
Abstract
Since the genetic basis for Down syndrome (DS) was described, understanding the causative relationship between genes at dosage imbalance and phenotypes associated with DS has been a principal goal of researchers studying trisomy 21 (Ts21). Though inferences to the gene-phenotype relationship in humans have been made, evidence linking a specific gene or region to a particular congenital phenotype has been limited. To further understand the genetic basis for DS phenotypes, mouse models with three copies of human chromosome 21 (Hsa21) orthologs have been developed. Mouse models offer access to every tissue at each stage of development, opportunity to manipulate genetic content, and ability to precisely quantify phenotypes. Numerous approaches to recreate trisomic composition and analyze phenotypes similar to DS have resulted in diverse trisomic mouse models. A murine intraspecies comparative analysis of different genetic models of Ts21 and specific DS phenotypes reveals the complexity of trisomy and important considerations to understand the etiology of and strategies for amelioration or prevention of trisomic phenotypes. By analyzing individual phenotypes in different mouse models throughout development, such as neurologic, craniofacial, and cardiovascular abnormalities, greater insight into the gene-phenotype relationship has been demonstrated. In this review we discuss how phenotype-based comparisons between DS mouse models have been useful in analyzing the relationship of trisomy and DS phenotypes.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Trisomy 21 (Ts21) is one of the most prevalent serious congenital malformations of genetic origin and the most common human aneuploidy compatible with survival. In the United States, 1 of every 733 live births has Ts21 (CDC 2006). Worldwide about 220,000 infants with Ts21 are born each year with phenotypes collectively referred to as Down syndrome (DS) (Christianson et al. 2006). Individuals with DS have subsets of approximately 80 clinical phenotypes, including cognitive impairment, craniofacial dysmorphology, congenital heart defects, gastrointestinal tract abnormalities, acute megakaryoblastic leukemia, immunologic defects, endocrine abnormalities, neuropathology leading to dementia, and dysmorphic physical features. To characterize the variability and origin of the many characteristic features of DS, multiple phenotypes have been studied during fetal and postnatal development (Delabar et al. 2006). The incidence and severity of specific DS phenotypes are influenced by genetic, environmental, and stochastic factors that occur throughout development and after birth (Cohen 1999; Epstein 2001).
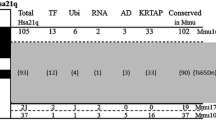
The long arm of human chromosome 21 (Hsa21) contains 33.7 Mb and approximately 230 genes that are homologous to syntenic regions of mouse chromosomes 16, 17, and 10 (Fig. 1) (Gardiner et al. 2003). The distal end of mouse chromosome 16 (Mmu16) contains 144 conserved and minimally conserved Hsa21 orthologs (Chromosome 21 gene function and pathway database, http://www.chr21db.cudenver.edu/) (Gardiner et al. 2003; Nikolaienko et al. 2005), and a number of segmental trisomy mouse models have been made with portions of this chromosomal region at dosage imbalance (Table 1). The most widely used and well-studied mouse model of trisomy and DS phenotypes is the Ts(1716)65Dn (hereafter Ts65Dn). This segmental trisomy model has a small translocation chromosome comprising the distal region of Mmu16 attached to the centromeric end of Mmu17 (Davisson et al. 1993; Reeves et al. 1995) and contains about half of the Hsa21 gene orthologs (Hattori et al. 2000). Ts65Dn mice show DS-related phenotypes, including reduced birth weight, cognitive and behavioral impairments, craniofacial abnormalities, perinatal lethality, cardiovascular malformations, and neurologic structural deficiencies (Baxter et al. 2000; Belichenko et al. 2004; Cooper et al. 2001; Holtzman et al. 1996; Lorenzi and Reeves 2006; Moore 2006; Richtsmeier et al. 2000; Roper et al. 2006b; Rueda et al. 2005). A number of phenotypes characterized in Ts65Dn mice have been used as a standard to compare the incidence and severity of trisomic phenotypes in other mouse models (Aldridge et al. 2007; Arron et al. 2006; Olson et al. 2004a, 2004b, 2007; Richtsmeier et al. 2002; Sago et al. 2000; Siarey et al. 2005). Other segmental trisomies of Mmu16 include Ts(12;16C-tel)1Cje and Dp(16Cbr1-ORF9)1Rhr (Ts1Cje and Ts1Rhr, respectively). Additional models can be made when a third copy of a gene or region is added to or subtracted from existing models. Both Ms1Cje/Ts65Dn and Ms1Rhr/Ts65Dn were produced by breeding the corresponding monosomy of newly developed Ts1Cje and Ts1Rhr trisomies to the existing Ts65Dn mouse (Olson et al. 2004a; Sago et al. 2000). Ts[Rb(12.Ts171665Dn)]2Cje (Ts2Cje) mice were identified after a fortuitous translocation of the T65Dn marker chromosome (Villar et al. 2005a). Owing to a conservation of genetic content and developmental processes in human and mouse, the mouse has served as an effective research model for many DS phenotypes.

Hsa21 and mouse homology, and trisomic mouse models. Homology of the long arm of Hsa21 is represented on Mmu16, 17, and 10. The trisomic mouse models show the trisomic Hsa21 homologous regions for each model (striped region for Ts16). Information is based on mouse build 36, http://www.ensembl.org/Mus_musculus/index.html, and build 42 of the human genome, http://www.ensembl.org/Homo_sapiens/index.html
Hsa21 genes have also been introduced into mouse cell lines to produce transchromosomic mice through microcell-mediated chromosome transfer and embryonic stem cell technology (O’Doherty et al. 2005; Shinohara et al. 2001). The recently developed Tc(Hsa21)1TybEmcf (Tc1) mouse has germline transmission of an almost intact Hsa21 (proximal and distal gaps omit approximately 10% of Hsa21 sequence and 8% of Hsa21 genes) but tissue-specific variability in cellular trisomy. Tc1 mice exhibit DS-like neurologic, behavioral, cardiovascular, and craniofacial abnormalities (O’Doherty et al. 2005).
Additional mouse models with three copies of segments of mouse chromosomes homologous to Hsa21 have been used to understand the gene-phenotype relationship of DS. These models contain dosage imbalance of some Hsa21 orthologs as well as large segments of nonhomologous genetic material. Trisomy 16 (Ts16) embryos are trisomic for all of Mmu16 (∼98 Mb), including the Hsa21 homologous region on the distal end of Mmu16, and also contain trisomic regions homologous to Hsa3, 8, 12, 6, and 22. Ts16 offspring die perinatally and this has limited studies in this model to developmental phenotypes, including edema and fetal cardiac, neurologic, and thymic abnormalities (Epstein et al. 1985; Hiltgen et al. 1996; Miyabara et al. 1982). The Ts(1617)43H (Ts43H) mouse is trisomic for 30 Mb of proximal Mmu17 and has been investigated for DS-related behavioral and gene expression phenotypes (Vacik et al. 2005). The breakpoint on the T43H segmental chromosome occurs in the Hsa21 homologous region and the Ts43H model is trisomic for at least nine Hsa21 orthologs found on Mmu17. Because of the large trisomic Mmu17 region, Ts43H has been estimated to have an overall greater number of genes at dosage imbalance than Ts65Dn or Tc1 mice. DS-like phenotypes identified in the Ts43H and Ts16 models could be due to three copies of Hsa21 orthologs, trisomy of large genomic regions, or trisomic heterogeneity that disturbs distinct pathways but leads to similar phenotypes.
