
Abstract
The clinical relevance of fungal infections has increased dramatically in recent decades as a consequence of the rise of immunocompromised populations, and efforts to understand the underlying mechanisms of protective immunity have attracted renewed interest. Here we review Dectin-1, a pattern recognition receptor involved in antifungal immunity, and discuss recent discoveries of polymorphisms in the gene encoding this receptor which result in human disease.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
The immune system of mammals and other vertebrates is characterized by two interrelated components, the innate and adaptive responses, both of which are required for the resolution of most infections. The innate immune system comprises cells such as macrophages, neutrophils, and dendritic cells, which are broadly distributed throughout the body, including at portals of pathogen entry, and are involved in the initial capture and presentation of microbial antigens. The field of innate immunology was revolutionized by Charlie Janeway Jr. with the theory of pattern recognition, which proposed that conserved structures of infectious organisms (the pathogen associated molecular patterns or PAMPs) were recognized by the immune system through a set of specialized germline-encoded receptors (the pattern recognition receptors or PRRs). Janeway’s theory not only suggested a general principle of innate immune recognition, it also provided a foundation for the current understanding of how it connects with the adaptive immune system (Medzhitov 2009). Accordingly, the field of immunology has witnessed the discovery of many different PRRs over the last few years, some of which can induce cellular responses and initiate the adaptive arm of the immune system. The adaptive response involves specific recognition of microbial antigens by lymphocyte receptors resulting in clonal expansion, cellular differentiation, the production of specific antibodies, and the development of immunological memory. Here we review recent insights regarding the innate recognition and response to fungal infections. We focus in particular on Dectin-1, a PRR that we and others have shown is important in antifungal defense in humans and mice. However, although beyond the scope of this review, the reader is reminded that there are also several polymorphisms in other genes (such as MBL and STAT3, for example) that have been linked with susceptibility to fungal infections (for reviews see Antachopoulos et al. 2007; Carvalho et al. 2010).
Pattern recognition receptors in cellular responses against fungal infections
PRRs can be broadly categorized based on their cellular expression—on the cell surface, in intracellular compartments, or secreted into the serum or tissue fluids; these groups can be further subdivided into families based on structure and function. PRRs account for the quick responses of innate immunity because once they recognize a PAMP, the PRR-expressing effector cells function immediately rather than after they have undergone somatic recombination and proliferation, as occurs in adaptive immunity. PRRs activate effector cells by driving key cellular functions, and the most important and widely studied PRRs in terms of fungal recognition are Toll-like receptors (TLRs) and C-type lectin receptors (CLRs).
Toll-like receptors
The TLR proteins are characterized by extracellular leucine-rich repeat motifs that are involved in ligand recognition. They also possess a cytoplasmic Toll/interleukin (IL-1) receptor (TIR) domain, which mediates intracellular signaling. Toll was first discovered in Drosophila melanogaster as a cell surface receptor involved in the regulation of development and subsequently shown to function in immunity (Hashimoto et al. 1988; Lemaitre et al. 1996). To date, 10 and 12 TLRs have been identified in humans and mice, respectively. Most TLRs have now been demonstrated as having PAMP “ligands,” including lipoproteins, lipids, proteins, and nucleic acids derived from a wide variety of microorganisms (Akira et al. 2006). Ligand recognition by TLR homo- or heterodimers leads to the recruitment of intracellular adaptors such as MyD88, Mal (also called TIRAP), TRIF, and TRAM. This activates signaling cascades that ultimately trigger transcription factors and induce TLR-specific patterns of gene expression and the production of various cytokines and chemokines.
A role for TLRs in antifungal defense was first suggested by the increased susceptibility of Toll-deficient Drosophila to infection with Aspergillus fumigatus (Lemaitre et al. 1996). It has since been shown that mice lacking the TLR adaptor MyD88 are highly susceptible to fungal infections, including Candida albicans, Aspergillus fumigatus, and Cryptococcus neoformans (Netea et al. 2006). The precise fungal ligands detected by TLRs are not very well characterized; however, it is known that phospholipomannans are recognized by TLR2 and glucuronoxylomannans and mannans are recognized by TLR4 (Netea et al. 2008).
It transpires that the involvement of individual TLRs in antifungal immunity is not as clear-cut as with the MyD88 adaptor, as conflicting results have been reported by different research groups. For example, initial studies suggested that TLR4-deficient mice were more susceptible to disseminated candidiasis. Other studies using models of intragastric infection or intravenous reinfection also showed that TLR4−/− mice were more susceptible to C. albicans (Bellocchio et al. 2004). In contrast, TLR4−/− mice were equally susceptible as wild-type mice in models of intravenous infection with C. albicans yeast (Murciano et al. 2006), and even survived longer in a model of intravenous infection with C. albicans hyphae (Bellocchio et al. 2004). A recent investigation that used a panel of different C. albicans strains suggests that TLR4 is important for the recognition of some, but not all, strains of C. albicans (Netea et al. 2010), and this variability in recognition is likely to account for the differences observed in the above studies. TLR2, TLR9, TLR1, and TLR6 have also been implicated in the recognition and response to fungal pathogens (Carvalho et al. 2010; van de Veerdonk et al. 2008). Although the precise contributions of individual TLRs is still unclear, an integrated model of fungal recognition involving TLRs and C-type lectins has been proposed and highlights the complex interaction that occurs between host PRRs and invading pathogens (Netea et al. 2008).
There are associations between genetic variations in TLRs and fungal diseases in humans (for review see Carvalho et al. 2010). For example, polymorphisms in TLR4 have been associated with susceptibility to invasive aspergillosis in certain transplant recipients and chronic cavitary pulmonary aspergillosis. Furthermore, TLR4 polymorphisms were shown to contribute to a higher risk of systemic Candida infection. An association between a TLR9 polymorphism and the development of allergic bronchopulmonary aspergillosis has also been reported. A global role of TLRs in host defense against fungal infections in humans, however, has been questioned by clinical observations that patients with a defect in MyD88 are not any more susceptible to fungal infections than the normal population (von Bernuth et al. 2008).
C-type lectin receptors
A noteworthy discovery in the field of innate immunology was the identification of C-type lectin receptors (CLRs) with the ability to induce intracellular signaling. These receptors have been divided into 17 groups, but of relevance here are the myeloid-expressed CLRs belonging to the Group II, V, and VI subgroups (Zelensky and Gready 2005). A number of these receptors, namely, DC-SIGN, the mannose receptor, Dectin-1, and Dectin-2, function in the recognition of fungal pathogens and have been reviewed elsewhere (Willment and Brown 2008). Of these receptors, Dectin-1 is the most extensively studied in terms of a role in fungal disease and is the focus of the remainder of this review.
