts65Dn model. We further consider the effect of the trisomy of genes located in the Ts65Dn Mmu17 region on their expression in one adult organ, the heart. Our results will help us better understand the translocated chromosomes and open the discussion on the limits of the use of these DS models compared to the new engineered mouse lines.
Materials and methods
Mouse lines
The mice were purchased from the Jackson Laboratory. Both the Ts1Cje line and the Ts65Dn line were kept on a F1 B6C3B genetic background in which the C3B are sighted C3H/HeH, a congenic line for the BALB/c allele at the Pde6b gene (Hoelter et al. 2008). They were bred under specific pathogen-free (SPF) conditions and were treated in compliance with the animal welfare policies of the French Ministry of Agriculture (law 87 848). YH, as the principal investigator in this study, was granted the accreditation 67–369 to perform the reported experiments.
Genotyping
For both mouse lines, genomic DNA was isolated from tail biopsies using a NaCl precipitation technique. Ts1Cje were genotyped using the protocol described by the Jackson Laboratory (stock No. 004861 JAX® Mice Database) by using two pairs of primers. One matches within the neomycin gene in Ts1Cje (IMR6916: CTTGGGTGGAGAGGCTATTC and IMR6917: AGGTGAGATGACAGGAGATC) and the other, used as an internal control, matches in the Tcrd gene (IMR8744: CAAATGTTGCTTGTCTGGTG and IMR8745: GTCAGTCGAGTGCACAGTTT).
Ts65Dn was genotyped using a quantitative PCR protocol. The technique is based on the ΔΔCT calculation method between a control gene present in two copies (ApoB) and a target gene present in three copies (Mx1). Two marked probes are used, one for the control gene ApoB: VIC-CCAATGGTCGGGCAC-MGB-NFQ®, and another for the target gene Mx1: 6-FAM-CCTGGTCGCTGTGCA-MGB-NFQ® (Applied Biosystems, Villebon-sur-Yvette, France), coupled with their respective primers already described (Liu et al. 2003). To avoid sampling variation, DNA concentration was homogenized and the TaqMan probes were labeled with two different fluorescent reporters (FAM and VIC) so that a multiplexed PCR could be used. The qPCR was done in triplicate and we tested a minimum of 15 individuals per experiment. In addition, the standard deviation of the threshold cycle (CT) triplicates should not exceed the value of 0.3 for each dye in order to be validated. After real-time PCR, the average change (∆) in CT of the target gene from that of the internal control gene was calculated (∆CT = mean triplicate CT for Mx1—mean triplicate CT for ApoB). As both diploid and trisomic samples are normally present in one qPCR experiment, the ∆CT values for each sample were classified into two categories of similar values. The mean ∆CT value for the group with the lowest ∆CT (considered as the diploid samples) was calculated per experiment and permitted us to calculate the ∆∆CT (∆∆CT = ∆CT of sample—mean ∆CT of diploid group). Finally, the copy number of the target gene was calculated using the 2(∆∆CT) formula. If the value of 2(∆∆CT) was close to 1, then the animal’s genotype was considered a wild-type, and if the value was close to 1.5, it was considered Ts65Dn.
Comparative genomic hybridization
To study the duplicated region in the Ts1Cje and Ts65Dn models, a comparative genomic hybridization (CGH) was undertaken on the whole genome using NimbleGen mouse HD2 oligonucleotide arrays. Comparative analysis was done using DNA extracts from one wild-type animal that were fluorescently labeled with Cy5 and from one animal bearing the duplication labeled with Cy3. After sonication and labeling, DNA was hybridized to the CGH array, followed by washing the slide according to the manufacturer’s instructions (Roche NimbleGen, Madison, WI, USA). Slides were scanned using a G2565 scanner at 3-μm resolution (Agilent Technologies, Palo Alto, CA, USA), and array images were analyzed using NimbleScan v2.5 software (Roche NimbleGen), with default parameters incorporating spatial correction. Arrays include 2,100,000 isothermal probes 50–75 bp in length with a median spacing of 1.1 kb throughout the genome. All the base pair coordinates mentioned here are indicated and refer to the UCSC NCBI37/mm9/July 2007 assembly.
Total RNA extraction
For Affymetrix arrays, hearts were isolated from Ts65Dn and wild-type mice (n = 5 per group) at 5 months old and flash frozen. Total RNA was prepared using Trizol (Invitrogen, Cergy Pontoise, France) according to the manufacturer’s instructions. Sample quality was checked using an Agilent 2100 Bioanalyzer (Agilent Technologies, Massy, France).
