
Abstract
This review highlights the utility of comparative genetics in understanding the molecular mechanisms that underlie neuronal migration. It is apparent from studies in humans, mice, and a fungus that nuclear migration is a key component of neuronal migration and that both are dependent on a dynamic microtubule network. In vertebrates regulation of this network involves a complex pathway that is dependent on extracellular guidance cues, membrane-bound receptors, intracellular signaling molecules, proteins associated with microtubules, and the components of microtubules themselves.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
It is undeniably exciting the first time, when peering down a microscope, you see the beauty of the cerebral cortex. Its six ordered layers like a medieval army awaiting a cognitive battle. This precise layering and, with it, the ability to entertain higher thought are consequences of neuronal migration. The cortex with its laminar structure is not the only brain region that relies on neuronal migration; the cellular architecture of the cerebellum, hippocampus, and colliculi is the product of a precise neuronal journey. Born in the proliferative ventricular zones, each neuron must migrate to its final destination. But how do they get there? This review focuses on the contribution of humans, mice, and a fungus to the understanding of the molecular mechanisms that underlie neuronal migration. The article begins with an introduction on neuronal migration, followed by assessment of the contribution made by spontaneous and transgenic mutant mice, then human disorders, and finally a look at rather an unlikely contributor, Aspergillus nidulans. It does not aim to discuss all genes required for neuronal migration but rather to highlight the value of comparative genetics in the understanding of this biological phenomenon.
Radial and tangential neuronal migration
Two types of neuronal migration predominate during embryonic stages: radial and tangential. Radial migration describes neurons that migrate in a direction perpendicular to the surface of the brain, guided by a scaffold of radial glial cells. In the cortex, radial glial cells, which are generated prior to neurogenesis, have a cell body in the ventricular zone and a process that reaches the surface of the brain. While initially thought to be static, it has recently been demonstrated that this population of cells undergoes mitosis producing new neurons (Noctor et al. 2001, 2002). Neurons that rely on radial migration are not limited to the cortex and include the pyramidal neurons of the hippocampus, the Purkinje cells in the cerebellum, and the majority of the cells in the superior colliculus. Tangential migration describes neurons that migrate in a direction parallel to the brain surface. Most of these neurons originate from the ganglionic eminences (GE) and are known to include cortical and hippocampal GABAnergic interneurons (Marin and Rubenstein 2001). Unlike radial migration, tangential migration does not rely on radial glia and instead is referred to as neurophilic. Neurons that migrate tangentially are known to employ each other (such as neurons destined for the olfactory bulb), axons, and extracellular guidance cues. Both radial and tangential migrations involve the extension of a leading neurite, somal translocation of the nucleus into that neurite, and the retraction of the trailing process (Lambert de Rouvroit and Goffinet 2001). The most important question for investigators in the field has been: What are the molecular mechanisms that drive this migration?
Spontaneous mouse mutants: reeler, scrambler, and yotari
The advent of genetic techniques such as positional cloning and exon trapping revolutionized the field of developmental neurobiology, bringing the mouse to the forefront (Rice and Curran 2001). The genes responsible for the neuronal migration phenotypes observed in the spontaneous mouse mutants—reeler, scrambler, and yotari—could now be identified (Rakic and Sidman 1973; Stanfield and Cowan 1979). The first to be cloned was a mutation in reelin, which was responsible for the inverted cortex and the disorganized hippocampus and cerebellum in the reeler mouse (D’Arcangelo et al. 1995). The gene reelin encodes a large extracellular protein that is expressed primarily in the Cajal-Retzius cells in the marginal zone of the cortex and the cerebellum (Hirotsune et al. 1995). This was the first protein identified in the molecular pathway required for neurons to migrate, and it emphasized the importance of extracellular guidance cues. The cloning of scrambler and yotari followed shortly afterward. Both of these mice, with similar phenotypes to the reeler mouse, have mutations in the disabled gene (Dab1), a tyrosine phosphorylated cytoplasmic protein (Sheldon et al. 1997). The similarity of the phenotypes in these mice provided strong evidence that the same pathway was involved.