Transgenic mouse models with single Hsa21 orthologous genes or regions at dosage imbalance have also been created (Altafaj et al. 2001; Ema et al. 1999; Kola and Hertzog 1997; Roubertoux et al. 2006; Smith et al. 1997). These models, along with mice that have a specific gene or region knocked out or deleted, provide valuable data for understanding gene function, especially when analyzed in parallel with segmental trisomy models. Because of differences in quantitative gene expression, absence of potential regulatory regions, and limitations of splice variants in transgenic mice, careful consideration must be given when analyzing these models. Although single trisomic genes may have a large effect on a specific phenotype, spatial and temporal gene expression must be accurately compared with segmental trisomy models as well as with individuals with DS.
The most useful intraspecies assessments compare well-defined phenotypes of trisomic and control euploid mice at similar ages or developmental stages, examine identical tissues, and utilize experimental procedures employing similar rigor and precise quantification (Sago et al. 1998). Because of genetic differences, it is likely that only a subset of DS-like phenotypes will be represented in a mouse model and each model must be thoroughly evaluated for phenotypes that recapitulate the human condition. Intraspecies comparisons must consider additional factors, including accurate gene expression, and different patterns of expression or function of human or mouse genes or regions in mouse models (Gardiner 2003; Reeves 2006). Intraspecies comparisons between strains should either use a similar genetic background (optimally using littermate euploid control animals) or provide methodologic means to adjust for differences in genetic background (Olson et al. 2004b; Roper and Reeves 2006; Sago et al. 2000) since it has been shown that different backgrounds may affect trisomic phenotypes (Villar et al. 2005b). Other inherent differences between trisomic strains include presence/absence of an extra centromere, potential trisomic maternal environment during development, and presence of trisomy in every cell (Box 1). Even with similar rigor, dissimilarities in findings may be the result of experimental and methodologic differences or phenotypic variation (Insausti et al. 1998; Lorenzi and Reeves 2006; Richtsmeier et al. 2000).

Intraspecies comparison: Interaction and modifier loci
The incidence and severity of phenotypes in individuals with DS is highly variable. The importance of heterotrisomy (inheritance of multiple nonhomologous alleles) in specific regions has been demonstrated to correlate with pathogenesis in trisomy (Baptista et al. 2000). Incidence and severity of traits also vary in DS mouse models. For example, mandibular traits of Ts65Dn were demonstrated to be more variable in Ts65Dn than euploid mice (Richtsmeier et al. 2000). Interacting loci from trisomic regions may be important in producing an equivalent DS-like phenotype. Attenuated phenotypes in different murine models when trisomic content is reduced is most likely the result of interacting factors in separate trisomic regions (Olson et al. 2004a, 2007; Sago et al. 1998, 2000). A single gene with major effect may be modified by other trisomic loci, and such interactions may represent genes defined as sufficient or necessary for a specific phenotype (Olson et al. 2004a, 2007; Salehi et al. 2006). Phenotypic variation also illustrates the possible importance of interacting loci and intervening nongenic sequences in determining phenotype (Antonarakis and Epstein 2006; Antonarakis et al. 2004).
Modifier loci from nontrisomic regions may also impact incidence and severity of phenotypes. Modifier loci such as GATA1 and CRELD1 are not located on Hsa21 but have been shown to have importance in DS childhood leukemia and heart abnormalities, respectively (Maslen et al. 2006; Vyas and Crispino 2007). Nontrisomic modifier loci may also be of importance in mouse models since many trisomic models cannot be inbred and therefore are maintained on a mixed background (O’Doherty et al. 2005); Paz-Miguel et al. 2001; Vacik et al. 2005). The effect of genetic background on the cardiovascular development of Ts16 embryos indicates that frequency and timing of abnormalities in pharyngeal arch arteries vary based on the genetic background in the four strains tested (Villar et al. 2005b). The importance of background and modifier loci needs to be investigated in future intraspecies studies.
Phenotypic analysis throughout development
Teaming comparative genomic analysis with developmental studies of DS models has the power to uncover the genesis of a specific phenotype by identifying where in development abnormal diverges from normal. Embryonic studies with Ts16 embryos provided the first insights into the mechanisms by which dosage imbalance of trisomic genes may impinge upon normal developmental processes in complex systems such as cardiovascular (Miyabara et al. 1982) and neurologic development (Ozand et al. 1984; Singer et al. 1984). Identifying the spatial, temporal, and molecular defects leading to an abnormal phenotype requires precise identification and quantification (e.g., cell number, volumetric analysis, complete histologic examination) since such differences may be small at their inception and may occur only in a subset of trisomic individuals. A phenotype-based analysis throughout development may not only lead to the identification of genes causing the phenotype, but may lead to identification of a particular pathway that may be important for therapeutic intervention. A phenotype-based intraspecies comparison may offer insight into the gene-phenotype interaction if development is different among trisomic models.
Developing trisomic humans and mice have been described as “developmentally delayed” (Holtzman et al. 1996; Reeves et al. 1995; Wisniewski 1990). In trisomic mouse models, this unfortunate nomenclature has often been used to describe developing and neonatal mice that are smaller than euploid littermates yet fails to define specific areas of dissimilar development. Ts16 embryos show reduced brain weight and disrupted neuronal transmitter system development (Ozand et al. 1984; Singer et al. 1984), reduced number and delayed maturation of thymocytes (Epstein et al. 1985), and reduced endocardial mesenchymal cells (Hiltgen et al. 1996) compared with euploid littermates. To date, little data reflect upon the delays associated with prenatal development in particular structures at specific embryonic stages in other trisomic models. It is unclear if all structures are delayed at the same developmental point or only those that later become dysmorphic. If there is a cellular paucity in a structure during development, is it only because of miscues in gene expression relating to those particular cells? Does a phenotypic change in a single population of cells or structure lead to abnormalities in other cells or structures? If a structure is “delayed,” does the window of opportunity for tissue interaction close and thus the derived tissues can never recover? Is each area of dysmorphology under separate genetic control? Do many developmental abnormalities combine to produce a significant phenotype? These questions remain to be answered by thorough investigation.
Hypotheses for mechanisms of gene action
Different hypotheses have been presented to account for the relationship between Ts21 and the occurrence of DS phenotypes. A number of the most common hypotheses are presented here, although components of these hypotheses may not be mutually exclusive. Several genes are likely to be involved and specific genes and genetic mechanisms could differ for an individual phenotype. The “gene dosage effects” model states that three copies of a particular gene or genes cause increased expression of specific transcripts that lead to DS phenotypes. The “amplified developmental instability” model proposes that it is not the presence of three copies of a particular gene but rather the presence of a threshold number of genes (no matter the genes) that leads to DS phenotypes (Patterson and Costa 2005). The “Down syndrome critical region” (DSCR) hypothesis states that genes in a defined region are responsible for a number of the essential DS phenotypes (Delabar et al. 1993; Korenberg et al. 1994). Gene expression analyses have led to hypotheses that the majority of genetic dysregulation is the product of trisomic genes (Mao et al. 2005) or that the increased expression of trisomic genes leads to a global destabilization in the expression patterns of many disomic genes (Saran et al. 2003). A few genes may have a major effect (Arron et al. 2006; Salehi et al. 2006) or an interaction of many genes with modest affect may underlie the etiology of a specific phenotype. Functional noncoding elements in the genome may also play an important role in DS phenotypes (Antonarakis and Epstein 2006; Antonarakis et al. 2004). Intraspecies comparisons of mice with differing trisomic content illustrate the complexity of the gene-phenotype relationship in DS, present evidence that integrates many of the ideas from these different hypotheses, and refines our understanding of the mechanisms of gene action leading to specific DS phenotypes.