Whole-genome expression arrays
Biotinylated cDNAs were prepared from the total RNAs previously mentioned and hybridized onto GeneChip Mouse GENE 1.0ST arrays (Affymetrix, Santa Clara, CA, USA). Chips were washed and scanned on the Affymetrix Complete GeneChip instrument system generating digitized image data files. Raw data were processed with the Robust Multiarray Average (RMA) algorithm developed by Irizarry et al. (2003), and values were log transformed using Partek (Partek Inc., St. Louis, MO) and GeneSpring software (Agilent Technologies, Massy, France).
Results
Refining the Ts1Cje translocation break reveals a truncation at the level of Dnahc11 on Mmu12
The trisomy of the Ts1Cje mouse line is the result of a reciprocal translocation between Mmu12 and Mmu16 (Huang et al. 1997; Sago et al. 1998). Thus, Ts1Cje animals carry an abnormal chromosome composed of the centromeric part of chromosome 12, fused to the telomeric fragment of chromosome 16. CGH experiments allowed us to locate the translocation breakpoint on chromosome 16 upstream of Sod1 and downstream of Tiam1 and between probes located at positions 90,183,499 and 90,194,499 (NCBI37/mm9; Fig. 1). Further analysis also reveals a small deletion of the telomeric part of chromosome 12, with a translocation breakpoint between positions 119,278,499 and 119,289,499, in the Dnahc11 gene, between exons 35 and 41. Therefore, approximately half of the gene is truncated in Ts1Cje, leading to a monosomy that encompasses all the genes located downstream, toward the telomeric end of Mmu12.

Representation of CGH profile in Ts1Cje and Ts65Dn mice. Plots are log2 transformations of the hybridization ratios of transgenic versus wild-type mouse DNA. The red rectangle indicates the area containing the breakpoint, with different enlargement. The location of the genes inside the interval is indicated at the bottom of the figure. a Results on Ts1Cje Mmu12 confirm the loss of one copy of the telomeric part, with a breakpoint at the Dnahc11 gene. b Results on Ts1Cje Mmu16 confirm a gain of copy, with a breakpoint just before the Sod1 gene
CGH analysis of the Ts65Dn mice highlights the duplication of the centromeric part of the Mmu17
The Ts65Dn mouse line also results from a reciprocal chromosomal translocation (Davisson et al. 1993). The animals carry a supernumerary chromosome made up of the centromeric part of Mmu17 to the end of which hangs up the telomeric part of Mmu16. For this line, CGH experiments revealed the translocation breakpoint on chromosome 16, in a gene-desert region between Ncam2 and Mrlp39, between the base pairs 84,353,499 and 84,364,499 (Fig. 2). The translocation breakpoint area on chromosome 17 localized between probes located at positions 9,421,499 and 9,432,499, resulting in a duplication of the centromeric part of Mmu17, encompassing Pde10a, up to 6530411M01Rik, a noncoding RNA gene, with 60 genes listed in the different databases, including Ensembl, MGI, and UCSC and in mm9 Refseq (Pruitt et al. 2003).

a Results on Ts65Dn Mmu17 confirm a gain of copy of centromeric sequences, with a breakpoint downstream of Pde10a. b Results on Ts65Dn Mmu16 confirm a gain of copy of a telomeric region, with a breakpoint proximal to the Mrlp39 gene
Cloning of the Ts65Dn breakpoint
To map the translocation breakpoint more precisely, we performed targeted studies in a window of 11,000 bp around the putative breakpoint as indicated by array CGH. As rearrangements are possible in the neighborhood of the breakpoint, 10 PCR primers were designed surrounding the interval, with five primers upstream (Up1–5) on Mmu17 and five primers downstream (Dw1–5) on Mmu16 (Fig. 3a), and used in different pairwise combinations. On chromosome 17, primers Up1, Up2, and Up3 amplified PCR fragments with primers Dw6 and Dw5, localizing the breakpoint between these markers. On the other hand, no amplification was obtained with primers Up4 and Up5 (Fig. 3b). The sequencing of the PCR fragments confirmed the localization of the breakpoint between primers Up3 and Up4, at position 9,426,821 on chromosome 17 and 84,351,351 on chromosome 16 (NCBI37/mm9; Fig. 4).