Transgenics: Vldlr, Apoer2, and Cdk5
The link between reelin and Dab1 was soon provided by a series of studies conducted by Trommsdorff et al. (1998, 1999). They undertook pull-down experiments that demonstrated an interaction between DAB1 and the cytoplasmic tails of the VLDLR and APOER2 receptors. Generation of Apoer2 and Vldlr knockout mice confirmed the importance of these receptors in neuronal migration. The mice had a loosely packed pyramidal cell layer in the CA1 region of the hippocampus, a less foliated cerebellum, and abnormal cellular distribution in the cortex. They speculated that these receptors interacted with reelin, which was supported by the findings of Howell et al. (1999) who showed that phosphorylation of DAB1 was increased by extracellular application of reelin. The phosphorylation of DAB1 has also shown to be regulated by another important protein, cyclin-dependent kinase 5 (CDK5). Mice lacking Cdk5 (or its activator p35), exhibit lamination defects similar to the reeler mouse (Chae et al. 1997; Ohshima et al. 1996). CDK5 phosphorylates DAB1 on serine 491 independent of reelin signaling, suggesting an intersection of pathways. CDK5 is thought to play an important role in the remodeling of the cytoskeleton required for specific modes of migration. Indeed one of the proteins. CDK5 is known to phosphorylate is doublecortin (DCX); a protein that was discovered by studying neuronal migration disorders in humans (Tanaka et al. 2004b; Xie et al. 2006).
The human mutations: DCX and LIS1
Disorders of neuronal migration are not limited to mice. In humans abnormalities of neuronal migration cause a range of diseases, most notably lissencephaly. Classic lissencephaly, a disorder of both tangential migration and radial migration, is characterized by a cortex that has only four layers and a brain that appears smooth with an absence of gyri and sulci (Dobyns and Truwit 1995) (Fig. 1). Genetic studies have resulted in the identification of several genes that are responsible for abnormal cortical migration in humans.

The consequences of nuclear and neuronal defects in humans, mice and Aspergillus. In humans neuronal migration defects can result in lissencephaly. A–B Lissencephalic individuals have a smooth cortex lacking the characteristic sulci and gyri observable in controls (A). The individual show in panel B has a hemizygous deletion of L1S1. C–D In mice neuronal migration abnormalities manifest themselves as a fractured pyramidal cell layer in the hippocampus (arrowed). Panel D shows a sagittal section of the hippocampus in the jenna mice, which harbour a S140G substitution in TUBA1. E–F In Aspergillus mutations that affect nuclear migration result in smaller less developed colonies. The colony shown in panel F, is a nudF mutant
In two landmark papers, des Portes et al. (1998) and Gleeson et al. (1998) reported that mutations in the X-linked gene doublecortin cause lissencephaly in males and a syndrome known as double-cortex in females. While initially thought to code for a putative signaling protein, it has since been demonstrated that DCX, a protein that is highly expressed in migrating and differentiating neurons, plays a key role in the stabilization of microtubules (Francis et al. 1999; Gleeson et al. 1999). Indeed, the mutations in DCX that cause lissencephaly have been shown to cluster in DCX’s two tubulin-binding domains (Sapir et al. 2000; Taylor et al. 2000). The creation of transgenic mice lacking Dcx have further demonstrated the importance of this gene for neuronal migration. Dcx knockout mice have a fractured pyramidal cell layer in the hippocampus and exhibit defects in tangential migration from the subventricular zone to the olfactory bulbs via the rostral migratory stream (Corbo et al. 2002; Koizumi et al. 2006). It has also been shown that migrating neuron populations derived from the medial ganglionic eminence in Dcx knockout mice show abnormalities in migratory dynamics (Kappeler et al. 2006).
Lissencephaly is also known to result from hemizygous deletions in a gene on chromosome 17, known as LIS1 or PAFAH1B1 (Reiner et al. 1993). LIS1 contains WD40 domains and is highly expressed in Cajal-Retzius cells and in the ventricular epithelium in the developing human cortex (Clark et al. 1997). Initially discovered because it catalyzes the inactivation of platelet-activating factor (Hattori et al. 1994), LIS1 has been shown to influence microtubule function. Cellular studies have demonstrated that overexpression of LIS1 increases retrograde movement of cytoplasmic dynein leading to the accumulation of microtubules (Smith et al. 2000). There is also evidence of an interaction between the reelin signaling pathway and LIS1. It has been shown that phosphorylated DAB1 binds to LIS1 and that compound mutant mice with mutations in both reelin and Lis1 exhibit enhanced layering defects in the cortex and hippocampus (Assadi et al. 2003).