Dectin-1
Structure and expression
Dectin-1 consists of a single extracellular C-type lectin-like domain (CTLD), a transmembrane region, and a cytoplasmic tail that contains a single tyrosine-based activation motif (Fig. 1). In both humans and mice, alternative splicing generates two major Dectin-1 isoforms and a number of minor isoforms (Heinsbroek et al. 2006; Willment et al. 2001). The two major isoforms, which are the only isoforms functional for β-glucan binding, differ with regard to the presence or absence of a stalk region and their ability to bind and induce cellular responses (Heinsbroek et al. 2006).

A schematic representation of the structure of Dectin-1
Originally thought to be a dendritic cell-specific receptor, Dectin-1 is now known to be expressed by many other cell types such as monocytes, macrophages, neutrophils, and a subset of T cells (Taylor et al. 2002). Dectin-1 (βGR) is also expressed on B cells, eosinophils, and mast cells in humans, and recent reports have also demonstrated expression of this receptor on murine microglia (Olynych et al. 2006; Shah et al. 2008; Willment et al. 2005). Whether the differences in expression between species are functionally significant is currently unclear. Consistent with a potential role in immune surveillance, this receptor is prominently expressed at portals of pathogen entry such as the lung and intestine (Reid et al. 2004; Taylor et al. 2002). The levels of Dectin-1 expression can be significantly influenced by various factors such as steroids, some cytokines, and microbial stimuli. For example, IL-4, IL-13, and GM-CSF (granulocyte–macrophage colony-stimulating factor) cause significant upregulation of Dectin-1 expression. On the other hand, IL-10, LPS, and dexamethasone trigger downregulation of Dectin-1 expression (Willment et al. 2003). In addition, the systemic administration of C. albicans resulted in an increase in Dectin-1 expression on leukocytes. In contrast, Dectin-1 expression was decreased during polymicrobial sepsis (Ozment-Skelton et al. 2009).
Dectin-1 mediated fungal β-glucan recognition
A number of endogenous and exogenous ligands have been reported for Dectin-1, but this receptor is best known for its ability to recognize fungal β-glucans. β-Glucans are carbohydrate PAMPs found predominantly in fungal cell walls, but they are also present in plants and some bacteria. The β-glucan components of fungal cell walls consist primarily of (1→3)-β-D-linked polymer backbones with (1→6)-β-linked side chains of varying length and distribution (Romani et al. 2004; Tsoni and Brown 2008). These carbohydrates are well known for their anti-infective and antitumorogenic activities, and the identification of Dectin-1 as a PRR that recognizes β-glucans has enabled significant advances in our understanding of the mechanisms underlying these activities. A number of other receptors have also been implicated in β-glucan recognition, namely, complement receptor 3 (CR3), lactosylceramide, langerin, and the scavenger receptors CD5, CL-P1, SCARF1, and CD36 (de Jong et al. 2010; Jang et al. 2009; Means et al. 2009; Ross et al. 1987; Vera et al. 2009; Zimmerman et al. 1998). We and others have shown that Dectin-1 is the primary receptor for β-glucans, at least on leukocytes. However, it should be noted that the other receptors may have significant roles in mediating responses to β-glucans, particularly in nonimmune cells.
Initially identified as a receptor that recognized an unidentified ligand on T lymphocytes (Ariizumi et al. 2000), Dectin-1 was reidentified as a receptor for β-glucans following a screen of a murine macrophage cDNA expression library with zymosan, a β-glucan-rich extract of S. cerevisiae (Brown and Gordon 2001). Biochemical characterization of the interaction has shown that Dectin-1 is highly specific for (1→3)-linked β-glucans and that its binding affinity for these carbohydrates is influenced by factors such as polymer length and number of branches (Adams et al. 2008; Goodridge et al. 2009b; Palma et al. 2006). The manner in which Dectin-1 recognizes β-glucans is still unclear as its CTLD lacks the residues typically known for carbohydrate binding and ligand recognition is metal ion-independent. However, at least two residues flanking a shallow groove on the protein surface, Trp221 and His223, have been implicated for β-glucan binding (Adachi et al. 2004).
Dectin-1 has been shown to interact with a number of fungal species, including Candida, Pneumocystis, Saccharomyces, Aspergillus, Coccidioides, and Penicillum, by way of its β-glucan specificity (Brown et al. 2003; Gantner et al. 2005; Gersuk et al. 2006; Nakamura et al. 2008; Saijo et al. 2007; Steele et al. 2003, 2005; Taylor et al. 2007; Viriyakosol et al. 2005). It has also been shown that Dectin-1 can recognize other nonfungal pathogens. For instance, a role for this receptor has been suggested in the recognition of mycobacteria (Lee et al. 2009; Rothfuchs et al. 2007; Shin et al. 2008; Yadav and Schorey 2006), which do not possess β-glucans, suggesting that there may still be unidentified exogenous ligands of Dectin-1 yet to be discovered.
Cellular responses induced by Dectin-1 following fungal β-glucan recognition
Dectin-1 recognition of β-glucans can result in the induction of a number of cellular responses, including ligand uptake by phagocytosis and endocytosis, dendritic cell maturation, the respiratory burst, the production of arachidonic acid metabolites, and the induction of numerous cytokines, including tumor necrosis factor (TNF), IL-10, IL-2, IL-23, and IL-6, as well as chemokines like CXCL2 (Brown 2006; Reid et al. 2009). In addition to triggering innate immune responses, ligation of Dectin-1 can also activate adaptive immune responses. For example, Dectin-1-mediated activation of dendritic cells in response to curdlan, a selective agonist of this receptor, has been shown to direct the differentiation of T-helper 17 (Th17) and T-helper 1 (Th1) CD4+ T cells in vitro (LeibundGut-Landmann et al. 2007). Furthermore, curdlan acted as an adjuvant for the priming of Th17 and Th1 CD4+ T cells in vivo (LeibundGut-Landmann et al. 2007). Dectin-1 has also been implicated in the expansion and function of regulatory T cells (Tregs) (Dillon et al. 2006; Karumuthil-Melethil et al. 2008), as well as in the conversion of selected populations of Tregs into IL-17, producing T cells that cannot be strictly classified as either Th17 cells or Tregs (Osorio et al. 2008). In addition, Dectin-1 signaling can induce CD8+ T-cells responses, as demonstrated by in vivo studies that showed that curdlan was found to efficiently prime cytotoxic T-cell responses (Leibundgut-Landmann et al. 2008). Signaling induced via Dectin-1 alone can directly trigger some of these responses; however, other responses such as the production of proinflammatory cytokines require, or are enhanced by, collaborative signaling with TLRs (Brown et al. 2003; Dennehy et al. 2008; Gantner et al. 2003). Yet another element that adds to the complexity of Dectin-1 signaling was revealed by studies showing that the ability of β-glucans to directly trigger these responses is cell-type dependent (Goodridge et al. 2009a; Rosas et al. 2008). In the following section we review the current understanding of the Dectin-1-mediated signaling pathways that underlie some of these responses.