Intraspecies comparison of structural and cognitive phenotypes
Ts21 is the most common genetic cause of mental retardation and cognitive impairment is found in all individuals with DS. By adulthood the brain is microcephalic with proportionately greater reductions in the hippocampus, prefrontal cortex, and cerebellum (Epstein 2001; Pennington et al. 2003). Gross structures as well as cellular components have been quantified in mouse models to investigate the association between trisomic brain structure and function (Belichenko et al. 2004; Cooper et al. 2001; Holtzman et al. 1996; Kurt et al. 2000; Reeves et al. 1995). A comparison of the cerebellum and the cerebrum in different trisomic models has suggested interesting connections between pathology and dosage imbalance. Ts65Dn mice display a small cerebellum, with a reduction in cerebellar granule cell density that recapitulates the deficiency seen in individuals with DS (Baxter et al. 2000). A diminution in Purkinje cell density is also characteristic of the adult Ts65Dn brain. Though there is no apparent reduction in size of the cerebrum, the shape of the Ts65Dn cerebrum is different compared with that of euploid littermates (Aldridge et al. 2007). Ts1Cje mice have a similar reduction in cerebellar volume as do Ts65Dn mice, but they have only a slight decrease in cerebellar granule cell density and no significant change in Purkinje cell density (Olson et al. 2004b). Ms1Cje/Ts65Dn mice do not display a significant reduction in cerebellar volume and have a significantly reduced cerebellar granule cell density but no reduction in Purkinje cell density. A direct comparison between Ts65Dn, Ts1Cje, and Ms1Cje/Ts65Dn mice revealed that cerebellar size deficit may not be directly related to the paucity in granule cell density. The Ts1Rhr cerebellum is slightly smaller but is not as reduced as either the Ts65Dn or the Ts1Cje cerebellum, with a shape distinctly different from euploid littermates (Aldridge et al. 2007; Olson et al. 2004b). Although cerebellar granule cell and Purkinje cell density is not significantly altered, both size and shape of the cerebrum differ between Ts1Rhr and euploid littermates (Aldridge et al. 2007). Limited studies of Tc1 brain structure showed a reduction in cerebellar volume and granule cell density when compared with those of euploid littermates O’Doherty et al. (2005). These analyses illustrate that specific brain phenotypes may be under different genetic control and that a similar pathology may result from heterogeneous sources or not be related to an obvious cellular deficiency. Different mouse models may be more useful for an in-depth study of brain phenotypes (e.g., Ts65Dn for Purkinje cell deficit and Ts1Rhr for cerebrum size deficit).
Long-term memory research has supported the notion of a hippocampal dysfunction related to DS (Pennington et al. 2003). The hippocampus is thought to be important in learning and memory, two parameters that are affected in individuals with DS. Total hippocampal volume is not changed in Ts65Dn mice, though reductions in dentate gyrus volume and granule cells have been documented (Insausti et al. 1998; Lorenzi and Reeves 2006; Rueda et al. 2005). Like Ts65Dn mice, Ts1Rhr total hippocampal volume is not decreased compared with that of euploid, although no quantification of Ts1Rhr-specific hippocampal regions and cellular constituents has been done (Olson et al. 2007). When survival and proliferation of Ts65Dn hippocampal granule cells were examined in adult mice, no significant difference was observed in either proliferation or survival of granule cells (Rueda et al. 2005). There were, however, a smaller number of proliferating cells in the dentate gyrus of older Ts65Dn mice. Fewer proliferating cells in adult trisomic brains coupled with the reduction of basal forebrain cholinergic neurons may lead to increased neurodegeneration of cells in the adult brain (Cooper et al. 2001). No reduction of volume of the dentate gyrus was seen in six-day-old Ts65Dn mice, but significantly fewer dentate gyrus granule cells and mitotic cells at six days were found when compared with euploid littermates. This result suggested that differences in neurogenesis of granule cells may contribute to the lower number of granule cells in the dentate gyrus of Ts65Dn mice (Lorenzi and Reeves 2006).
Changes in hippocampal structure may affect learning and memory as well as long-term synaptic plasticity (Galdzicki and Siarey 2003). Ts65Dn mice have been characterized as hyperactive and with deficits in learning and memory as defined by the Morris water maze (Escorihuela et al. 1995; Holtzman et al. 1996; Reeves et al. 1995). Ts65Dn mice generally decrease the time taken to locate both visible and hidden platforms in successive trials (nonspatial and spatial learning, respectively), although their improvement is significantly less than that of euploid littermates. In the probe trial test, Ts65Dn mice spend less time in the quadrant that contained the platform. Ts65Dn, Ts1Cje, and Ms1Cje/Ts65Dn mice were generated to correlate the genetic contributions of different Hsa21 homologous regions with behavioral characteristics associated with DS (Sago et al. 2000). Although none of the strains were different from euploid controls in the visible platform test, Ts65Dn, Ts1Cje, and Ms1Cje/Ts65Dn were all deficient in the hidden platform test compared with euploid controls. By comparison, Ts65Dn mice were the most, Ts1Cje were slightly less, and Ms1Cje/Ts65Dn were somewhat less impaired. Ts65Dn mice also spent less time in a specific quadrant test, whereas Ts1Cje mice were a little more like euploid littermates. In contrast with Ts65Dn, Ts1Cje mice were found to be hypoactive and Ms1Cje/Ts65Dn mice were not different than euploid mice in their activity. The most significant differences between Ts65Dn and Ts1Cje mice were observed in the reverse probe dwell and crossing tests examining cognitive flexibility. Overall, learning defects in Ts1Cje mice were similar to but slightly less than those seen in Ts65Dn mice. Ms1Cje/Ts65Dn mice showed little impairment in most tasks and deficits were less than those seen in Ts65Dn mice.
Similar results were seen when synaptic plasticity was compared in Ts65Dn and Ts1Cje strains. Long-term potentiation (LTP) and long-term depression (LTD) are physiologic measures associated with learning and memory (Antonarakis and Epstein 2006). Comparing Ts65Dn and Ts1Cje mice bred onto a similar background (but not littermates) showed differences between the two strains in hippocampal electrophysiology (Siarey et al. 1997, 1999, 2005). Ts65Dn mice showed reduction of LTP in CA1 and dentate gyrus areas and increased LTD in CA1 (Kleschevnikov et al. 2004; Siarey et al. 1997, 1999). LTP was decreased and LTD was increased in Ts1Cje mice but the overall changes in long-term synaptic plasticity were less dramatic than those in Ts65Dn mice. The implications from this study were that the contribution from the trisomic segment common to Ts65Dn and Ts1Cje was significant for synaptic plasticity but other important interacting genes are part of the additional trisomic region in Ts65Dn mice.
Interestingly, Tc1 mice with an entirely different genetic background had a significantly reduced LTP in the dentate gyrus and a trend toward hyperactivity. These results reflect the robustness of these phenotypes even with different potential modifying loci, transchromosomal dissimilarities, including the human origin of the trisomy, and possible trisomic mosaicism (O’Doherty et al. 2005). Other tests examining learning and memory still need to be addressed with the Tc1 model (Reeves 2006). Ts1Rhr mice showed no deficiencies in either Morris water maze or synaptic plasticity when compared to euploid controls (Olson et al. 2007). Ts43H mice were not found to be hyperactive but had spatial learning defects in the Morris maze test comparable to those seen in Ts65Dn mice (Vacik et al. 2005). Hyperactivity differences in Ts43H may be due to necessary Hsa21 orthologs not included in the Ts43H mice and the spatial learning deficits may mean additional Hsa21 orthologs or trisomic heterogeneity may play an important role in these traits.