Tubulin and lissencephaly
The importance of microtubules in neuronal migration is further emphasized by our recent discovery that mutations in α-tubulin cause abnormalities in neuronal migration (Keays et al. 2007). Microtubules are formed by the polymerization of heterodimers consisting of α- and β-tubulin. We found that an abnormal radial neuronal migration phenotype in the ENU-induced mouse mutant jenna was attributable to a substitution in the GTP binding site of α-tubulin (TUBA1) that affected heterodimerization with β-tubulin. Consequences of impaired neuronal migration in this mouse were notable defects in hippocampal pyramidal cell lamination (Fig. 1) and wavelike perturbations in layers II/III and IV of the cortex. The jenna mouse mutant showed striking similarity to the Lis1 and Dcx knockout mice (Corbo et al. 2002; Hirotsune et al. 1998), leading us to speculate that mutations in the human homolog of Tuba1 might also cause lissencephaly. This was found to be the case, and to date approximately ten de novo mutations have been found that result in cortical anomalies in humans (K. Poirier et al., personal communication).
So far we can see that the study of human and mouse mutants has unraveled a pathway that involves extracellular guidance cues, intracellular signaling molecules, and a number of proteins that are associated with cytoskeleton stabilization and modulation (Fig. 2). How do these mutations operate to affect how a neuron actually moves? Insight into the mechanism was to come from an unlikely source: Aspergillus nidulans.

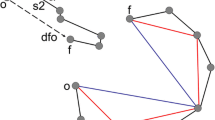
Molecular pathway associated with neuronal migration. This diagram shows an outline of the molecular pathway required for neuronal migration. Extracellular reelin binds to the membrane-bound receptors APOER2 and VLDLR, which stimulate the intracellular signaling molecule DAB1. Phosphorylated DAB1 interacts with LIS1. CDK5, thought to act via a parallel but intersecting pathway, is activated by P35 and phosphorylates the microtubule stabilizer DCX and NDEL1. NDEL1 complexes with LIS1 and dynein, which act to sustain microtubule bundles and facilitate nuclear migration
Aspergillus nidulans and nuclear migration
Aspergillus nidulans is a filamentous soil fungus. Beginning its existence as a single spore, it undergoes several rounds of nuclear division (resulting in about 4 nuclei), followed by the formation of a projection known as a germ tube. Nuclei then migrate along the germ tube, with each nucleus moving a different distance so the nuclei are separated equally, prior to cellular division (Xiang and Morris 1999) (Fig. 3). Polarized growth of the germ tube then continues, creating an extending tip called a hyphae that undergoes branching and growth. The ability to view this process with a light microscope established Aspergillus nidulans as a model organism to study the biology of nuclear migration.

Nuclear migration in Aspergillus. A Following several rounds of nuclear division (resulting in about 4 nuclei), each nucleus migrates a different distance along the germ tube prior to cellular division. B While nuclear division is normal, nuclear migration fails in the Nud mutants, with the nuclei remaining in the spore
Pioneering experiments with this species were undertaken by Morris. They employed UV radiation to identify a host of nuclear migration mutants, labeling them nuclear distribution (nud) mutants (Morris 1975). These mutants showed normal germination and nuclear division, but the nuclei failed to migrate into the germ tube at restrictive temperatures. The mutants include nudA, nudC, nudE, nudF, and nudG. They were to be informative in the study of neuronal migration.
Comparative studies showed that NudF, a WD-repeat protein, is 42% identical in amino acid sequence to LIS1 (Xiang et al. 1995), the same gene that causes lissencephaly in humans. It was this observation that gave rise to the hypothesis that nuclear migration is required for neuronal migration (Xiang et al. 1995). Other nud genes also have mammalian homologs. The homolog of nudA is cytoplasmic dynein heavy chain and that of nudG is cytoplasmic dynein light chain (Xiang et al. 1994). The dyneins are microtubule-dependent motor proteins that are involved in the motility of a wide variety of organelles. It has been demonstrated that in vertebrates LIS1 colocalizes and interacts with cytoplasmic dynein heavy chain, and both are highly expressed in postmitotic migrating neurons in the cortex (Niethammer et al. 2000; Smith et al. 2000). Mice with ENU-induced mutations in cytoplasmic dynein heavy chain are a model for motor neuron degeneration but also exhibit defects in the migration of facial motor neurons; their cell bodies fail to migrate to their final destination in the hindbrain (Hafezparast et al. 2003). Moreover, disruption of cytoplasmic dynein results in impaired motility in a cellular assay for neuronal migration (Shu et al. 2004; Tanaka et al. 2004a). The dyneins and their role as microtubule motors are clearly important for nuclear and neuronal migration.