Dectin-1 intracellular signaling
Dectin-1 signaling is mediated by a sequence within the cytoplasmic tail that resembles an immunoreceptor tyrosine-based activation motif (ITAM). Traditional ITAMs present in T-cell receptors, B-cell receptors, and Fc receptors contain an amino acid sequence comprising duplicate YXXL/I motifs (YXXL/IX6-12YXXL/I), where Y is tyrosine, L is leucine, I is isoleucine, and X denotes any amino acid. Phosphorylation of both tyrosines by Src family kinases is required for recruitment of Syk family kinases and subsequent downstream signaling (Underhill and Goodridge 2007). The cytoplasmic tail of Dectin-1 also contains two tyrosines: the membrane-proximal tyrosine is located in a YXXL motif and the membrane-distal one is found in a motif containing an additional amino acid (YxxxL and YxxxI in humans and mice, respectively) (Brown 2006). Dectin-1 also becomes tyrosine phosphorylated following ligand binding, but in contrast to traditional ITAM receptors, phosphorylation of only the membrane-proximal tyrosine is required for Dectin-1 signaling (Fuller et al. 2007; Rogers et al. 2005). This single-tyrosine-based motif (YxxL) is now known as an ITAM-like motif or a hemITAM. Dectin-1 was the first receptor shown to signal via this pathway, which although quite similar to ITAM signaling, is unique in its dependence on only a single tyrosine. Other receptors, specifically the C-type lectins CLEC-2 and CLEC-9A, have subsequently been shown to also signal via an ITAM-like motif (Fuller et al. 2007; Huysamen et al. 2008). Although the exact nature of the interaction between Syk and Dectin-1 is unclear, a model suggesting that Syk bridges two monophosphorylated molecules has been proposed (Brown 2006; Fuller et al. 2007; Goodridge et al. 2009b; Rogers et al. 2005). Indeed, experimental evidence strongly suggests that the related ITAM-like-containing receptor CLEC-2 regulates Syk through dimerization, with each of the Syk SH2 domains binding to a phosphorylated YxxL sequence in the individual cytosolic tails of two CLEC-2 proteins (Hughes et al. 2010).
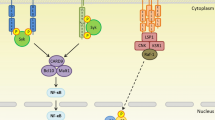
Ligand binding and the subsequent recruitment of Syk to Dectin-1 result in activation of PLCγ2, leading to the engagement of the caspase recruitment domain (CARD)-containing protein CARD9 (Gross et al. 2006; Tassi et al. 2009; Xu et al. 2009). CARD9, which then assembles with BCL10 and MALT1, has been identified as a key downstream adaptor required to link Dectin-1/Syk ligation to canonical NF-κB activation (Gross et al. 2006; Hara et al. 2007) (Fig. 2). Work by Gringhuis et al. (2009) also demonstrated Dectin-1 as the first PRR to induce noncanonical NF-κB activation. In addition, these authors showed that the stimulation of Dectin-1 with curdlan or C. albicans induced a second Syk-independent signaling pathway mediated by the serine-threonine kinase Raf-1. This pathway integrates with the Syk pathway at the point of NF-KB activation and might be involved in the pathogenesis of fungal infections (Gringhuis et al. 2009). Dectin-1 has also been shown to trigger NFAT activation, which regulates the induction of early growth response (Egr) family transcription factors Egr-2 and Egr-3 (Goodridge et al. 2007). Evidence also exists for Syk-dependent pathways that do not rely on CARD9 for cytokine production, e.g., activation of the MAP kinase ERK (Slack et al. 2007).

Signaling pathway induced by Dectin-1. Upon ligand binding, Dectin-1 becomes tyrosine-phosphorylated by Src kinases, thereby providing a docking site for Syk which initiates downstream signaling. The downstream signaling is effected by molecules such as CARD9, Bcl10, and MALT1, which lead to NF-κB activation and cytokine production. Dectin-1 can also activate NFAT and noncanonical NF-κB in a CARD9-Bcl10-MALT1-independent manner. Stimulation of Dectin-1 with β-glucans can also induce a second Syk-independent signaling pathway mediated by the serine-threonine kinase Raf-1
Dectin-1 in defense against fungal pathogens and effects of genetic polymorphisms/deficiency in mice and humans
Over the last few decades the growth of immunocompromised populations, such as individuals infected with HIV and transplant recipients, has resulted in an increased clinical relevance of fungal diseases. Thus, fungal research has attracted renewed interest, particularly efforts that seek to understand the mechanisms underlying protective immunity. In this section we discuss the role of Dectin-1 in antifungal immunity and highlight studies that demonstrate the consequences of Dectin-1 deficiency in fungal infections.
As mentioned previously, the recognition of fungal β-glucans by Dectin-1 results in a variety of cellular responses, some of which are host protective such as fungal uptake and killing, and the production of inflammatory cytokines and chemokines. Interestingly, Dectin-1 also induces the production of IL-10, an anti-inflammatory cytokine whose role during fungal infection is unclear. The presence of IL-10 has traditionally been thought of as disadvantageous to the host during fungal infection; however, recent investigations have led to the proposal that the inhibitory action of IL-10 on leukocyte activation may be important for limiting host damage during severe inflammation (Romani and Puccetti 2006). It has also been shown that IL-10 is involved in the development of Tregs, whose modulatory actions are beneficial in mucosal and cutaneous infections and which are known to be essential for the induction of long-term immunity to C. albicans (Netea et al. 2004). In contrast, it has been proposed that in situations such as disseminated candidiasis, where the pathogen penetrates the mucosa and disseminates through the bloodstream, the presence of Tregs is detrimental to the host (Netea et al. 2004).
We have also mentioned that Dectin-1 can drive other aspects of adaptive immunity, including Th1, Th17, and CTL responses, although there have been no studies looking specifically at the roles of these Dectin-1-mediated responses in antifungal immunity. Nevertheless, it is generally accepted that a Th1 response is required for protection against fungal infection in healthy hosts (Romani 2004). On the other hand, whether a Th17 response is required for efficient antifungal immunity is a more contentious issue, with contradictory reports demonstrating Th17 cells as both beneficial and detrimental to the host during systemic infections with Candida (Bozza et al. 2008; Huang et al. 2004; Romani 2004; Saijo et al. 2010; Zelante et al. 2007). In contrast, Th17 cells are known to be important during mucosal infections with Candida (Conti et al. 2009). The specific role of CTLs in direct antifungal responses is also undefined; however, it has been shown that CD8+ T cells are activated during fungal infections and can play a protective role in some cases (Leigh et al. 2006; Lindell et al. 2005; Marquis et al. 2006; Wuthrich et al. 2003).