Localisation of PCR primers for breakpoint sequencing on Mmu17 and Mmu16. a Black lines symbolize the sequence, gray rectangles correspond to the probe array with their location (UCSC NCBI37/mm9/July 2007 assembly), and between brackets is the result of the Cy3/Cy5 ratio in terms of copy number. Full triangles represent the PCR primers. b Electrophoresis of PCR products obtained with different PCR primers on the Mmu17 (primer Up) and Mmu16 (primer Dw). T trisomic DNA; w wild-type DNA. M1 is GeneRuler™ DNA ladder mix (Jena Biosience, Jena, Germany) and M2 is Lambda/HindIII DNA

Genomic composition of the Ts65Dn minichromosome. a An exhaustive list of genes located on the Mmu1716 Ts65Dn according to RefSeq, Ensembl, MGI, and UCSC databases. b Sequence of the Ts65Dn breakpoint. Red and black characters represent Mmu17 and Mmu16 sequences, respectively. The breakpoint is indicated by a slash. Underlined text shows the primer sequences used in Ts65Dn PCR genotyping protocol. c PCR genotyping of the Ts65Dn mice. The forward primer Fw_wt/Ts65Dn is used to amplify both the amplicon specific for the transgenic chromosome (with primer reverse RevTs65Dn) and the wt amplicon, with a reverse primer located on Mmu17 (GGGCAACACTGGATCAATC). After electrophoresis, two bands (396 and 290 bp) are detected by PCR, with DNA isolated from the Ts65Dn animals (Ts) and only one control band (290 bp) from the wild-type animals (wt)
Developing a robust PCR protocol for Ts65Dn genotyping
With the translocation breakpoint identified, we designed a new genotyping PCR protocol. Two primers were selected on both sides of the breakpoint to amplify a fragment of 396 bp (with forward primer Fw_wtTs65Dn: GACTTAGTAAGAGCAAGTGGC and reverse primer Rev_Ts65Dn: AGGTAGAAAGATGTGAGGACAC), and a third primer was designed on the reverse strand of chromosome 17 to amplify a fragment of 290 bp (GGGCAACACTGGATCAATC). After multiplex PCR with these three primers, the wild-type (wt) individuals showed amplification of one band corresponding to Mmu17 (290 bp) and the Ts65Dn individuals showed the amplification of two bands (290 bp) and one additional band specific of the translocated Mmu1716 Ts65Dn chromosome (396 bp; Fig. 4c).
Using this new method, we have genotyped 166 individuals and compared the results with the qPCR method. After the first qPCR run, the genotype was not determined for 5.4% of samples, mostly because of intermediate, ambiguous ∆∆Ct values between 0.25 and 0.35 (values of approximately 0 indicate a wt animal, while values of approximately 0.5 indicate a Ts65Dn animal). Thus, these genotypes had to be checked again using qPCR. If the second estimated value was still between 0.25 and 0.35, the genotype was not determined and the animal was discarded. With the PCR protocol, the genotype was determined for all the animals in only one step, with 100% concordance with the final result generated by qPCR, and no ambiguous genotypes.
Overexpression of trisomic Mmu17 genes in the Ts65Dn mice
To determine if the presence of additional copies of Mmu17 in the Ts65Dn have an impact on gene expression levels, we performed microarray expression profiling on mRNA derived from heart tissue of Ts65Dn and wt mice. The results showed upregulation of some genes in this organ (Fig. 5). Among the 35 genes from the trisomic Mmu17 interval found on the Affymetrix array, 80% (28) were expressed in heart and 62.8% (22) showed overexpression in Ts65Dn versus wt mice (fold change >1.2) (Fig. 5). A similar percentage of genes and ratio of expression were observed for genes located on the Mmu16 trisomic region which are expressed in the heart (Table 1). Functional Annotation Clustering was performed using Database for Annotation, Visualization and Integrated Discovery (DAVID) Bioinformatics, with all genes present in three copies on Mmu17. This tool provides mainly typical batch annotation and gene-GO term enrichment analysis to highlight the most relevant Gene Ontology (GO) terms associated with a given genes list (Huang et al. 2009a, b). The result of this analysis contains four clusters (Table 2). The most enriched cluster concerns genes involved in protein localization and transport. The second significant cluster concern genes involved in membrane fraction, cytoskeleton, and nucleotide or ion binding.

Representation of the expression profile of Mmu17 triplicate genes on the Ts65Dn minichromosome. The centromeric genes appear overexpressed up to Pde10a, the last trisomic gene located upstream of the breakpoint on the Mmu17
Discussion
We report here how CGH experiments carried out on the Ts65Dn and the Ts1Cje DS mouse models reveal the chromosomal changes in these transgenic lines. We designed a new robust PCR genotyping protocol and explored further the effect of the newly described aneuploidy in the Ts65Dn on gene expression.