The structural and functional neuroanatomical studies reveal that the relationship between trisomic gene content and DS-related phenotypes may be unique to each phenotype. In general, models with less trisomic genetic content exhibit an attenuated phenotype, including cerebellar size, Morris swim maze, and synaptic plasticity. Analyses with Ts65Dn, Ts1Cje, Ts1Rhr, and Ms1Rhr/Ts65Dn mice have demonstrated that the DSCR concept is incorrect. The comparative studies between segmental trisomic mice illustrate that the interaction between genes and/or regions in two different areas may be important to both the incidence and the severity of structural and functional neurologic phenotypes, thus superseding the “one gene-one phenotype” hypothesis.
Intraspecies comparison of craniofacial phenotypes
One of the most commonly associated phenotypes of Ts21 is the distinct craniofacial features seen in all individuals with DS. Using a sophisticated 3D skull analysis of Ts65Dn mice, Richtsmeier et al. (2000) showed that Ts65Dn mice exhibit craniofacial abnormalities that resemble those found in individuals with DS. Similar features included microcephaly, brachycephaly, small flattened face, reduced interorbital distance, and both a small maxilla and mandible. In Ts1Cje mice, more than 80% of the measurements were conserved between the two models and affected the same bones of the skull (Richtsmeier et al. 2002). Ts65Dn mice, however, had brachycephaly and bones were affected to a slightly greater degree. Both mouse models displayed a smaller mandible than euploid littermates, with the reduction in size specific to the coronoid and angular processes, and also had slight morphometric changes that are specific to each strain.
In contrast with Ts65Dn and Ts1Cje mice, Ts1Rhr mice, with the putative DSCR at dosage imbalance, had a larger skull and an overall rostrocaudal elongation of the skull compared with euploid littermates (Olson et al. 2004a). The mandible of Ts1Rhr mice was larger and had a different shape than Ts65Dn and Ts1Cje mice, with differences concentrated in the condyle, inferior ramus, and incisive alveolar. Ms1Rhr/Ts65Dn mice, with all of the genes at dosage imbalance the same as Ts65Dn except in the DSCR, showed similar but attenuated effects on the skull as seen in Ts65Dn and Ts1Cje mice. From these results, the DSCR was shown to contain genes that were not sufficient and largely not necessary to cause DS-like craniofacial abnormalities.
Light microscopy showed no gross differences in the craniofacial structure of Tc1 mice (O’Doherty et al. 2005). Simple vector measurements showed no differences in the skull of Tc1 mice but indicated reduction in mandibular structure compared with euploid littermates. The mandibular differences specified trisomic effects in the coronoid and angular processes similar to Ts65Dn and Ts1Cje mice. Differences in methodologies measuring craniofacial structure may account for additional abnormalities not ascertained in Tc1 mice. Alternatively, genes important in craniofacial structure may not have the same expression in the transchromosomic model as found in segmental trisomy models.
At birth, Ts65Dn mice have differences in craniofacial structure in the anterior face, anterior and posterior neurocranium, palate, and mandible (Hill et al. 2007). Although some differences in dysmorphologies were identified between adult and newborn Ts65Dn mice, an analysis of postnatal growth patterns between trisomic and euploid mice showed that many of these early changes led to differences seen in adult Ts65Dn mice. It has also been hypothesized that slight craniofacial differences seen in Ts65Dn and Ts1Cje mice could be due to developmental differences between the strains (Richtsmeier et al. 2002). Similar developmental hypotheses could be extended to each different model, with differences in the dosage imbalance of certain genes or regions causing small developmental alterations of craniofacial structure.
Intraspecies comparisons of cardiac phenotypes
The congenital heart defects (CHD) present in 50% of DS neonates include atrial, ventricular, and atrioventricular (AV) septal and valvar defects (Freeman et al. 1998; Wessels et al. 2003). These malformations are typically attributed to AV canal abnormalities with failure of proper endocardial cushion formation or fusion in the inner curvature of the heart tube of DS individuals. However, complex malformations also involving the outflow tract (e.g., tetralogy of Fallot) contribute to the cardiac phenotype in many DS cases (Freeman et al. 1998; Wessels et al. 2003). In a study of DS neonates undergoing both cardiac physical exam and echocardiography within a month of birth, 66% of this DS group had detectable cardiac anomalies. AVSD was identified in one third of the cases with abnormal echocardiographic findings, while tetralogy of Fallot was found in one fifth of these DS CHD cases (McElhinney et al. 2002). Aberrant formation or maintenance of the aortic arch arteries, manifested through persistent ductus arteriosus or aberrant right subclavian artery, also occur at a higher frequency in DS fetuses and neonates than in the general population (Chaoui et al. 2005; McElhinney et al. 2002). Studies of DS individuals with CHD, especially those with segmental trisomy of only part of Hsa21, have been used to investigate the molecular basis of the cardiac malformations. Several candidate genes mapped to Hsa21 that may contribute to a cardiac phenotype have been identified: COLVI (Davies et al. 1994), DSCAM (Barlow et al. 2001), and DSCR1 (Rothermel et al. 2000). As yet, no one gene has been found to cause the complex, variable cardiac anomalies. The presence of an extra copy of one or more interacting genes from the distal region of Hsa21 may lead to disruption in the process of septation. Murine models in which only a subset of the candidate genes is triplicated may implicate or exclude several of the current candidate genes as the primary factors determining the predisposition to cardiac malformations.
The presence or absence of cardiovascular malformation has been carefully characterized in some but not all DS models. Cardiovascular abnormalities in Ts16 show complete penetrance, are apparent as septation occurs, and in some ways parallel the heart defects seen in DS, with more than half of the embryos displaying a common AV canal (Miyabara et al. 1982). The cardiovascular phenotype of Ts16 embryos also demonstrates the limitations of using whole chromosomal interspecies comparisons to identify the critical genes at dosage imbalance with analysis of complex structures derived from multiple tissue types. In analyzing the Ts16 cardiac phenotype, misalignment of the endocardial cushions, disruptions in neural crest, and loss of extracardiac mesoderm that typically contribute to septation are all posited to contribute to the observed atrioventricular, conotruncal, and atrial septal defects (Waller et al. 2000; Webb et al. 1997). Yet the right aortic arch and persistent truncus arteriosus identified in Ts16 embryos also resemble the cardiovascular phenotype of DiGeorge syndrome, a human syndrome associated with deletion of genetic material from Hsa22q11 (Waller et al. 2000). Murine genes orthologous to the DiGeorge region of Hsa22 are found on the proximal region of Mmu16 and are triplicated in Ts16, but not in trisomies containing only distal Mmu16 (Ts65Dn, Ts1Cje, and derived lines). Therefore, the severe cardiac phenotypes characterized in Ts16 may be viewed as a combination of the mechanisms that contribute to cardiac phenotypes in both DiGeorge and DS. Attempts to eliminate the cardiovascular phenotype of the Ts16 mice with reduction of one candidate gene (Dscr1) to diploid levels did not significantly alter the cardiac phenotype (Lange et al. 2005). Ts65Dn lacks the syntenic Hsa22 region of Mmu16, yet the cardiac abnormalities identified thus far in the segmental trisomy include right aortic arch and intracardiac septal defects (Moore 2006). The low frequency of gross cardiac anomalies, coupled with selective loss of trisomic neonates, prevented identification of the cardiac phenotype in the Ts65Dn mice. This DS model had been reported to lack any cardiac phenotype, so identification of a cardiovascular phenotype, albeit at a low rate, indicates the care that must be taken in characterization of each potential phenotype in each DS model. Though Ts65Dn mice do not have the severe phenotype of complete AV canal commonly associated with DS, the etiology of defects in the great vessels arise with abnormal formation and/or regression of the aortic arch arteries. Some aspects of DS CHD, such as tetralogy of Fallot, persistent ductus arteriosus, and aberrant right subclavian artery, also have their origins in aortic arch architecture. Therefore, shared elements of DS, Ts65Dn, and Ts16 cardiac phenotypes suggest some component of the aortic arch and outflow tract malformations may be attributed to the Hsa21 orthologs on distal Mmu16 .