Interestingly, Dectin-2, another Syk/CARD9-coupled CLR, has also been shown to mediate dendritic cell activation and induction of Th17 immunity in response to C. albicans (Robinson et al. 2009). Furthermore, in the final stages of writing this review, an investigation using Dectin-2-deficient mice reported that this receptor was important in host defense against systemic C. albicans in mice (Saijo et al. 2010).
It is important to note that fungal morphology is a major determinant in the detection of cell wall β-glucans, as the exposure of these carbohydrates can vary from one fungal morphotype to another. For example, it has been shown that Dectin-1 recognizes C. albicans yeast but not filamentous forms (Gantner et al. 2005). Thus, the ability of C. albicans to switch between different forms may be a key virulence strategy to evade host immune responses (Calderone and Fonzi 2001; Lo et al. 1997; Saville et al. 2003). In fact, C. albicans mutants that lack the ability to switch from yeast to filamentous forms have been reported to be avirulent in mouse models (Lo et al. 1997). In A. fumigatus, swollen conidia and early germlings displaying surface β-glucans are recognized by Dectin-1, hyphal forms are recognized to a lesser degree, and resting conidia are not recognized (Gersuk et al. 2006; Hohl et al. 2005; Steele et al. 2005; Torosantucci et al. 2005). It has also been suggested that fungal pathogens actively mask their β-glucans to avoid immune recognition by Dectin-1 (Gantner et al. 2005; Wheeler and Fink 2006), prompting investigations into drugs, such as caspofungin, which enhance the exposure of β-glucans and improve antifungal responses (Hohl et al. 2008; Lamaris et al. 2008; Wheeler and Fink 2006; Wheeler et al. 2008).
Increasing evidence from in vivo studies, although not entirely consistent, suggests an important role for Dectin-1 in antifungal immunity. Studies in our laboratory have shown that the deletion of Clec7a, the gene encoding Dectin-1, significantly increased susceptibility of mice to systemic infection with C. albicans (Taylor et al. 2007). Loss of Dectin-1 in this model resulted in increased fungal burdens and much lower survival times. A peritoneal infection model revealed that the Dectin-1 knockout mice had fewer recruited cells than wild types, including neutrophils and inflammatory monocytes, which correlated with defects in the production of cytokines and chemokines such as TNF, IL-6, CCL2, CCL3, and GM-CSF. This study suggested a fundamental role of β-glucan recognition by Dectin-1 in antifungal immunity and the requirement for Dectin-1-dependent signaling for the induction of protective immune responses (Taylor et al. 2007). In contrast to this study, Saijo et al. showed that Dectin-1-deficient mice were not more susceptible than wild-type mice to infection with C. albicans (Saijo et al. 2007). We suspect that the inconsistencies between these two studies arose as a result of the use of different fungal strains.
Nevertheless, other studies have supported a role for Dectin-1 in antifungal immunity. In a model of oral candidiasis, Dectin-1-deficient mice showed increased susceptibility, with enhanced dissemination and decreased survival times (Hise et al. 2009). Furthermore, mice deficient in the downstream signaling component CARD9 were similarly more susceptible to systemic C. albicans infection (Gross et al. 2006). Dectin-1 is also required during infection with other fungal pathogens; e.g., Dectin-1-deficient mice displayed increased susceptibility to A. fumigatus infection which correlated with impaired cytokine production and fungal killing (Steele et al. 2005; Werner et al. 2009). Furthermore, in a model of intranasal infection with Pneumocystis carinii, Dectin-1-deficient mice were found to be more susceptible than wild-type mice in the early stages of infection (Saijo et al. 2007).
Recently, an investigation by Ferwerda et al. provided definitive evidence of the role of Dectin-1 during fungal infections in humans. This study identified and described a polymorphism of human Dectin-1 in four members of a Dutch family who were affected by either recurrent vulvovaginal candidiasis or onychomycosis (fungal infection of the nail), or both (Ferwerda et al. 2009). The polymorphism was characterized by an early-stop-codon mutation (Y238X) in the CTLD of Dectin-1, resulting in defective expression and lack of β-glucan recognition by phagocytes. In addition, the mutation resulted in impaired production of cytokines, including IL-17. However, the uptake and killing of C. albicans by neutrophils was not affected. This suggests that alternative receptor pathways can mediate these activities in the absence of Dectin-1, and this is likely to provide protection against systemic fungal infection in these individuals (Ferwerda et al. 2009).
This study also indicated that there are gene-dose effects associated with the Y238X mutation. For example, the homozygous daughters were 10-12 years of age at onset of symptoms, but the heterozygous mother and father were 40 and 55 years of age, respectively. Furthermore, heterozygotes had an intermediate production of proinflammatory cytokines upon stimulation with C. albicans or β-glucan and were statistically significantly more often colonized with C. albicans than patients with wild-type Dectin-1 (Plantinga et al. 2009). In terms of the frequency of the mutation, analysis indicated its presence only in populations from Africa and western Eurasia (allele frequency of 3-7%), with the highest prevalence (nearly 40%) found in the San population in South Africa (Ferwerda et al. 2009).
Plantinga et al. also investigated the association of the Dectin-1 polymorphism with C. albicans colonization in patients undergoing hematopoietic stem cell transplantation (HSCT) (Plantinga et al. 2009). Treatment of patients who have hematological malignancies with HSCT is accompanied by a high incidence of invasive fungal infections which cause considerable morbidity and mortality (Barnes and Marr 2007). The study by Plantinga et al. demonstrated that the Y238X Dectin-1 polymorphism is associated with increased susceptibility to oral and gastrointestinal colonization with Candida species in HSCT recipients (Plantinga et al. 2009). These findings affirm the importance of Dectin-1 in protection against fungal infections in immunocompromised patients, including those undergoing transplantation. The authors propose the implementation of an approach in which patients with the polymorphism are considered for antifungal prophylaxis to help prevent systemic candidiasis.
A further study investigated whether the Dectin-1 early-stop-codon polymorphism could have an impact on the immunological response following transplantation (van der Velden et al. 2010). The authors specifically screened for association between the Dectin-1 polymorphism and the incidence of graft-versus-host disease (GvHD) following stem cell transplantation. This study found that in patients colonized with Candida, the presence of the Dectin-1 polymorphism reduced the incidence of acute GvHD. Interestingly, recent studies have suggested a role for Th17 responses in the pathogenesis of GvHD (Carlson et al. 2009; Elmaagacli et al. 2008; Kappel et al. 2009). Considering that C. albicans is known to induce Th17 responses in mice and humans, the authors suggest that the link between Candida colonization and acute GvHD may be the induction of Th17 responses by the fungus (van der Velden et al. 2010). Their results suggest that despite increased colonization, defective Dectin-1 signaling prevented an increase in Th17-mediated acute GvHD. This study therefore established a link between Candida colonization and acute GvHD in humans.