Our results confirm and more accurately locate the chromosomal breakpoint on the Mmu12 of the Ts1Cje, far upstream of the Sod1 gene on the Mmu16. The Ts1Cje mouse line carries a DS trisomy for 67 Mmu16 genes homologous to Hsa21 plus a monosomy of the 7 most telomeric genes on Mmu12 in a region homologous to Hsa7 as described previously (Laffaire et al. 2009). The deletion on Mmu12 will affect seven annotated genes located in the interval (Dnach11, Sp4, Sp8, Abcb5, Itgb8, Macc1, and Tmem196) but could also affect additional genes found upstream through position effects or removal of regulatory elements. Indeed, gene expression analyses had already shown that in the cerebellum of the Ts1Cje mouse, Sp4 was downregulated and that the neighboring Cdca7l gene, located upstream of Dnach11, had reduced expression, certainly as a consequence of change in the chromatin structure (Laffaire et al. 2009). This monosomic gene in the Ts1Cje might affect neuronal proliferation, maturation, or development of the nervous system. Indeed, the transcription factor Sp4 regulates dendritic patterning during cerebellar development (Ramos et al. 2007) and could reduce postnatal cell proliferation in the hippocampus (Zhou et al. 2007). Sp8, the ortholog of the Drosophila transcription factor buttonhead, is involved in the development of rostromedial forebrain and in the specification of neuronal subpopulations (Zembrzycki et al. 2007), whereas Itgβ8-null mice develop severe deficits in neurogenesis and neurovascular physiology (Mobley et al. 2009). Dnahc11 drives correct left–right (LR) determination (Supp et al. 1997) and whose mutation affects the hippocampal circuitry and induces impaired spatial learning and working memory (Goto et al. 2010).
We also describe in more detail the translocated chromosome in the Ts65Dn mice, defining a series of 60 annotated genes nonhomologous to Hsa21 from a segment of about 10 Mb of the Mmu17 which are present in three copies in this mouse line. An exhaustive list of genes was determined using the four databases (MGI www.jax, UCSC, Ensembl, and RefSeq/MM9) and is given in Fig. 4. Interestingly, a few genes were found in only one or two databases, and Tcp10a and Tcp10b were included in the segment in UCSC and not in MGI or in Ensembl. Eight annotations corresponded to predicted genes of which four have homologous counterpart in the human genome. The segment encompasses a region homologous to Hsa6 but its sequence and synteny has been partly altered compare to human. Expression assays showed increased expression of these genes in the Ts65Dn heart, which by GO analysis are involved in several pathways. It is difficult to evaluate the impact on the observed Ts65Dn phenotypes. Indeed, some of the Mmu17 trisomic genes could interfere with various processes altered in the Ts65Dn. Tiam2 is a gene closely related to its homologous Tiam1, which maps to chromosome 21. Both Tiam1 and Tiam2 have important functions in neurite outgrowth, development and remodeling of synaptic connections (Terawaki et al. 2010). As the function of Tiam2 is similar to that of Tiam1, Ts65Dn mice might overamplify phenotypes due to increased copy of both genes. Similarly, synaptojanin 2 (Synj2) is located in the Mmu17 interval and its closest family member, Synj1, is found in the Mmu16 homologous region. Overexpression of Synj1 is known to modify the metabolism of PtdIns(4,5)P2 in the brain and could contribute to brain dysfunction and cognitive disabilities in DS (Voronov et al. 2008). An isoform of Synj2 was shown to be expressed predominantly in nerve terminals and colocalized with Synj1. Moreover, there is evidence that Synj1 and Synj2 have similar biochemical and protein interactions (Nemoto et al. 2001). Because Synj2 is duplicated in the Ts65Dn mice, the impact of Synj function could be amplified. Indeed, as Dyrk1A, a protein kinase, is known to inhibit synaptojanin 1 by phosphorylation, the additional copy of Synj2 may compensate the Dyrk1a effect (Adayev et al. 2006), which is also located on Hsa21 and overexpressed in Ts65Dn mice. Similarly, Istn1, which interacts directly with synaptojanin 1 (Hussain et al. 2001), or Dscr1, which mediates inhibition of calcineurin, normally stimulates synaptojanin 1 during nerve terminal depolarization by dephosphorylation, and their interaction could be altered by the overexpression of Synj2 (Lee et al. 2004; Rothermel et al. 2003).