The cardiac phenotype of the Tc1 mouse at embryonic day 14.5 (E14.5) resembled the abnormalities typically seen in DS CHD (O’Doherty et al. 2005). Seven of 11 Tc1 mice had an interventricular septal defect (one with overriding aorta), while one had unfused AV cushions. It should be noted that 20% of the euploid mice also had an interventricular septal defect and no later stages of development were presented, although the transmission rate in Tc1 on the F1 background was approximately 40%, lower than the 50% expected. Because failure of the cardiovascular system to form and function properly contributes to perinatal lethality in mouse and man, the non-Mendelian trisomic transmission rates seen in multiple DS models may indicate perinatal lethality of the most severely affected trisomic embryos due to cardiac or other anomalies.
Other researchers have begun to use the DS model mice as a primary line from which other transgenic lines may arise or be specifically created (Lange et al. 2005; Olson et al. 2004a; Salehi et al. 2006; Villar et al. 2005a). Definitive characterization of the cardiac phenotype in the Ts65Dn mice and other DS models as primary lines is crucial for ascertaining the effect of further genetic modifications produced by secondary modifications to these lines. The power of mouse models is that the genomic complement can be exactly manipulated and defined; we must therefore be just as exacting in identifying or, just as important, excluding the presence of DS phenotypes in the mice. Comparison of trisomic gene content and severity of cardiac phenotypes between Ts16, Ts65Dn, and other murine DS models may narrow the candidate regions of Mmu16, and hence Hsa21, responsible for different components (such as AV canal vs. outflow tract defects) of the complex and variable forms of DS CHD.
Gene expression analyses in trisomic mice
Powerful tools to analyze gene expression patterns have revealed a multitude of changes attributed to triplication of genes, yet results underscore the complexity of analyzing a multiphenotype, multigene syndrome such as DS. In humans, microarray analysis of fetal and adult tissue as well as cell lines derived from individuals with Ts21 show higher average gene expression from Hsa21 genes (FitzPatrick et al. 2002; Giannone et al. 2004; Mao et al. 2003, 2005). The dysregulation did not include all genes sampled from Hsa21, and secondary effects of increased transcript levels were noted on genes of nontrisomic chromosomes. Tissue-dependent patterns of overexpression were observed, further illustrating the complexity of correlating trisomic genes with transcript overexpression (Mao et al. 2005).
Gene expression studies in mouse models have shown similar complexity of results and interpretation. Although varied tissues and ages of mice were examined, analyses of neonatal and adult tissues of both Ts65Dn and Ts1Cje mice showed a near average 1.5-fold overexpression of triplicated genes (Amano et al. 2004; Dauphinot et al. 2005; Kahlem et al. 2004; Lyle et al. 2004; Saran et al. 2003). In addition, no average overexpression of genes found in two copies was seen either from nontriplicated regions of Mmu16 or the disomic chromosomes. Although average expression of trisomic genes was generally 1.5-fold, some genes in three copies were underexpressed, overexpressed to a greater degree, or unchanged. Differences in gene expression were specific to the tissue and developmental stage of the sample. Significant dysregulation of gene expression in disomic genes was reported in a number of studies, including one study that illustrated a global secondary disruption of gene expression due to trisomy (Saran et al. 2003). In Tc1 E14.5 embryos, microarray analysis on human arrays showed 39 of 131 Hsa21 genes and only 9 of 22,078 non-Hsa21 probe sets were overexpressed compared with euploid littermates (O’Doherty et al. 2005). When gene expression was examined in Ts43H mice, 20 brain-specific genes at dosage imbalance gave an average of 1.2-fold increased expression of euploid, with expression of only two genes reaching 1.5-fold expression (Vacik et al. 2005). In addition, 12 genes on the nontrisomic portion of chromosome 17 had expression levels that were 90% of euploid level. Brains from Ts2Cje mice exhibited a 1.5-fold expression level of specific trisomic genes comparable to Ts65Dn and different from euploid. Further data and analyses in both humans and mice are needed to reach biologically significant conclusions (Antonarakis and Epstein 2006; Reeves 2006).
Applications of an intraspecies approach
A phenotype-based intraspecies comparison of mice will help to elucidate how Ts21 leads to DS phenotypes. Three recent examples that examined development of DS-like phenotypes in mouse models implicated particular genes and pathways that may be important in specific phenotypes. These studies also illustrate the complexity of the gene-phenotype relationship in DS.
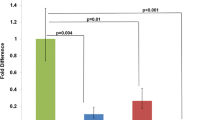
Previous observations in Ts65Dn mice showed an age-related atrophy and loss of basal forebrain cholinergic neurons (BFCNs) in the medial septal nucleus (Holtzman et al. 1996). In Ts65Dn mice, although nerve growth factor (NGF) levels were greater than normal, NGF retrograde transport was severely reduced. Normal size and number of BFCNs were found after delivering NGF directly to the BFCN cell bodies (Cooper et al. 2001). Retrograde transport of NGF in Ts1Cje mice was about 70% of control and significantly greater than Ts65Dn mice, but no significant differences in size and number of BFCNs nor the abnormal axonal phenotype were observed in Ts1Cje mice (Salehi et al. 2006). Protein levels of full-length App, triplicated in Ts65Dn but not in Ts1Cje mice, were linked to the abnormal retrograde transport of NGF. NGF transport, however, was not completely returned to normal in either Ts1Cje or Ts65Dn mice with only two functioning copies of App. From these experiments it was concluded that abnormal dosage of App combined with the trisomy of other regions was an important factor in the deficient transport of NGF and cholinergic neurodegeneration.
Mice with defects in the NFAT signaling pathway display many phenotypic similarities to DS, including neurologic, craniofacial, and endocardial cushion abnormalities (Arron et al. 2006). Though not all phenotypes occur in every model, Nfatc2-/-; Nfatc4-/- double knockout mice display aspects of brachycephaly, midface hypoplasia, and dysmorphic mandible. Dscr1, an inhibitor of calcineurin/NFATc signaling, triplicated in Ts65Dn and Ts1Cje mice, and expressed in higher levels in DS fetuses, was selected as a candidate gene for craniofacial defects. Dyrk1a was also selected as a candidate gene and was shown to regulate the calcineurin/NFAT signaling pathway in response to fibroblast growth factor 8 (FGF8). Dyrk1a and Dscr1 were shown to synergistically block NFAT-dependent transcription. Transgenic overexpression of Dyrk1a alone and with Dscr1 led to vascular defects and a failure in heart valve elongation, respectively. Interestingly, cortical neurons of Ts1Cje E13.5 embryos showed an increase of Dyrk1a expression but whole heads of E11.5 and postnatal day 1 (P1) hippocampal neurons did not have increased Dyrk1a or Dscr1 protein levels or alterations in phosphorylation of NFATc. The authors conclude that during brief developmental periods an increased dosage of Dscr1 and Dyrk1a reduces NFAT transcriptional activity and leads to mild versions of NFATc phenotypes.