As discussed previously, CARD9 is a major component of the downstream signaling pathway of Dectin-1, and recently Glocker et al. (2009) described a mutation causing a loss of function of CARD9 that was associated with an increased susceptibility to chronic mucocutaneous candidiasis. In addition, the CARD9−/− patients had significantly reduced numbers of Th17 cells. This study further supports the role of Dectin-1/Syk/CARD9 signaling in the differentiation of Th17 cells and in antifungal immunity.
Dectin-1 polymorphisms in other human diseases
The effects of Dectin-1 polymorphisms have also been studied in other human diseases and are briefly described here. First, Dectin-1-mediated responses have been implicated in driving autoimmunity. Fungal particles such as zymosan and β-glucans can induce inflammation and autoimmune arthritis in mice, and Yoshitomi et al. (2005) have shown that blocking Dectin-1 prevented β-glucan-induced autoimmune arthritis in genetically susceptible mice. Therefore, in order to determine if Dectin-1 is associated with rheumatoid arthritis (RA) in humans, Plantinga et al. (2010) investigated clinical parameters of inflammation and bone destruction in arthritis patients, including those bearing the heterozygous Dectin-1 polymorphism. This study showed that partial Dectin-1 deficiency had no association with disease susceptibility or with the degree of inflammation and bone destruction in RA patients.
Mutations in CARD9 have been associated with inflammatory bowel disease (IBD) (Zhernakova et al. 2008), and consequently the involvement of the Dectin-1 polymorphism in IBD was also investigated. Although Dectin-1 expression was found to be elevated on macrophages, neutrophils, and other cells involved in the inflammatory reaction in IBD lesions, the Dectin-1 polymorphism was found not to be a major susceptibility factor for the development of this disease (de Vries et al. 2009).
Conclusion
The discovery of Dectin-1 has revolutionized our understanding of molecular mechanisms underlying innate recognition of fungi. The study of this receptor has revealed novel insights into the intracellular signaling pathways and cellular responses induced by fungal β-glucans. Definitive evidence for its protective role in fungal disease has come from studies of patients harboring a Dectin-1 polymorphism that results in defective expression of the protein. Finally, given the current clinical evidence for the role of this receptor in fungal diseases, looking at ways of restoring the defective responses that are normally induced by Dectin-1 may provide new avenues for therapeutic intervention.
References
Adachi Y, Ishii T, Ikeda Y, Hoshino A, Tamura H et al (2004) Characterization of beta-glucan recognition site on C-type lectin, dectin 1. Infect Immun 72:4159–4171
Adams EL, Rice PJ, Graves B, Ensley HE, Yu H et al (2008) Differential high-affinity interaction of dectin-1 with natural or synthetic glucans is dependent upon primary structure and is influenced by polymer chain length and side-chain branching. J Pharmacol Exp Ther 325:115–123
Akira S, Uematsu S, Takeuchi O (2006) Pathogen recognition and innate immunity. Cell 124:783–801
Antachopoulos C, Walsh TJ, Roilides E (2007) Fungal infections in primary immunodeficiencies. Eur J Pediatr 166:1099–1117
Ariizumi K, Shen GL, Shikano S, Xu S, Ritter R III et al (2000) Identification of a novel, dendritic cell-associated molecule, dectin-1, by subtractive cDNA cloning. J Biol Chem 275:20157–20167
Barnes PD, Marr KA (2007) Risks, diagnosis and outcomes of invasive fungal infections in haematopoietic stem cell transplant recipients. Br J Haematol 139:519–531
Bellocchio S, Montagnoli C, Bozza S, Gaziano R, Rossi G et al (2004) The contribution of the Toll-like/IL-1 receptor superfamily to innate and adaptive immunity to fungal pathogens in vivo. J Immunol 172:3059–3069
Bozza S, Zelante T, Moretti S, Bonifazi P, DeLuca A et al (2008) Lack of Toll IL-1R8 exacerbates Th17 cell responses in fungal infection. J Immunol 180:4022–4031
Brown GD (2006) Dectin-1: a signalling non-TLR pattern-recognition receptor. Nat Rev 6:33–43
Brown GD, Gordon S (2001) Immune recognition. A new receptor for beta-glucans. Nature 413:36–37
Brown GD, Herre J, Williams DL, Willment JA, Marshall AS et al (2003) Dectin-1 mediates the biological effects of beta-glucans. J Exp Med 197:1119–1124
Calderone RA, Fonzi WA (2001) Virulence factors of Candida albicans. Trends Microbiol 9:327–335
Carlson MJ, West ML, Coghill JM, Panoskaltsis-Mortari A, Blazar BR et al (2009) In vitro-differentiated TH17 cells mediate lethal acute graft-versus-host disease with severe cutaneous and pulmonary pathologic manifestations. Blood 113:1365–1374
Carvalho A, Cunha C, Pasqualotto AC, Pitzurra L, Denning DW et al (2010) Genetic variability of innate immunity impacts human susceptibility to fungal diseases. Int J Infect Dis 14:e460–e468
Conti HR, Shen F, Nayyar N, Stocum E, Sun JN et al (2009) Th17 cells and IL-17 receptor signaling are essential for mucosal host defense against oral candidiasis. J Exp Med 206:299–311
de Jong MA, Vriend LE, Theelen B, Taylor ME, Fluitsma D et al (2010) C-type lectin Langerin is a beta-glucan receptor on human Langerhans cells that recognizes opportunistic and pathogenic fungi. Mol Immunol 47(6):1216–1225
de Vries HS, Plantinga TS, van Krieken JH, Stienstra R, van Bodegraven AA et al (2009) Genetic association analysis of the functional c.714T>G polymorphism and mucosal expression of dectin-1 in inflammatory bowel disease. PLoS One 4:e7818
Dennehy KM, Ferwerda G, Faro-Trindade I, Pyz E, Willment JA et al (2008) Syk kinase is required for collaborative cytokine production induced through Dectin-1 and Toll-like receptors. Eur J Immunol 38:500–506
Dillon S, Agrawal S, Banerjee K, Letterio J, Denning TL et al (2006) Yeast zymosan, a stimulus for TLR2 and dectin-1, induces regulatory antigen-presenting cells and immunological tolerance. J Clin Invest 116:916–928