Several authors also compared the results obtained on these two models to evaluate the contribution of genotype to the DS phenotype. Now they will have to consider that the two models have clear differences, not only with their control littermate but also due to additional modification of the translocated chromosome. Ts1Cje mice do not carry a triplication of the chromosome 17 centromeric area, and, conversely, Ts65Dn mice do not have a deletion of the telomeric chromosome 12 region. These differences may also have an impact on the observed phenotype differences. It would be interesting to carry out a similar study on the Rb(12.Ts171665Dn)2Cje (Ts2Cje) (Villar et al. 2005), an alternative model of Ts65Dn. This would allow a comparison of the genomic composition of this model relative to Ts65Dn, specifically if the part of Mmu17 is identical between the two and if the translocation involves losses of material as in the Ts1Cje model.
Knowledge of the sequence of the translocation breakpoint allowed the design of a robust, faster, and cheaper PCR genotyping protocol, providing simpler access to this mouse line for laboratories not equipped with qPCR equipment, which was a limiting factor. Thus, research aiming at dissecting DS mechanisms must take into account advantages and disadvantages of the various existing models. Taking into account the mapping of the breakpoint, there are several ways to confirm results obtained with the Ts65Dn and Ts1Cje. Indeed, one can compare consequences of larger segmental duplication mouse models like Ts1Yu (Li et al. 2007) by reducing gene dosage using a specific knockout of gene of interest (Hill et al. 2009; Salehi et al. 2006) or by making a rescue with a monosomic model (Duchon et al. 2011; Olson et al. 2004). In such a rescue experiment, one might still consider multiple-gene interactions with residual trisomic Mmu17 centromeric or monosomic Mmu12 telomeric genes in the Ts65Dn and Ts1Cje, respectively.
Additional DS mouse models have been developed over the last few years using chromosomal engineering and alternative methods (Brault et al. 2007; Duchon et al. 2008). These new models carry a well-defined segmental duplication without additional chromosomal rearrangement. Nevertheless, these models do not possess, like the Ts65Dn model and human pathology, a free segregating chromosome. The impact of such an independent minichromosome on cellular division (proliferation) and on the organization of the nuclear chromatin (gene expression) should not be minimized. Thus, a new model such as the Tc1 mouse model, with a supernumerary chromosome without extra rearrangement (O’Doherty et al. 2005), would certainly help us understand further the DS physiopathology.
References
Adayev T, Chen-Hwang MC, Murakami N, Wang R, Hwang YW (2006) MNB/DYRK1A phosphorylation regulates the interactions of synaptojanin 1 with endocytic accessory proteins. Biochem Biophys Res Commun 351:1060–1065
Belichenko PV, Masliah E, Kleschevnikov AM, Villar AJ, Epstein CJ, Salehi A, Mobley WC (2004) Synaptic structural abnormalities in the Ts65Dn mouse model of Down syndrome. J Comp Neurol 480:281–298
Belichenko PV, Kleschevnikov AM, Masliah E, Wu CB, Takimoto-Kimura R, Salehi A, Mobley WC (2009) Excitatory-inhibitory relationship in the fascia dentata in the Ts65Dn mouse model of Down syndrome. J Comp Neurol 512:453–466
Braudeau J, Delatour B, Duchon A, Lopes-Pereira P, Dauphinot L, de Chaumont F, Olivo-Marin JC, Dodd RH, Herault Y, Potier MC (2011) Specific targeting of the GABA-A receptor α5 subtype by a selective inverse agonist restores cognitive deficits in Down syndrome mice. J Psychopharmacol 25:1030–1042
Brault V, Pereira P, Duchon A, Hérault Y (2006) Modeling chromosomes in mouse to explore the function of genes, genomic disorders, and chromosomal organization. PLoS Genet 2:e86
Brault V, Besson V, Magnol L, Duchon A, Hérault Y (2007) Cre/loxP-mediated chromosome engineering of the mouse genome. Handb Exp Pharmacol (178):29–48