The above example illustrates the complexity of intraspecies comparison in DS mouse models. Ts1Rhr mice (trisomic for Dyrk1a but not Dscr1) display craniofacial defects that are distinctly different from Ts65Dn and Ts1Cje mice (Olson et al. 2004a). Ms1Rhr/Ts65Dn mice (trisomic for all genes in Ts65Dn except those found in the “DSCR,” including Dscr1) exhibit slightly attenuated craniofacial abnormalities compared to Ts65Dn and Ts1Cje and do not have brachycephaly. Furthermore, Tc1 mice have only two copies of Dscr1 and mandibular abnormalities similar to Ts65Dn mice. A meta-analysis of all strains on a similar genetic background, at similar developmental timepoints, using stringent methodologic analyses will be useful to understand the complete role of Dyrk1a and Dscr1 in craniofacial structural abnormalities. Similar arguments could be made for the role of these genes in heart defects because of a recent report finding heart defects in Ts65Dn mice (Moore 2006). Dysregulation of multiple pathways may lead to similar DS-like phentoypes and it will be important to understand which pathways are important in Ts21.
Ts65Dn and Ts1Cje mice were used to examine the origin of the cerebellar size deficit and paucity of granule cells (Baxter et al. 2000; Olson et al. 2004b). Reduction in size of the Ts65Dn cerebellum was observed throughout development until and including P6 (Roper et al. 2006b). A granule cell deficiency was seen throughout development and traced to a deficit in mitosis of granule cell precursors at the day of birth. The mitotic deficit was linked to a decreased response by trisomic granule cell precursors to sonic hedgehog (Shh), a molecule important in proliferation of granule cell precursors. Treatment of newborn mice with a Shh pathway agonist overcame deficits in mitosis and the number of granule cell precursors six days later. Though the pathogenesis of the cerebellar deficit was described and linked to a cellular mechanism, the trisomic genetic mechanism leading to the cellular deficit is still unknown. Though Shh is not found on Hsa21, it is possible that it may be linked to many DS phenotypes (Roper et al. 2006b).
In these examples a single gene or pathway may provide a major factor in the development of a DS-like phenotype. As noted from the microarray studies, triplication of genes and/or genomic regions may lead to dysregulation of disomic genes and a number of different pathways that may appear unrelated to the initial trisomic insult. A number of distinct pathways, however, can produce a single phenotype or be used to correct a phenotype; therefore, therapy derived for a particular phenotype may involve a pathway that may be only indirectly disturbed by trisomy. Correlation of gene misexpression with trisomic phenotypes is among the next challenges in understanding DS phenotypes.
Conclusions
Comparative analyses of DS mouse models provide a powerful tool to identify genes that are misregulated by trisomy, place these genes in pathways leading to phenotypic abnormalities, and understand regulatory networks, including compensation and interaction. In the future, additional models need to be developed with three copies of Hsa21 homologous regions not found in current models. Existing models can be modified by adding or subtracting genes or regions to isolate specific gene-phenotype relationships. In future comparative studies, it is important to examine tissues at multiple timepoints during development and to use identical and robust methodologies. Intraspecies phenotype-based analyses show promise in finding ways to define phenotypic etiology and to ameliorate or prevent specific DS phenotypes.
References
Aldridge K, Reeves RH, Olson LE, Richtsmeier JT (2007) Differential effects of trisomy on brain shape and volume in related aneuploid mouse models. Am J Med Genet A 143(10):1060–1070
Altafaj X, Dierssen M, Baamonde C, Marti E, Visa J, et al. (2001) Neurodevelopmental delay, motor abnormalities and cognitive deficits in transgenic mice overexpressing Dyrk1A (minibrain), a murine model of Down’s syndrome. Hum Mol Genet 10(18):1915–1923
Amano K, Sago H, Uchikawa C, Suzuki T, Kotliarova SE, et al. (2004) Dosage-dependent over-expression of genes in the trisomic region of Ts1Cje mouse model for Down syndrome. Hum Mol Genet 13(13):1333–1340
Antonarakis SE, Epstein CJ (2006) The challenge of Down syndrome. Trends Mol Med 12(10):473–479
Antonarakis SE, Lyle R, Dermitzakis ET, Reymond A, Deutsch S (2004) Chromosome 21 and down syndrome: from genomics to pathophysiology. Nat Rev Genet 5(10):725–738
Arron JR, Winslow MM, Polleri A, Chang CP, Wu H, et al. (2006) NFAT dysregulation by increased dosage of DSCR1 and DYRK1A on chromosome 21. Nature 441(7093):595–600
Baptista MJ, Fairbrother UL, Howard CM, Farrer MJ, Davies GE, et al. (2000) Heterotrisomy, a significant contributing factor to ventricular septal defect associated with Down syndrome? Hum Genet 107(5):476–482
Barlow GM, Chen XN, Shi ZY, Lyons GE, Kurnit DM, et al. (2001) Down syndrome congenital heart disease: a narrowed region and a candidate gene. Genet Med 3(2):91–101
Baxter LL, Moran TH, Richtsmeier JT, Troncoso J, Reeves RH (2000) Discovery and genetic localization of Down syndrome cerebellar phenotypes using the Ts65Dn mouse. Hum Mol Genet 9(2):195–202
Belichenko PV, Masliah E, Kleschevnikov AM, Villar AJ, Epstein CJ, et al. (2004) Synaptic structural abnormalities in the Ts65Dn mouse model of Down Syndrome. J Comp Neurol 480(3):281–298
Centers for Disease Control, Prevention (CDC) (2006) Improved national prevalence estimates for 18 selected major birth defects - United States, 1999–2001. MMWR Morb Mortal Wkly Rep 54(51):1301–1305
Chaoui R, Heling KS, Sarioglu N, Schwabe M, Dankof A, et al. (2005) Aberrant right subclavian artery as a new cardiac sign in second- and third-trimester fetuses with Down syndrome. Am J Obstet Gynecol 192(1):257–263
Christianson A, Howson CP, Modell B (2006) 2006 March of Dimes Global Report on Birth Defects: The Hidden Toll of Dying and Disabled Children (White Plains, NY: March of Dimes Birth Defects Foundation), 98 pp
Cohen WI (1999) Health care guidelines for individuals with Down syndrome: 1999 revision. Down Syndrome Q 4:1–15
Cooper JD, Salehi A, Delcroix JD, Howe CL, Belichenko PV, et al. (2001) Failed retrograde transport of NGF in a mouse model of Down’s syndrome: reversal of cholinergic neurodegenerative phenotypes following NGF infusion. Proc Natl Acad Sci U S A 98(18):10439–10444
Dauphinot L, Lyle R, Rivals I, Dang MT, Moldrich RX, et al. (2005) The cerebellar transcriptome during postnatal development of the Ts1Cje mouse, a segmental trisomy model for Down syndrome. Hum Mol Genet 14(3):373–384