Elmaagacli AH, Koldehoff M, Landt O, Beelen DW (2008) Relation of an interleukin-23 receptor gene polymorphism to graft-versus-host disease after hematopoietic-cell transplantation. Bone Marrow Transplant 41:821–826
Ferwerda B, Ferwerda G, Plantinga TS, Willment JA, van Spriel AB et al (2009) Human dectin-1 deficiency and mucocutaneous fungal infections. N Engl J Med 361:1760–1767
Fuller GL, Williams JA, Tomlinson MG, Eble JA, Hanna SL et al (2007) The C-type lectin receptors CLEC-2 and Dectin-1, but not DC-SIGN, signal via a novel YXXL-dependent signaling cascade. J Biol Chem 282:12397–12409
Gantner BN, Simmons RM, Canavera SJ, Akira S, Underhill DM (2003) Collaborative induction of inflammatory responses by dectin-1 and Toll-like receptor 2. J Exp Med 197:1107–1117
Gantner BN, Simmons RM, Underhill DM (2005) Dectin-1 mediates macrophage recognition of Candida albicans yeast but not filaments. EMBO J 24:1277–1286
Gersuk GM, Underhill DM, Zhu L, Marr KA (2006) Dectin-1 and TLRs permit macrophages to distinguish between different Aspergillus fumigatus cellular states. J Immunol 176:3717–3724
Glocker EO, Hennigs A, Nabavi M, Schaffer AA, Woellner C et al (2009) A homozygous CARD9 mutation in a family with susceptibility to fungal infections. N Engl J Med 361:1727–1735
Goodridge HS, Simmons RM, Underhill DM (2007) Dectin-1 stimulation by Candida albicans yeast or zymosan triggers NFAT activation in macrophages and dendritic cells. J Immunol 178:3107–3115
Goodridge HS, Shimada T, Wolf AJ, Hsu YM, Becker CA et al (2009a) Differential use of CARD9 by dectin-1 in macrophages and dendritic cells. J Immunol 182:1146–1154
Goodridge HS, Wolf AJ, Underhill DM (2009b) Beta-glucan recognition by the innate immune system. Immunol Rev 230:38–50
Gringhuis SI, den Dunnen J, Litjens M, van der Vlist M, Wevers B et al (2009) Dectin-1 directs T helper cell differentiation by controlling noncanonical NF-kappaB activation through Raf-1 and Syk. Nat Immunol 10:203–213
Gross O, Gewies A, Finger K, Schafer M, Sparwasser T et al (2006) Card9 controls a non-TLR signalling pathway for innate anti-fungal immunity. Nature 442:651–656
Hara H, Ishihara C, Takeuchi A, Imanishi T, Xue L et al (2007) The adaptor protein CARD9 is essential for the activation of myeloid cells through ITAM-associated and Toll-like receptors. Nat Immunol 8:619–629
Hashimoto C, Hudson KL, Anderson KV (1988) The Toll gene of Drosophila, required for dorsal-ventral embryonic polarity, appears to encode a transmembrane protein. Cell 52:269–279
Heinsbroek SE, Taylor PR, Rosas M, Willment JA, Williams DL et al (2006) Expression of functionally different dectin-1 isoforms by murine macrophages. J Immunol 176:5513–5518
Hise AG, Tomalka J, Ganesan S, Patel K, Hall BA et al (2009) An essential role for the NLRP3 inflammasome in host defense against the human fungal pathogen Candida albicans. Cell Host Microbe 5:487–497
Hohl TM, Van Epps HL, Rivera A, Morgan LA, Chen PL et al (2005) Aspergillus fumigatus triggers inflammatory responses by stage-specific beta-glucan display. PLoS Pathog 1:e30
Hohl TM, Feldmesser M, Perlin DS, Pamer EG (2008) Caspofungin modulates inflammatory responses to Aspergillus fumigatus through stage-specific effects on fungal beta-glucan exposure. J Infect Dis 198:176–185
Huang W, Na L, Fidel PL, Schwarzenberger P (2004) Requirement of interleukin-17A for systemic anti-Candida albicans host defense in mice. J Infect Dis 190:624–631
Hughes CE, Pollitt AY, Mori J, Eble JA, Tomlinson MG et al (2010) CLEC-2 activates Syk through dimerization. Blood 115:2947–2955
Huysamen C, Willment JA, Dennehy KM, Brown GD (2008) CLEC9A is a novel activation C-type lectin-like receptor expressed on BDCA3+ dendritic cells and a subset of monocytes. J Biol Chem 283:16693–16701
Jang S, Ohtani K, Fukuoh A, Yoshizaki T, Fukuda M et al (2009) Scavenger receptor collectin placenta 1 (CL-P1) predominantly mediates zymosan phagocytosis by human vascular endothelial cells. J Biol Chem 284:3956–3965
Kappel LW, Goldberg GL, King CG, Suh DY, Smith OM et al (2009) IL-17 contributes to CD4-mediated graft-versus-host disease. Blood 113:945–952
Karumuthil-Melethil S, Perez N, Li R, Vasu C (2008) Induction of innate immune response through TLR2 and dectin 1 prevents type 1 diabetes. J Immunol 181:8323–8334
Lamaris GA, Lewis RE, Chamilos G, May GS, Safdar A et al (2008) Caspofungin-mediated beta-glucan unmasking and enhancement of human polymorphonuclear neutrophil activity against Aspergillus and non-Aspergillus hyphae. J Infect Dis 198:186–192
Lee HM, Shin DM, Choi DK, Lee ZW, Kim KH et al (2009) Innate immune responses to Mycobacterium ulcerans via toll-like receptors and dectin-1 in human keratinocytes. Cell Microbiol 11:678–692
LeibundGut-Landmann S, Gross O, Robinson MJ, Osorio F, Slack EC et al (2007) Syk- and CARD9-dependent coupling of innate immunity to the induction of T helper cells that produce interleukin 17. Nat Immunol 8:630–638
Leibundgut-Landmann S, Osorio F, Brown GD, Reis e Sousa C (2008) Stimulation of dendritic cells via the dectin-1/Syk pathway allows priming of cytotoxic T-cell responses. Blood 112:4971–4980
Leigh JE, McNulty KM, Fidel PL Jr (2006) Characterization of the immune status of CD8+ T cells in oral lesions of human immunodeficiency virus-infected persons with oropharyngeal candidiasis. Clin Vaccine Immunol 13:678–683
Lemaitre B, Nicolas E, Michaut L, Reichhart JM, Hoffmann JA (1996) The dorsoventral regulatory gene cassette spatzle/Toll/cactus controls the potent antifungal response in Drosophila adults. Cell 86:973–983
Lindell DM, Moore TA, McDonald RA, Toews GB, Huffnagle GB (2005) Generation of antifungal effector CD8+ T cells in the absence of CD4+ T cells during Cryptococcus neoformans infection. J Immunol 174:7920–7928
Lo HJ, Kohler JR, DiDomenico B, Loebenberg D, Cacciapuoti A et al (1997) Nonfilamentous C. albicans mutants are avirulent. Cell 90:939–949