Christianson A, Howson C, Modell B (2006) March of dimes global report on birth defects: the hidden toll of dying and disabled children. March of Dimes Birth Defects Foundation, White Plains, NY, p 29
Contestabile A, Benfenati F, Gasparini L (2010) Communication breaks-Down: from neurodevelopment defects to cognitive disabilities in Down syndrome. Prog Neurobiol 91:1–22
Costa ACS, Grybko MJ (2005) Deficits in hippocampal CA1 LTP induced by TBS but not HFS in the Ts65Dn mouse: a model of Down syndrome. Neurosci Lett 382:317–322
Costa ACS, Scott-McKean JJ, Stasko MR (2008) Acute injections of the NMDA receptor antagonist memantine rescue performance deficits of the Ts65Dn mouse model of Down syndrome on a fear conditioning test. Neuropsychopharmacology 33:1624–1632
Davisson MT, Schmidt C, Akeson EC (1990) Segmental trisomy of murine chromosome 16: a new model system for studying Down syndrome. Prog Clin Biol Res 360:263–280
Davisson MT, Schmidt C, Reeves RH, Irving NG, Akeson EC, Harris BS, Bronson RT (1993) Segmental trisomy as a mouse model for Down syndrome. Prog Clin Biol Res 384:117–133
Dierssen M, Herault Y, Estivill X (2009) Aneuploidy: from a physiological mechanism of variance to Down syndrome. Physiol Rev 89:887–920
Duchon A, Besson V, Pereira PL, Magnol L, Hérault Y (2008) Inducing segmental aneuploid mosaicism in the mouse through targeted asymmetric sister chromatid event of recombination. Genetics 180:51–59
Duchon A, Pothion S, Brault V, Sharp AJ, Tybulewicz VLJ, Fisher EMC, Herault Y (2011) The telomeric part of the human chromosome 21 from Cstb to Prmt2 is not necessary for the locomotor and short-term memory deficits observed in the Tc1 mouse model of Down syndrome. Behav Brain Res 217:271–281
Fernandez F, Morishita W, Zuniga E, Nguyen J, Blank M, Malenka RC, Garner CC (2007) Pharmacotherapy for cognitive impairment in a mouse model of Down syndrome. Nat Neurosci 10:411–413
Goto K, Kurashima R, Gokan H, Inoue N, Ito I, Watanabe S (2010) Left-right asymmetry defect in the hippocampal circuitry impairs spatial learning and working memory in IV mice. PLoS One 5(11):e15468
Hill CA, Sussan TE, Reeves RH, Richtsmeier JT (2009) Complex contributions of Ets2 to craniofacial and thymus phenotypes of trisomic “Down syndrome” mice. Am J Med Genet A 149A:2158–2165
Hoelter SM, Dalke C, Kallnik M, Becker L, Horsch M, Schrewe A, Favor J, Klopstock T, Beckers J, Ivandic B, Gailus-Durner V, Fuchs H, de Angelis MH, Graw J, Wurst W (2008) “Sighted C3H” mice: a tool for analysing the influence of vision on mouse behaviour? Front Biosci 13:5810–5823
Huang da W, Sherman BT, Lempicki RA (2009a) Bioinformatics enrichment tools: paths toward the comprehensive functional analysis of large gene lists. Nucleic Acids Res 37:1–13
Huang da W, Sherman BT, Lempicki RA (2009b) Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc 4:44–57
Huang TT, Yasunami M, Carlson EJ, Gillespie AM, Reaume AG, Hoffman EK, Chan PH, Scott RW, Epstein CJ (1997) Superoxide-mediated cytotoxicity in superoxide dismutase-deficient fetal fibroblasts. Arch Biochem Biophys 344:424–432
Huret JL, Sinet PM (2000) Trisomie 21. Atlas of Genetics and Cytogenetics in Oncology and Haematology. Available at http://AtlasGeneticsOncology.org/Educ/PolyTri21Fr.html
Hussain NK, Jenna S, Glogauer M, Quinn CC, Wasiak S, Guipponi M, Antonarakis SE, Kay BK, Stossel TP, Lamarche-Vane N, McPherson PS (2001) Endocytic protein intersectin-I regulates actin assembly via Cdc42 and N-WASP. Nat Cell Biol 3:927–932
Irizarry RA, Hobbs B, Collin F, Beazer-Barclay YD, Antonellis KJ, Scherf U, Speed TP (2003) Exploration, normalization, and summaries of high density oligonucleotide array probe level data. Biostatistics 4:249–264
Kleschevnikov AM, Belichenko PV, Villar AJ, Epstein CJ, Malenka RC, Mobley WC (2004) Hippocampal long-term potentiation suppressed by increased inhibition in the Ts65Dn mouse, a genetic model of Down syndrome. J Neurosci 24:8153–8160