Davies GE, Howard CM, Farrer MJ, Coleman MM, Cullen LM, et al. (1994) Unusual genotypes in the COL6A1 gene in parents of children with trisomy 21 and major congenital heart defects. Hum Genet 93(4):443–446
Davisson MT, Schmidt C, Reeves RH, Irving NG, Akeson EC, et al. (1993) Segmental trisomy as a mouse model for Down syndrome. Prog Clin Biol Res 384:117–133
Davisson M, Akeson E, Schmidt C, Harris B, Farley J, et al. (2007) Impact of trisomy on fertility and meiosis in male mice. Hum Reprod 22(2):468–476
Delabar JM, Theophile D, Rahmani Z, Chettouh Z, Blouin JL, et al. (1993) Molecular mapping of twenty-four features of Down syndrome on chromosome 21. Eur J Hum Genet 1(2):114–124
Delabar JM, Aflalo-Rattenbac R, Creau N (2006) Developmental defects in trisomy 21 and mouse models. ScientificWorldJournal 6:1945–1964
Ema M, Ikegami S, Hosoya T, Mimura J, Ohtani H, et al. (1999) Mild impairment of learning and memory in mice overexpressing the mSim2 gene located on chromosome 16: an animal model of Down’s syndrome. Hum Mol Genet 8(8):1409–1415
Epstein CJ (2001) Down Syndrome (Trisomy 21). In Scriver CR, Beaudet A, Sly WS, Valle D, eds. The Metabolic & Molecular Bases of Inherited Disease, (New York: McGraw-Hill), pp 1223–1256
Epstein CJ, Hofmeister BG, Yee D, Smith SA, Philip R, et al. (1985) Stem cell deficiencies and thymic abnormalities in fetal mouse trisomy 16. J Exp Med 162(2):695–712
Escorihuela RM, Fernandez-Teruel A, Vallina IF, Baamonde C, Lumbreras MA, et al. (1995) A behavioral assessment of Ts65Dn mice: a putative Down syndrome model. Neurosci Lett 199(2):143–146
FitzPatrick DR, Ramsay J, McGill NI, Shade M, Carothers AD, et al. (2002) Transcriptome analysis of human autosomal trisomy. Hum Mol Genet 11(26):3249–3256
Freeman SB, Taft LF, Dooley KJ, Allran K, Sherman SL, et al. (1998) Population-based study of congenital heart defects in Down syndrome. Am J Med Genet 80(3):213–217
Galdzicki Z, Siarey RJ (2003) Understanding mental retardation in Down’s syndrome using trisomy 16 mouse models. Genes Brain Behav 2(3):167–178
Gardiner K (2003) Predicting pathway perturbations in Down syndrome. J Neural Transm Suppl (67):21–37
Gardiner K, Fortna A, Bechtel L, Davisson MT (2003) Mouse models of Down syndrome: how useful can they be? Comparison of the gene content of human chromosome 21 with orthologous mouse genomic regions. Gene 318:137–147
Giannone S, Strippoli P, Vitale L, Casadei R, Canaider S, et al. (2004) Gene expression profile analysis in human T lymphocytes from patients with Down Syndrome. Ann Hum Genet 68(Pt 6):546–554
Hattori M, Fujiyama A, Taylor TD, Watanabe H, Yada T, et al. (2000) The DNA sequence of human chromosome 21. Nature 405(6784):311–319
Hill CA, Reeves RH, Richtsmeier JT (2007) Effects of aneuploidy on skull growth in a mouse model of Down syndrome. J Anat 210(4):394–405
Hiltgen GG, Markwald RR, Litke LL (1996) Morphogenetic alterations during endocardial cushion development in the trisomy 16 (Down syndrome) mouse. Pediatr Cardiol 17(1):21–30
Holtzman DM, Santucci D, Kilbridge J, Chua-Couzens J, Fontana DJ, et al. (1996) Developmental abnormalities and age-related neurodegeneration in a mouse model of Down syndrome. Proc Natl Acad Sci U S A 93(23):13333–13338
Insausti AM, Megias M, Crespo D, Cruz-Orive LM, Dierssen M, et al. (1998) Hippocampal volume and neuronal number in Ts65Dn mice: a murine model of Down syndrome. Neurosci Lett 253(3):175–178
Kahlem P, Sultan M, Herwig R, Steinfath M, Balzereit D, et al. (2004) Transcript level alterations reflect gene dosage effects across multiple tissues in a mouse model of down syndrome. Genome Res 14(7):1258–1267
Kleschevnikov AM, Belichenko PV, Villar AJ, Epstein CJ, Malenka RC, et al. (2004) Hippocampal long-term potentiation suppressed by increased inhibition in the Ts65Dn mouse, a genetic model of Down syndrome. J Neurosci 24(37):8153–8160
Kola I, Hertzog PJ (1997) Animal models in the study of the biological function of genes on human chromosome 21 and their role in the pathophysiology of Down syndrome. Hum Mol Genet 6(10):1713–1727
Korenberg JR, Chen XN, Schipper R, Sun Z, Gonsky R, et al. (1994) Down syndrome phenotypes: the consequences of chromosomal imbalance. Proc Natl Acad Sci U S A 91(11):4997–5001
Kurt MA, Davies DC, Kidd M, Dierssen M, Florez J (2000) Synaptic deficit in the temporal cortex of partial trisomy 16 (Ts65Dn) mice. Brain Res 858(1):191–197
Lange AW, Rothermel BA, Yutzey KE (2005) Restoration of DSCR1 to disomy in the trisomy 16 mouse model of Down syndrome does not correct cardiac or craniofacial development anomalies. Dev Dyn 233(3):954–963
Lorenzi HA, Reeves RH (2006) Hippocampal hypocellularity in the Ts65Dn mouse originates early in development. Brain Res 1104(1):153–159
Lyle R, Gehrig C, Neergaard-Henrichsen C, Deutsch S, Antonarakis SE (2004) Gene expression from the aneuploid chromosome in a trisomy mouse model of down syndrome. Genome Res 14(7):1268–1274
Mao R, Zielke CL, Zielke HR, Pevsner J (2003) Global up-regulation of chromosome 21 gene expression in the developing Down syndrome brain. Genomics 81(5):457–467
Mao R, Wang X, Spitznagel EL Jr, Frelin LP, Ting JC, et al. (2005) Primary and secondary transcriptional effects in the developing human Down syndrome brain and heart. Genome Biol 6(13):R107
Maslen CL, Babcock D, Robinson SW, Bean LJ, Dooley KJ, et al. (2006) CRELD1 mutations contribute to the occurrence of cardiac atrioventricular septal defects in Down syndrome. Am J Med Genet A 140(22):2501–2505
McElhinney DB, Straka M, Goldmuntz E, Zackai EH (2002) Correlation between abnormal cardiac physical examination and echocardiographic findings in neonates with Down syndrome. Am J Med Genet 113(3):238–241
Miyabara S, Gropp A, Winking H (1982) Trisomy 16 in the mouse fetus associated with generalized edema and cardiovascular and urinary tract anomalies. Teratology 25(3):369–380
Moore CS (2006) Postnatal lethality and cardiac anomalies in the Ts65Dn Down syndrome mouse model. Mamm Genome 17(10):1005–1012
Nikolaienko O, Nguyen C, Crinc LS, Cios KJ, Gardiner K (2005) Human chromosome 21/Down syndrome gene function and pathway database. Gene 364:90–98
O’Doherty A, Ruf S, Mulligan C, Hildreth V, Errington ML, et al. (2005) An aneuploid mouse strain carrying human chromosome 21 with Down syndrome phenotypes. Science 309(5743):2033–2037
Olson LE, Richtsmeier JT, Leszl J, Reeves RH (2004a) A chromosome 21 critical region does not cause specific Down syndrome phenotypes. Science 306(5696):687–690
Olson LE, Roper RJ, Baxter LL, Carlson EJ, Epstein CJ, et al. (2004b) Down syndrome mouse models Ts65Dn, Ts1Cje, and Ms1Cje/Ts65Dn exhibit variable severity of cerebellar phenotypes. Dev Dyn 230(3):581–589
