
Abstract
Objective
To develop and characterize a clinically applicable, fast and efficient method for stem cell labeling with ferucarbotran and protamine for depiction with clinical MRI.
Methods
The hydrodynamic diameter, zeta potential and relaxivities of ferucarbotran and varying concentrations of protamine were measured. Once the optimized ratio was found, human mesenchymal stem cells (MSCs) were labeled at varying incubation times (1–24 h). Viability was assessed via Trypan blue exclusion testing. 150,000 labeled cells in Ficoll solution were imaged with T1-, T2- and T2*-weighted sequences at 3 T, and relaxation rates were calculated.
Results
Varying the concentrations of protamine allows for easy modification of the physicochemical properties. Simple incubation with ferucarbotran alone resulted in efficient labeling after 24 h of incubation while assisted labeling with protamine resulted in similar results after only 1 h. Cell viability remained unaffected. R2 and R2* relaxation rates were drastically increased. Electron microscopy confirmed intracellular iron oxide uptake in lysosomes. Relaxation times correlated with results from ICP-AES.
Conclusion
Our results show internalization of ferucarbotran can be accelerated in MSCs with protamine, an approved heparin antagonist and potentially clinically applicable uptake-enhancing agent.
Similar content being viewed by others

Introduction
Cell replacement therapy is a promising therapeutic approach for diseases such as type 1 diabetes mellitus [1], Parkinson’s disease [2] or myocardial infarction (MI) [3]. Stem cells play an important role as a source of cells destined for implantation. Mesenchymal stem cells (MSCs) are non-hematopoietic stem cells derived from bone marrow and have the distinct advantages of being well characterized, easily obtained and efficiently expanded in vitro compared with other stem cell populations [4]. Patients who received autologous MSCs after an acute MI showed favorable left ventricular remodeling and better ejection fractions than patients who received a placebo [5]. Unfortunately, detecting what happens to engrafted cells after transplantation remains difficult. Magnetic resonance (MR) imaging can provide a non-invasive, real-time diagnostic method for tracking implanted cells [6].
In order to detect implanted cells via MR imaging, they must be labeled with a contrast agent. Most labeling studies have been conducted using superparamagnetic iron oxide particles (SPIO) or ultrasmall SPIOs (USPIO), which function by creating local field inhomogeneities that cause decreased signal on T2- and T2*-weighted images [7]. Iron oxide-based contrast agents show high MR sensitivity and good biocompatibility [8]. Cell labeling can be achieved by simple incubation with contrast agents [9–11]. Enhanced uptake can be achieved by linking SPIOs to HIV Tat peptides [12] or by using poly-l-lysine (PLL) [10], lipofectamine [10] or dendrimers [13]. While cellular uptake using these agents is greatly increased, these agents are unfortunately not approved by health authorities.
Protamine is a low molecular weight polycationic peptide that is approved as an antidote for heparin anticoagulation [14]. It is also used as a transfection agent for DNA [15] and oligonucleotides [16]. Arbab et al. have shown that protamine can aid in the uptake of ferumoxides for the application of cell labeling [17]. Ferucarbotran (Resovist) is a SPIO with a carboxydextran coating, a net negative charge and a hydrodynamic diameter slightly smaller than ferumoxides. It is currently in clinical use in Europe for MR examination of focal liver lesions and possesses advantages compared with other iron-based contrast agents, such as its ability to be administered in a bolus. It has been shown that the cellular uptake in monocytes of ferucarbotran is greater and its induction of apoptosis is lower when compared with ferumoxides [18]. Thus, it seems that the combination of protamine and ferucarbotran is promising in order to develop a clinically applicable, fast and efficient method for stem cell labeling for subsequent depiction with MRI.
Materials and methods
Contrast agent
Ferucarbotran (Resovist, Bayer Schering Pharma AG, Berlin, Germany) are SPIO particles that are coated with carboxydextran [19]. It has been approved for the MR characterization of focal liver lesions in Europe since 2001. The contrast agent has an r1 relaxivity of 8.7 (8.2–9.2) mM−1s−1, r2 relaxivity of 61 (54–68) mM−1s−1 (at 37°C and 1.5 T in water) and an overall hydrodynamic diameter of 62 nm [19, 20]. The carboxydextran coating ensures optimal dispersion of the nanoparticles within aqueous solutions and bestows upon the complex a net negative charge. Contrast agent was provided as a 1.4-ml ready-to-inject syringe of 0.5 M iron.
Protamine is a group of small (molecular weight 4000–4250) cationic peptides with a high arginine content [14]. It occurs naturally in sperm where it assists in forming a compact structure by binding DNA. Clinically, it is used as an antidote to heparin–induced anticoagulation and as a complexing agent where it prolongs the duration of action of insulin (in the form of NPH insulin). Protamine sulphate stock solution of 50 mg in 5 ml saline solution (0.9%) was obtained (American Pharmaceutical Partners Inc., Schaumberg, IL).
Complex formation between ferucarbotran and protamine was performed by mixing constant concentrations of ferucarbotran (iron concentration of 50 μg/ml) and various concentrations of protamine (0, 1, 3, 5, 7, 10 μg/ml). Respective substances were added directly to the labeling media [Dulbecco’s Modified Eagle Medium (DMEM) + 25% fetal calf serum (FCS)], vigorously shaken and left for 10 min, after which the complexes were utilized for further experiments.