Laffaire J, Rivals I, Dauphinot L, Pasteau F, Wehrle R, Larrat B, Vitalis T, Moldrich RX, Rossier J, Sinkus R, Herault Y, Dusart I, Potier MC (2009) Gene expression signature of cerebellar hypoplasia in a mouse model of Down syndrome during postnatal development. BMC Genomics 10:15
Lee SY, Wenk MR, Kim Y, Naim AC, De Camilli P (2004) Regulation of synaptojanin 1 by cyclin-dependent kinase 5 at synapses. Proc Natl Acad Sci USA 101:546–551
Li Z, Yu T, Morishima M, Pao A, LaDuca J, Conroy J, Nowak N, Matsui S-I, Shiraishi I, Yu YE (2007) Duplication of the entire 22.9 Mb human chromosome 21 syntenic region on mouse chromosome 16 causes cardiovascular and gastrointestinal abnormalities. Hum Mol Genet 16:1359–1366
Liu DP, Schmidt C, Billings T, Davisson MT (2003) Quantitative PCR genotyping assay for the Ts65Dn mouse model of Down syndrome. Biotechniques 35:1170–1174, 1176
Lorenzi H, Duvall N, Cherry SM, Reeves RH, Roper RJ (2010) PCR prescreen for genotyping the Ts65Dn mouse model of Down syndrome. Biotechniques 48:35–37
Mobley AK, Tchaicha JH, Shin J, Hossain MG, McCarty JH (2009) Beta 8 integrin regulates neurogenesis and neurovascular homeostasis in the adult brain. J Cell Sci 122:1842–1851
Moldrich RX, Dauphinot L, Laffaire J, Rossier J, Potier MC (2007) Down syndrome gene dosage imbalance on cerebellum development. Prog Neurobiol 82:87–94
Moran TH, Capone GT, Knipp S, Davisson MT, Reeves RH, Gearhart JD (2002) The effects of piracetam on cognitive performance in a mouse model of Down’s syndrome. Physiol Behav 77:403–409
Nemoto Y, Wenk MR, Watanabe M, Daniell L, Murakami T, Ringstad N, Yamada H, Takei K, De Camilli P (2001) Identification and characterization of a synaptojanin 2 splice isoform predominantly expressed in nerve terminals. J Biol Chem 276:41133–41142
O’Doherty A, Ruf S, Mulligan C, Hildreth V, Errington ML, Cooke S, Sesay A, Modino S, Vanes L, Hernandez D, Linehan JM, Sharpe PT, Brandner S, Bliss TVP, Henderson DJ, Nizetic D, Tybulewicz VLJ, Fisher EMC (2005) An aneuploid mouse strain carrying human chromosome 21 with Down syndrome phenotypes. Science 309:2033–2037
Olson LE, Richtsmeier JT, Leszl J, Reeves RH (2004) A chromosome 21 critical region does not cause specific Down syndrome phenotypes. Science 306:687–690
Pereira PL, Magnol L, Sahún I, Brault V, Duchon A, Prandini P, Gruart A, Bizot JC, Chadefaux-Vekemans B, Deutsch S, Trovero F, Delgado-García JM, Antonarakis SE, Dierssen M, Herault Y (2009) A new mouse model for the trisomy of the Abcg1–U2af1 region reveals the complexity of the combinatorial genetic code of down syndrome. Hum Mol Genet 18:4756–4769
Popov VI, Kleschevnikov AM, Klimenko OA, Stewart MG, Belichenko PV (2011) Three-dimensional synaptic ultrastructure in the dentate gyrus and hippocampal area CA3 in the Ts65Dn mouse model of Down syndrome. J Comp Neurol 519:1338–1354
Pruitt KD, Tatusova T, Maglott DR (2003) NCBI reference sequence project: update and current status. Nucleic Acids Res 31:34–37
Ramos B, Gaudilliere B, Bonni A, Gill G (2007) Transcription factor Sp4 regulates dendritic patterning during cerebellar maturation. Proc Natl Acad Sci USA 104:9882–9887
Reeves RH, Irving NG, Moran TH, Wohn A, Kitt C, Sisodia SS, Schmidt C, Bronson RT, Davisson MT (1995) A mouse model for Down syndrome exhibits learning and behaviour deficits. Nat Genet 11:177–184
Roper RJ, Baxter LL, Saran NG, Klinedinst DK, Beachy PA, Reeves RH (2006) Defective cerebellar response to mitogenic Hedgehog signaling in Down [corrected] syndrome mice. Proc Natl Acad Sci USA 103:1452–1456
Rothermel BA, Vega RB, Williams RS (2003) The role of modulatory calcineurin-interacting proteins in calcineurin signaling. Trends Cardiovasc Med 13:15–21
Rueda N, Florez J, Martinez-Cue C (2008) Chronic pentylenetetrazole but not donepezil treatment rescues spatial cognition in Ts65Dn mice, a model for Down syndrome. Neurosci Lett 433:22–27