Marquis M, Lewandowski D, Dugas V, Aumont F, Senechal S et al (2006) CD8+ T cells but not polymorphonuclear leukocytes are required to limit chronic oral carriage of Candida albicans in transgenic mice expressing human immunodeficiency virus type 1. Infect Immun 74:2382–2391
Means TK, Mylonakis E, Tampakakis E, Colvin RA, Seung E et al (2009) Evolutionarily conserved recognition and innate immunity to fungal pathogens by the scavenger receptors SCARF1 and CD36. J Exp Med 206:637–653
Medzhitov R (2009) Approaching the asymptote: 20 years later. Immunity 30:766–775
Murciano C, Villamon E, Gozalbo D, Roig P, O’Connor JE et al (2006) Toll-like receptor 4 defective mice carrying point or null mutations do not show increased susceptibility to Candida albicans in a model of hematogenously disseminated infection. Med Mycol 44:149–157
Nakamura K, Miyazato A, Koguchi Y, Adachi Y, Ohno N et al (2008) Toll-like receptor 2 (TLR2) and dectin-1 contribute to the production of IL-12p40 by bone marrow-derived dendritic cells infected with Penicillium marneffei. Microbes Infect 10:1223–1227
Netea MG, Sutmuller R, Hermann C, Van der Graaf CA, Van der Meer JW et al (2004) Toll-like receptor 2 suppresses immunity against Candida albicans through induction of IL-10 and regulatory T cells. J Immunol 172:3712–3718
Netea MG, Ferwerda G, van der Graaf CA, Van der Meer JW, Kullberg BJ (2006) Recognition of fungal pathogens by toll-like receptors. Curr Pharm Des 12:4195–4201
Netea MG, Brown GD, Kullberg BJ, Gow NA (2008) An integrated model of the recognition of Candida albicans by the innate immune system. Nat Rev Microbiol 6:67–78
Netea MG, Gow NA, Joosten LA, Verschueren I, van der Meer JW et al (2010) Variable recognition of Candida albicans strains by TLR4 and lectin recognition receptors. Med Mycol (in press)
Olynych TJ, Jakeman DL, Marshall JS (2006) Fungal zymosan induces leukotriene production by human mast cells through a dectin-1-dependent mechanism. J Allergy Clin Immunol 118:837–843
Osorio F, LeibundGut-Landmann S, Lochner M, Lahl K, Sparwasser T et al (2008) DC activated via dectin-1 convert Treg into IL-17 producers. Eur J Immunol 38:3274–3281
Ozment-Skelton TR, deFluiter EA, Ha T, Li C, Graves BM et al (2009) Leukocyte Dectin-1 expression is differentially regulated in fungal versus polymicrobial sepsis. Crit Care Med 37:1038–1045
Palma AS, Feizi T, Zhang Y, Stoll MS, Lawson AM et al (2006) Ligands for the beta-glucan receptor, Dectin-1, assigned using “designer” microarrays of oligosaccharide probes (neoglycolipids) generated from glucan polysaccharides. J Biol Chem 281:5771–5779
Plantinga TS, van der Velden WJ, Ferwerda B, van Spriel AB, Adema G et al (2009) Early stop polymorphism in human DECTIN-1 is associated with increased Candida colonization in hematopoietic stem cell transplant recipients. Clin Infect Dis 49:724–732
Plantinga TS, Fransen J, Takahashi N, Stienstra R, van Riel PL et al (2010) Functional consequences of DECTIN-1 early stop codon polymorphism Y238X in rheumatoid arthritis. Arthritis Res Ther 12:R26
Reid DM, Montoya M, Taylor PR, Borrow P, Gordon S et al (2004) Expression of the beta-glucan receptor, Dectin-1, on murine leukocytes in situ correlates with its function in pathogen recognition and reveals potential roles in leukocyte interactions. J Leukoc Biol 76:86–94
Reid DM, Gow NA, Brown GD (2009) Pattern recognition: recent insights from Dectin-1. Curr Opin Immunol 21:30–37
Robinson MJ, Osorio F, Rosas M, Freitas RP, Schweighoffer E et al (2009) Dectin-2 is a Syk-coupled pattern recognition receptor crucial for Th17 responses to fungal infection. J Exp Med 206:2037–2051
Rogers NC, Slack EC, Edwards AD, Nolte MA, Schulz O et al (2005) Syk-dependent cytokine induction by Dectin-1 reveals a novel pattern recognition pathway for C type lectins. Immunity 22:507–517
Romani L (2004) Immunity to fungal infections. Nat Rev Immunol 4:1–23
Romani L, Puccetti P (2006) Protective tolerance to fungi: the role of IL-10 and tryptophan catabolism. Trends Microbiol 14:183–189
Romani L, Montagnoli C, Bozza S, Perruccio K, Spreca A et al (2004) The exploitation of distinct recognition receptors in dendritic cells determines the full range of host immune relationships with Candida albicans. Int Immunol 16:149–161
Rosas M, Liddiard K, Kimberg M, Faro-Trindade I, McDonald JU et al (2008) The induction of inflammation by dectin-1 in vivo is dependent on myeloid cell programming and the progression of phagocytosis. J Immunol 181:3549–3557
Ross GD, Cain JA, Myones BL, Newman SL, Lachmann PJ (1987) Specificity of membrane complement receptor type three (CR3) for beta-glucans. Complement 4:61–74
Rothfuchs AG, Bafica A, Feng CG, Egen JG, Williams DL et al (2007) Dectin-1 interaction with Mycobacterium tuberculosis leads to enhanced IL-12p40 production by splenic dendritic cells. J Immunol 179:3463–3471
Saijo S, Fujikado N, Furuta T, Chung SH, Kotaki H et al (2007) Dectin-1 is required for host defense against Pneumocystis carinii but not against Candida albicans. Nat Immunol 8:39–46
Saijo S, Ikeda S, Yamabe K, Kakuta S, Ishigame H et al (2010) Dectin-2 recognition of alpha-mannans and induction of Th17 cell differentiation is essential for host defense against Candida albicans. Immunity 32(5):681–691
Saville SP, Lazzell AL, Monteagudo C, Lopez-Ribot JL (2003) Engineered control of cell morphology in vivo reveals distinct roles for yeast and filamentous forms of Candida albicans during infection. Eukaryot Cell 2:1053–1060
Shah VB, Huang Y, Keshwara R, Ozment-Skelton T, Williams DL et al (2008) Beta-glucan activates microglia without inducing cytokine production in Dectin-1-dependent manner. J Immunol 180:2777–2785
Shin DM, Yang CS, Yuk JM, Lee JY, Kim KH et al (2008) Mycobacterium abscessus activates the macrophage innate immune response via a physical and functional interaction between TLR2 and dectin-1. Cell Microbiol 10:1608–1621