nudE, Nde1, and Ndel1
There is growing evidence that the interaction between dynein and LIS1 is influenced by another nud homolog: nudE. nudE mutants display impaired nuclear migration and reduced colony growth. They also suppress the more severe phenotype in nudF mutants, improving growth and nuclear distribution (Efimov and Morris 2000). In vertebrates there are two nudE homologs (with very similar names), Nde1 and Nde1 like (Ndel1), that are known to interact with LIS1. Each is clearly involved in neuronal migration; however, it seems that Ndel1 plays a more important role. Nde1 mice, which exhibit a modest neuronal migration defect, are most noted for their small cerebral cortex and mitotic defects in cortical progenitors (Feng and Walsh 2004). This contrasts with Ndel1 mutant mice, which display a dosage-dependent neuronal migration phenotype (Sasaki et al. 2005). Functional studies on NDEL1 have shown that it is phosphorylated by CDK5 and forms a complex with LIS1 and dynein that sustains microtubule bundles and facilitates nuclear migration (Niethammer et al. 2000). It is of further interest that RNAi knockdown of Ndel1 in dissociated mouse cortical neurons results in a disruption of nuclear translocation by uncoupling the centrosome and nucleus (Shu et al. 2004). It is therefore apparent that both nuclear and neuronal migrations require a complex that involves LIS1, NDEL1, and cytoplasmic dynein (Table 1).
Tubulins in Aspergillus
We have seen that mutations in tubulin result in abnormal neuronal migration phenotypes in mice and humans. The tubulins are similarly important for nuclear migration in Aspergillus. Morris showed that nuclear migration was arrested by treatment with benomyl, an inhibitor of microtubule polymerization. Moreover, it was found that a mutant line resistant to benomyl treatment harbored a mutation in β-tubulin (benA), leading to the conclusion that nuclear movement was mediated by this protein (Oakley and Morris 1980). A mutation in one of two α-tubulin genes in Aspergillus has also been found to suppress the abnormal nuclear migration phenotypes observed in the nudA, nudF, nudC, and nudG mutants. This suppressor mutation is thought to act by destabilizing microtubules as it confers sensitivity to cold and benomyl (Willins et al. 1995). It is not known exactly how destabilization of microtubules rescues nuclear migration phenotypes in Aspergillus; nonetheless, it again emphasizes the importance of the tubulins in nuclear migration.
Conclusion
It is apparent from studies conducted in Aspergillus, mice, and humans that nuclear migration is a key component of neuronal migration and that both are highly dependent on a dynamic microtubule network. In vertebrates regulation of this network involves a complex pathway that is dependent on extracellular guidance cues (reelin), membrane-bound receptors (APOER2, VLDLR), intracellular signaling molecules (DAB1, CDK5, P35), proteins associated with microtubules (DCX, LIS1, NDEL1, dyneins), and the components of microtubules themselves (TUBA). The studies conducted in Aspergillus are of particular note. Few would have predicted that studying a simple fungus would help understand the human cerebral cortex, arguably the most complex structure on the planet.
References
Assadi AH, Zhang G, Beffert U, McNeil RS, Renfro AL, et al. (2003) Interaction of reelin signaling and Lis1 in brain development. Nat Genet 35(3):270–276
Chae T, Kwon YT, Bronson R, Dikkes P, Li E, et al. (1997) Mice lacking p35, a neuronal specific activator of Cdk5, display cortical lamination defects, seizures, and adult lethality. Neuron 18(1):29–42
Clark GD, Mizuguchi M, Antalffy B, Barnes J, Armstrong D (1997) Predominant localization of the LIS family of gene products to Cajal-Retzius cells and ventricular neuroepithelium in the developing human cortex. J Neuropathol Exp Neurol 56(9):1044–1052
Corbo JC, Deuel TA, Long JM, LaPorte P, Tsai E, et al. (2002) Doublecortin is required in mice for lamination of the hippocampus but not the neocortex. J Neurosci 22(17):7548–7557