Olson LE, Roper RJ, Sengstaken C, Peterson E, Aquino V, et al. (2007) Trisomy for the Down syndrome “critical region” is necessary but not sufficient for brain phenotypes of trisomic mice. Hum Mol Genet 16(7):774–782
Ozand PT, Hawkins RL, Collins RM Jr, Reed WD, Baab PJ, et al. (1984) Neurochemical changes in murine trisomy 16: delay in cholinergic and catecholaminergic systems. J Neurochem 43(2):401–408
Patterson D, Costa AC (2005) Down syndrome and genetics - a case of linked histories. Nat Rev Genet 6(2):137–147
Paz-Miguel JE, Pardo-Manuel de Villena F, Sanchez-Velasco P, Leyva-Cobian F (2001) H2-haplotype-dependent unequal transmission of the 17(16) translocation chromosome from Ts65Dn females. Mamm Genome 12(1):83–85
Pennington BF, Moon J, Edgin J, Stedron J, Nadel L (2003) The neuropsychology of Down syndrome: evidence for hippocampal dysfunction. Child Dev 74(1):75–93
Reeves RH (2006) Down syndrome mouse models are looking up. Trends Mol Med 12(6):237–240
Reeves RH, Irving NG, Moran TH, Wohn A, Kitt C, et al. (1995) A mouse model for Down syndrome exhibits learning and behaviour deficits. Nat Genet 11(2):177–184
Richtsmeier JT, Baxter LL, Reeves RH (2000) Parallels of craniofacial maldevelopment in Down syndrome and Ts65Dn mice. Dev Dyn 217(2):137–145
Richtsmeier JT, Zumwalt A, Carlson EJ, Epstein CJ, Reeves RH (2002) Craniofacial phenotypes in segmentally trisomic mouse models for Down syndrome. Am J Med Genet 107(4):317–324
Roper RJ, Reeves RH (2006) Understanding the basis for Down syndrome phenotypes. PLoS Genet 2(3):e50
Roper RJ, St John HK, Philip J, Lawler A, Reeves RH (2006a) Perinatal loss of Ts65Dn Down syndrome mice. Genetics 172(1):437–443
Roper RJ, Baxter LL, Saran NG, Klinedinst DK, Beachy PA, et al. (2006b) Defective cerebellar response to mitogenic Hedgehog signaling in Down [corrected] syndrome mice. Proc Natl Acad Sci U S A 103(5):1452–1456
Rothermel B, Vega RB, Yang J, Wu H, Bassel-Duby R, et al. (2000) A protein encoded within the Down syndrome critical region is enriched in striated muscles and inhibits calcineurin signaling. J Biol Chem 275(12):8719–8725
Roubertoux PL, Bichler Z, Pinoteau W, Jamon M, Seregaza Z, et al. (2006) Pre-weaning sensorial and motor development in mice transpolygenic for the critical region of trisomy 21. Behav Genet 36(3):377–386
Rueda N, Mostany R, Pazos A, Florez J, Martinez-Cue C (2005) Cell proliferation is reduced in the dentate gyrus of aged but not young Ts65Dn mice, a model of Down syndrome. Neurosci Lett 380(1–2):197–201
Sago H, Carlson EJ, Smith DJ, Kilbridge J, Rubin EM, et al. (1998) Ts1Cje, a partial trisomy 16 mouse model for Down syndrome, exhibits learning and behavioral abnormalities. Proc Natl Acad Sci U S A 95(11):6256–6261
Sago H, Carlson EJ, Smith DJ, Rubin EM, Crnic LS, et al. (2000) Genetic dissection of region associated with behavioral abnormalities in mouse models for Down syndrome. Pediatr Res 48(5):606–613
Salehi A, Delcroix JD, Belichenko PV, Zhan K, Wu C, et al. (2006) Increased App expression in a mouse model of Down’s syndrome disrupts NGF transport and causes cholinergic neuron degeneration. Neuron 51(1):29–42
Saran NG, Pletcher MT, Natale JE, Cheng Y, Reeves RH (2003) Global disruption of the cerebellar transcriptome in a Down syndrome mouse model. Hum Mol Genet 12(16):2013–2019
Shinohara T, Tomizuka K, Miyabara S, Takehara S, Kazuki Y, et al. (2001) Mice containing a human chromosome 21 model behavioral impairment and cardiac anomalies of Down’s syndrome. Hum Mol Genet 10(11):1163–1175
Siarey RJ, Stoll J, Rapoport SI, Galdzicki Z (1997) Altered long-term potentiation in the young and old Ts65Dn mouse, a model for Down Syndrome. Neuropharmacology 36(11–12):1549–1554
Siarey RJ, Carlson EJ, Epstein CJ, Balbo A, Rapoport SI, et al. (1999) Increased synaptic depression in the Ts65Dn mouse, a model for mental retardation in Down syndrome. Neuropharmacology 38(12):1917–1920
Siarey RJ, Villar AJ, Epstein CJ, Galdzicki Z (2005) Abnormal synaptic plasticity in the Ts1Cje segmental trisomy 16 mouse model of Down syndrome. Neuropharmacology 49(1):122–128
Singer HS, Tiemeyer M, Hedreen JC, Gearhart J, Coyle JT (1984) Morphologic and neurochemical studies of embryonic brain development in murine trisomy 16. Brain Res 317(2):155–166
Smith DJ, Stevens ME, Sudanagunta SP, Bronson RT, Makhinson M, et al. (1997) Functional screening of 2 Mb of human chromosome 21q22.2 in transgenic mice implicates minibrain in learning defects associated with Down syndrome. Nat Genet 16(1):28–36
Vacik T, Ort M, Gregorova S, Strnad P, Blatny R, et al. (2005) Segmental trisomy of chromosome 17: a mouse model of human aneuploidy syndromes. Proc Natl Acad Sci U S A 102(12):4500–4505
Villar AJ, Belichenko PV, Gillespie AM, Kozy HM, Mobley WC, et al. (2005a) Identification and characterization of a new Down syndrome model, Ts[Rb(12.1716)]2Cje, resulting from a spontaneous Robertsonian fusion between T(171)65Dn and mouse chromosome 12. Mamm Genome 16(2):79–90
Villar AJ, Kim J, De Blank P, Gillespie AM, Kozy HM, et al. (2005b) Effects of genetic background on cardiovascular anomalies in the Ts16 mouse. Dev Dyn 232(1):131–139
Vyas P, Crispino JD (2007) Molecular insights into Down syndrome-associated leukemia. Curr Opin Pediatr 19(1):9–14
Waller BR 3rd, McQuinn T, Phelps AL, Markwald RR, Lo CW, et al. (2000) Conotruncal anomalies in the trisomy 16 mouse: an immunohistochemical analysis with emphasis on the involvement of the neural crest. Anat Rec 260(3):279–293
Webb S, Brown NA, Anderson RH (1997) Cardiac morphology at late fetal stages in the mouse with trisomy 16: consequences for different formation of the atrioventricular junction when compared to humans with trisomy 21. Cardiovasc Res 34(3):515–524
Wessels MW, Los FJ, Frohn-Mulder IM, Niermeijer MF, Willems PJ, et al. (2003) Poor outcome in Down syndrome fetuses with cardiac anomalies or growth retardation. Am J Med Genet A 116(2):147–151
Wisniewski KE (1990) Down syndrome children often have brain with maturation delay, retardation of growth, and cortical dysgenesis. Am J Med Genet Suppl 7:274–281
Acknowledgment
C. S. Moore was supported by NIH grant HL081099.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License ( https://creativecommons.org/licenses/by-nc/2.0 ), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
Moore, C.S., Roper, R.J. The power of comparative and developmental studies for mouse models of Down syndrome. Mamm Genome 18, 431–443 (2007). https://doi.org/10.1007/s00335-007-9030-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00335-007-9030-8