Sago H, Carlson EJ, Smith DJ, Kilbridge J, Rubin EM, Mobley WC, Epstein CJ, Huang TT (1998) Ts1Cje, a partial trisomy 16 mouse model for Down syndrome, exhibits learning and behavioral abnormalities. Proc Natl Acad Sci USA 95:6256–6261
Salehi A, Delcroix JD, Belichenko PV, Zhan K, Wu CB, Valletta JS, Takimoto-Kimura R, Kleschevnikov AM, Sambamurti K, Chung PP, Xia WM, Villar A, Campbell WA, Kulnane LS, Nixon RA, Lamb BT, Epstein CJ, Stokin GB, Goldstein LSB, Mobley WC (2006) Increased App expression in a mouse model of Down’s syndrome disrupts NGF transport and causes cholinergic neuron degeneration. Neuron 51:29–42
Salehi A, Faizi M, Colas D, Valletta J, Laguna J, Takimoto-Kimura R, Kleschevnikov A, Wagner SL, Aisen P, Shamloo M, Mobley WC (2009) Restoration of norepinephrine-modulated contextual memory in a mouse model of Down syndrome. Sci Transl Med 1:7ra17
Supp DM, Witte DP, Potter SS, Brueckner M (1997) Mutation of an axonemal dynein affects left-right asymmetry in inversus viscerum mice. Nature 389:963–966
Terawaki S, Kitano K, Mori T, Zhai Y, Higuchi Y, Itoh N, Watanabe T, Kaibuchi K, Hakoshima T (2010) The PHCCEx domain of Tiam1/2 is a novel protein- and membrane-binding module. EMBO J 29:236–250
Villar AJ, Belichenko PV, Gillespie AM, Kozy HM, Mobley WC, Epstein CJ (2005) Identification and characterization of a new Down syndrome model, Ts Rb(12.17(16)) 2Cje, resulting from a spontaneous Robertsonian fusion between T(17(16))65Dn and mouse Chromosome 12. Mamm Genome 16:79–90
Voronov SV, Frere SG, Giovedi S, Pollina EA, Borel C, Zhang H, Schmidt C, Akeson EC, Wenk MR, Cimasoni L, Arancio O, Davisson MT, Antonarakis SE, Gardiner K, De Camilli P, Di Paolo G (2008) Synaptojanin 1-linked phosphoinositide dyshomeostasis and cognitive deficits in mouse models of Down’s syndrome. Proc Natl Acad Sci USA 105:9415–9420
Yu T, Li Z, Jia Z, Clapcote SJ, Liu C, Li S, Asrar S, Pao A, Chen R, Fan N, Carattini-Rivera S, Bechard AR, Spring S, Henkelman RM, Stoica G, Matsui S-I, Nowak NJ, Roder JC, Chen C, Bradley A, Yu YE (2010a) A mouse model of Down syndrome trisomic for all human chromosome 21 syntenic regions. Hum Mol Genet 19:2780–2791
Yu T, Liu CH, Belichenko P, Clapcote SJ, Li SM, Pao AN, Kleschevnikov A, Bechard AR, Asrar S, Chen RQ, Fan N, Zhou ZY, Jia ZP, Chen C, Roder JC, Liu B, Baldini A, Mobley WC, Yu YE (2010b) Effects of individual segmental trisomies of human chromosome 21 syntenic regions on hippocampal long-term potentiation and cognitive behaviors in mice. Brain Res 1366:162–171
Zembrzycki A, Griesel G, Stoykova A, Mansouri A (2007) Genetic interplay between the transcription factors Sp8 and Emx2 in the patterning of the forebrain. Neural Dev 2:8
Zhou X, Qyang Y, Kelsoe JR, Masliah E, Geyer MA (2007) Impaired postnatal development of hippocampal dentate gyrus in Sp4 null mutant mice. Genes Brain Behav 6:269–276
Acknowledgments
We thank members of the research group, of the IGBMC laboratory, of the ICS, and of the AnEUploidy consortium (www.aneuploidy.org), particularly Veronique Brault and Emilie Velot for their helpful comments. We are grateful to the caretakers of the ICS and to the people of the transcriptomic platform at the IGBMC for their help. We thank the National Centre for Scientific Research, the INSERM, the University of Strasbourg, and the European commission with the AnEUploidy project (LSHG-CT-2006-037627) for support.
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License (https://creativecommons.org/licenses/by-nc/2.0), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
Duchon, A., Raveau, M., Chevalier, C. et al. Identification of the translocation breakpoints in the Ts65Dn and Ts1Cje mouse lines: relevance for modeling down syndrome. Mamm Genome 22, 674–684 (2011). https://doi.org/10.1007/s00335-011-9356-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00335-011-9356-0