D’Arcangelo G, Miao GG, Chen SC, Soares HD, Morgan JI, et al. (1995) A protein related to extracellular matrix proteins deleted in the mouse mutant reeler. Nature 374(6524):719–723
des Portes V, Pinard JM, Billuart P, Vinet MC, Koulakoff A, et al. (1998) A novel CNS gene required for neuronal migration and involved in X-linked subcortical laminar heterotopia and lissencephaly syndrome. Cell 92(1):51–61
Dobyns WB, Truwit CL (1995) Lissencephaly and other malformations of cortical development: 1995 update. Neuropediatrics 26(3):132–147
Efimov VP, Morris NR (2000) The LIS1-related NUDF protein of Aspergillus nidulans interacts with the coiled-coil domain of the NUDE/RO11 protein. J Cell Biol 150(3):681–688
Feng Y, Walsh CA (2004) Mitotic spindle regulation by Nde1 controls cerebral cortical size. Neuron 44(2):279–293
Francis F, Koulakoff A, Boucher D, Chafey P, Schaar B, et al. (1999) Doublecortin is a developmentally regulated, microtubule-associated protein expressed in migrating and differentiating neurons. Neuron 23(2):247–256
Gleeson JG, Allen KM, Fox JW, Lamperti ED, Berkovic S, et al. (1998) Doublecortin, a brain-specific gene mutated in human X-linked lissencephaly and double cortex syndrome, encodes a putative signaling protein. Cell 92(1):63–72
Gleeson JG, Lin PT, Flanagan LA, Walsh CA (1999) Doublecortin is a microtubule-associated protein and is expressed widely by migrating neurons. Neuron 23(2):257–271
Hafezparast M, Klocke R, Ruhrberg C, Marquardt A, Ahmad-Annuar A, et al. (2003) Mutations in dynein link motor neuron degeneration to defects in retrograde transport. Science 300(5620):808–812
Hattori M, Adachi H, Tsujimoto M, Arai H, Inoue K (1994) Miller-Dieker lissencephaly gene encodes a subunit of brain platelet-activating factor acetylhydrolase [corrected]. Nature 370(6486):216–218
Hirotsune S, Takahara T, Sasaki N, Hirose K, Yoshiki A, et al. (1995) The reeler gene encodes a protein with an EGF-like motif expressed by pioneer neurons. Nat Genet 10(1):77–83
Hirotsune S, Fleck MW, Gambello MJ, Bix GJ, Chen A, et al. (1998) Graded reduction of Pafah1b1 (Lis1) activity results in neuronal migration defects and early embryonic lethality. Nat Genet 19(4):333–339
Howell BW, Herrick TM, Cooper JA (1999) Reelin-induced tyrosine [corrected] phosphorylation of disabled 1 during neuronal positioning. Genes Dev 13(6):643–648
Kappeler C, Saillour Y, Baudoin JP, Tuy FP, Alvarez C, et al. (2006) Branching and nucleokinesis defects in migrating interneurons derived from doublecortin knockout mice. Hum Mol Genet 15(9):1387–1400
Keays DA, Tian G, Poirier K, Huang GJ, Siebold C, et al. (2007) Mutations in alpha-tubulin cause abnormal neuronal migration in mice and lissencephaly in humans. Cell 128(1):45–57
Koizumi H, Higginbotham H, Poon T, Tanaka T, Brinkman BC, et al. (2006) Doublecortin maintains bipolar shape and nuclear translocation during migration in the adult forebrain. Nat Neurosci 9(6):779–786
Lambert de Rouvroit C, Goffinet AM (2001) Neuronal migration. Mech Dev 105(1–2):47–56
Marin O, Rubenstein JL (2001) A long, remarkable journey: tangential migration in the telencephalon. Nat Rev Neurosci 2(11):780–790
Morris NR (1975) Mitotic mutants of Aspergillus nidulans. Genet Res 26(3):237–254
Niethammer M, Smith DS, Ayala R, Peng J, Ko J, et al. (2000) NUDEL is a novel Cdk5 substrate that associates with LIS1 and cytoplasmic dynein. Neuron 28(3):697–711
Noctor SC, Flint AC, Weissman TA, Dammerman RS, Kriegstein AR (2001) Neurons derived from radial glial cells establish radial units in neocortex. Nature 409(6821):714–720
Noctor SC, Flint AC, Weissman TA, Wong WS, Clinton BK, et al. (2002) Dividing precursor cells of the embryonic cortical ventricular zone have morphological and molecular characteristics of radial glia. J Neurosci 22(8):3161–3173
Oakley BR, Morris NR (1980) Nuclear movement is beta-tubulin-dependent in Aspergillus nidulans. Cell 19(1):255–262