Slack EC, Robinson MJ, Hernanz-Falcon P, Brown GD, Williams DL et al (2007) Syk-dependent ERK activation regulates IL-2 and IL-10 production by DC stimulated with zymosan. Eur J Immunol 37:1600–1612
Steele C, Marrero L, Swain S, Harmsen AG, Zheng M et al (2003) Alveolar macrophage-mediated killing of Pneumocystis carinii f. sp. muris involves molecular recognition by the Dectin-1 beta-glucan receptor. J Exp Med 198:1677–1688
Steele C, Rapaka RR, Metz A, Pop SM, Williams DL et al (2005) The beta-glucan receptor dectin-1 recognizes specific morphologies of Aspergillus fumigatus. PLoS Pathog 1:e42
Tassi I, Cella M, Castro I, Gilfillan S, Khan WN et al (2009) Requirement of phospholipase C-gamma2 (PLCgamma2) for Dectin-1-induced antigen presentation and induction of TH1/TH17 polarization. Eur J Immunol 39:1369–1378
Taylor PR, Brown GD, Reid DM, Willment JA, Martinez-Pomares L et al (2002) The beta-glucan receptor, dectin-1, is predominantly expressed on the surface of cells of the monocyte/macrophage and neutrophil lineages. J Immunol 169:3876–3882
Taylor PR, Tsoni SV, Willment JA, Dennehy KM, Rosas M et al (2007) Dectin-1 is required for beta-glucan recognition and control of fungal infection. Nat Immunol 8:31–38
Torosantucci A, Bromuro C, Chiani P, De Bernardis F, Berti F et al (2005) A novel glyco-conjugate vaccine against fungal pathogens. J Exp Med 202:597–606
Tsoni SV, Brown GD (2008) beta-Glucans and dectin-1. Ann N Y Acad Sci 1143:45–60
Underhill DM, Goodridge HS (2007) The many faces of ITAMs. Trends Immunol 28:66–73
van de Veerdonk FL, Kullberg BJ, van der Meer JW, Gow NA, Netea MG (2008) Host–microbe interactions: innate pattern recognition of fungal pathogens. Curr Opin Microbiol 11:305–312
van der Velden WJ, Plantinga TS, Feuth T, Donnelly JP, Netea MG et al (2010) The incidence of acute graft-versus-host disease increases with Candida colonization depending the dectin-1 gene status. Clin Immunol 136(2):302–306
Vera J, Fenutria R, Canadas O, Figueras M, Mota R et al (2009) The CD5 ectodomain interacts with conserved fungal cell wall components and protects from zymosan-induced septic shock-like syndrome. Proc Natl Acad Sci USA 106:1506–1511
Viriyakosol S, Fierer J, Brown GD, Kirkland TN (2005) Innate immunity to the pathogenic fungus Coccidioides posadasii is dependent on Toll-like receptor 2 and Dectin-1. Infect Immun 73:1553–1560
von Bernuth H, Picard C, Jin Z, Pankla R, Xiao H et al (2008) Pyogenic bacterial infections in humans with MyD88 deficiency. Science 321:691–696
Werner JL, Metz AE, Horn D, Schoeb TR, Hewitt MM et al (2009) Requisite role for the dectin-1 beta-glucan receptor in pulmonary defense against Aspergillus fumigatus. J Immunol 182:4938–4946
Wheeler RT, Fink GR (2006) A drug-sensitive genetic network masks fungi from the immune system. PLoS Pathog 2:e35
Wheeler RT, Kombe D, Agarwala SD, Fink GR (2008) Dynamic, morphotype-specific Candida albicans beta-glucan exposure during infection and drug treatment. PLoS Pathog 4:e1000227
Willment JA, Brown GD (2008) C-type lectin receptors in antifungal immunity. Trends Microbiol 16:27–32
Willment JA, Gordon S, Brown GD (2001) Characterization of the human beta-glucan receptor and its alternatively spliced isoforms. J Biol Chem 276:43818–43823
Willment JA, Lin HH, Reid DM, Taylor PR, Williams DL et al (2003) Dectin-1 expression and function are enhanced on alternatively activated and GM-CSF-treated macrophages and are negatively regulated by IL-10, dexamethasone, and lipopolysaccharide. J Immunol 171:4569–4573
Willment JA, Marshall AS, Reid DM, Williams DL, Wong SY et al (2005) The human beta-glucan receptor is widely expressed and functionally equivalent to murine Dectin-1 on primary cells. Eur J Immunol 35:1539–1547
Wuthrich M, Filutowicz HI, Warner T, Deepe GS Jr, Klein BS (2003) Vaccine immunity to pathogenic fungi overcomes the requirement for CD4 help in exogenous antigen presentation to CD8+ T cells: implications for vaccine development in immune-deficient hosts. J Exp Med 197:1405–1416
Xu S, Huo J, Lee KG, Kurosaki T, Lam KP (2009) Phospholipase Cgamma2 is critical for Dectin-1-mediated Ca2+ flux and cytokine production in dendritic cells. J Biol Chem 284:7038–7046
Yadav M, Schorey JS (2006) The beta-glucan receptor dectin-1 functions together with TLR2 to mediate macrophage activation by mycobacteria. Blood 108:3168–3175
Yoshitomi H, Sakaguchi N, Kobayashi K, Brown GD, Tagami T et al (2005) A role for fungal {beta}-glucans and their receptor Dectin-1 in the induction of autoimmune arthritis in genetically susceptible mice. J Exp Med 201:949–960
Zelante T, De Luca A, Bonifazi P, Montagnoli C, Bozza S et al (2007) IL-23 and the Th17 pathway promote inflammation and impair antifungal immune resistance. Eur J Immunol 37:2695–2706
Zelensky AN, Gready JE (2005) The C-type lectin-like domain superfamily. FEBS J 272:6179–6217
Zhernakova A, Festen EM, Franke L, Trynka G, van Diemen CC et al (2008) Genetic analysis of innate immunity in Crohn’s disease and ulcerative colitis identifies two susceptibility loci harboring CARD9 and IL18RAP. Am J Hum Genet 82:1202–1210
Zimmerman JW, Lindermuth J, Fish PA, Palace GP, Stevenson TT et al (1998) A novel carbohydrate-glycosphingolipid interaction between a beta-(1–3)-glucan immunomodulator, PGG-glucan, and lactosylceramide of human leukocytes. J Biol Chem 273:22014–22020
Acknowledgments
We thank Wellcome Trust and South African National Research Foundation for funding.
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License (https://creativecommons.org/licenses/by-nc/2.0), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
Marakalala, M.J., Kerrigan, A.M. & Brown, G.D. Dectin-1: a role in antifungal defense and consequences of genetic polymorphisms in humans. Mamm Genome 22, 55–65 (2011). https://doi.org/10.1007/s00335-010-9277-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00335-010-9277-3