Ohshima T, Ward JM, Huh CG, Longenecker G, Veeranna, et al. (1996) Targeted disruption of the cyclin-dependent kinase 5 gene results in abnormal corticogenesis, neuronal pathology and perinatal death. Proc Natl Acad Sci U S A 93(20):11173–11178
Rakic P, Sidman RL (1973) Weaver mutant mouse cerebellum: defective neuronal migration secondary to abnormality of Bergmann glia. Proc Natl Acad Sci U S A 70(1):240–244
Reiner O, Carrozzo R, Shen Y, Wehnert M, Faustinella F, et al. (1993) Isolation of a Miller-Dieker lissencephaly gene containing G protein beta-subunit-like repeats. Nature 364(6439):717–721
Rice DS, Curran T (2001) Role of the reelin signaling pathway in central nervous system development. Annu Rev Neurosci 24:1005–1039
Sapir T, Horesh D, Caspi M, Atlas R, Buirgess HA, et al. (2000) Doublecortin mutations cluster in evolutionarily conserved functional domains. Hum Mol Genet 9(5):703–712
Sasaki S, Mori D, Toyo-oka K, Chen A, Carrett-Beal L, et al. (2005) Complete loss of Ndel1 results in neuronal migration defects and early embryonic lethality. Mol Cell Biol 25(17):7812–7827
Sheldon M, Rice DS, D’Arcangelo G, Yoneshima H, Nakajima K, et al. (1997) Scrambler and yotari disrupt the disabled gene and produce a reeler-like phenotype in mice. Nature 389(6652):730–733
Shu T, Ayala R, Nguyen MD, Xie Z, Gleeson JG, et al. (2004) Ndel1 operates in a common pathway with LIS1 and cytoplasmic dynein to regulate cortical neuronal positioning. Neuron 44(2):263–277
Smith DS, Niethammer M, Ayala R, Zhou Y, Gambello MJ, et al. (2000) Regulation of cytoplasmic dynein behaviour and microtubule organization by mammalian Lis1. Nat Cell Biol 2(11):767–775
Stanfield BB, Cowan WM (1979) The morphology of the hippocampus and dentate gyrus in normal and reeler mice. J Comp Neurol 185(3):393–422
Tanaka T, Serneo FF, Higgins C, Gambello MJ, Wynshaw-Boris A, et al. (2004a) Lis1 and doublecortin function with dynein to mediate coupling of the nucleus to the centrosome in neuronal migration. J Cell Biol 165(5):709–721
Tanaka T, Serneo FF, Tseng HC, Kulkarni AB, Tsai LH, et al. (2004b) Cdk5 phosphorylation of doublecortin ser297 regulates its effect on neuronal migration. Neuron 41(2):215–227
Taylor KR, Holzer AK, Bazan JF, Walsh CA, Gleeson JG (2000) Patient mutations in doublecortin define a repeated tubulin-binding domain. J Biol Chem 275(44):34442–34450
Trommsdorff M, Borg JP, Margolis B, Herz J (1998) Interaction of cytosolic adaptor proteins with neuronal apolipoprotein E receptors and the amyloid precursor protein. J Biol Chem 273(50):33556–33560
Trommsdorff M, Gotthardt M, Hiesberger T, Shelton J, Stockinger W, et al. (1999) Reeler/Disabled-like disruption of neuronal migration in knockout mice lacking the VLDL receptor and ApoE receptor 2. Cell 97(6):689–701
Willins DA, Xiang X, Morris NR (1995) An alpha tubulin mutation suppresses nuclear migration mutations in Aspergillus nidulans. Genetics 141(4):1287–1298
Xiang X, Morris NR (1999) Hyphal tip growth and nuclear migration. Curr Opin Microbiol 2(6):636–640
Xiang X, Beckwith SM, Morris NR (1994) Cytoplasmic dynein is involved in nuclear migration in Aspergillus nidulans. Proc Natl Acad Sci U S A 91(6):2100–2104
Xiang X, Osmani AH, Osmani SA, Xin M, Morris NR (1995) NudF, a nuclear migration gene in Aspergillus nidulans, is similar to the human LIS-1 gene required for neuronal migration. Mol Biol Cell 6(3):297–310
Xie Z, Samuels BA, Tsai LH (2006) Cyclin-dependent kinase 5 permits efficient cytoskeletal remodeling—a hypothesis on neuronal migration. Cereb Cortex 16 Suppl 1:i64–i68
Acknowledgments
The author thanks Fiona Francis for her comments on the manuscript. He is also indebted to Nadia Bahi-Buisson and Xin Xiang for the MRI and Aspergillus images.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License ( https://creativecommons.org/licenses/by-nc/2.0 ), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
Keays, D.A. Neuronal migration: unraveling the molecular pathway with humans, mice, and a fungus. Mamm Genome 18, 425–430 (2007). https://doi.org/10.1007/s00335-007-9034-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00335-007-9034-4