Physicochemical properties
Measurement of size, zeta potential and relaxivity of the contrast agent protamine complexes was performed. The hydrodynamic diameter of the complexes was determined by dynamic light scattering with a Zetasizer 3000 HAS (Malvern Instruments, Herrenberg, Germany). The measurement was performed in labeling media (DMEM + 25% FCS) in order to correctly measure the effective hydrodynamic diameter as would be pertinent to a cell. Five individual measurements were performed on each sample.
The surface charge of the complexes was determined by measuring the electrophoretic mobility in a microelectrophoresis flow cell using a Zetasizer 3000 HAS. To negate the effect of charged particles in the labeling solution, preparation and measurement of the complexes were performed in distilled water. Zeta potentials were measured at least ten times on each sample.
A 1.5-T clinical system (Philips Achieva, Philips Healthcare, Best, The Netherlands) was used for measuring the relaxivity of ferucarbotran/protamine complexes in labeling media. Samples with 1.5 ml of the complex were imaged in Eppendorf tubes placed inside a wrist coil (Medical Advances Inc, Milwaukee, WI). For the simultaneous measurement of T1 and T2, the standard Philips MIX sequence was used. This sequence is an inversion recovery (IR) sequence interleaved with a spin echo (SE) sequence. A 180°–90° pulse pair, separated by the inversion delay TI of 100 ms, is followed by a 90° SE excitation pulse after the SE repetition time TRSE of 650 ms. These three pulses are continuously repeated after the IR repetition time TRIR of 1,500 ms. After every 90° excitation pulse, 20 180° refocusing pulses generate 20 spin echoes (TE 7 ms). The echo trains determine T2, while the ratios of IR to SE signals determine T1 as described in [21]. A 3D sequence with five slices was used to minimize artifacts caused by the otherwise systematically smaller flip angles at the edges of the 2D slices. Only the central three slices were used for parameter quantification. T2* was calculated from the FID of a 2D multi GE sequence using 37 gradient echoes spaced 2.06 ms apart with the first echo situated at 6 ms, TR 1,000 ms and alpha 90°. T2* was determined from the FID assuming a mono-exponential decay of the signal. To obtain relaxivities, the relaxation rates (1/Tx) were plotted against the concentration of iron in the complex using a linear least-square fit.
Cell culture and cell labeling
Mesenchymal stem cell culture was initiated by bone marrow aspirates from a 20-year-old male patient without bone marrow disorder who was admitted to our institution for trauma surgery. The patient gave consent to donating bone marrow via needle aspiration from the posterior iliac crest during surgery. The study was approved by the Institutional Review Board. Preparation of MSCs was done as described in [22] and cultured using DMEM media (Invitrogen, Carlsbad, CA) supplemented with 10% FCS (HyClone, Logan, UT) and 1% penicillin/streptomycin (Mediatech, Manassas, VA) under standard cell culture conditions (37°C, 5% CO2). MSCs were plated at 80% confluency and let to adhere for one day. Labeling was performed using either 50 μg iron per ml ferucarbotran in DMEM or 50 μg iron per ml ferucarbotran + 5 μg/ml protamine in DMEM + 25% FCS. To investigate whether the addition of FCS might result in efficient uptake, labeling was also performed with 50 μg iron per ml ferucarbotran in DMEM + 25% FCS. Flasks were incubated under standard cell culture conditions for 1, 2, 6, 10 or 24 h. Cells were then washed three times by sedimentation (25°C, 400 ×g, 5 min) with phosphate buffered saline (PBS, Invitrogen) and counted in a Neubauer counting chamber. Trypan blue exclusion testing was performed. Cell counts of 150,000 were resuspended in 400 μl Ficoll solution and transferred into 2.0 ml cylindrical tubes for MR imaging. The isotonic Ficoll solution was prepared with a density of 1.07 g/ml and preserved the viability of the labeled cells during imaging [23].
MR imaging and data analysis
MR images of labeled cells were obtained using a clinical 3-T MR scanner (Signa EXCITE HD 3 T, GE Medical Systems, Milwaukee, WI) and a standard circularly polarized quadrature knee coil (Clinical MR Solutions, Brookfield, WI). T1- and T2-weighted spin echo (SE) sequences were obtained with varying TR (4,000, 1,000, 500, 250 ms) and TE (60, 45, 30, 15 ms). T2*-weighted gradient echo (GE) sequences were obtained with a flip angle of 30°, a TR of 500 ms and varying TE (31.2, 20.8, 15.6, 10.4, 5.2 ms). Images were acquired with a field of view (FOV) of 120 × 120 mm, a matrix of 256 × 196 pixels, a slice thickness of 1 and 5 mm and one acquisition. To avoid susceptibility artifacts from surrounding air, all probes were immersed in water at room temperature. MR images were transferred as DICOM images to a SUN/SPARC workstation (Sun Microsystems, Mountain View, CA) and processed by a self-written IDL program (Interactive Data Language by Research Systems, Boulder, CO). T1 and T2 relaxation times of the cell samples were calculated assuming monoexponential decay and using a non-linear function least-square curve fitting on a pixel-by-pixel basis. T1 relaxation times were calculated using SE images with a fixed TE of 15 ms. T2 relaxation times were calculated using SE images with a fixed TR of 4,000 ms and variable TE values of 60, 45, 30 and 15. T2* relaxation times were calculated using GE images with a fixed TR of 500 ms and variable TE values of 31.2, 20.8, 15.6, 10.4 and 5.2 ms. The signal intensity for each pixel as a function of time was expressed as follows: SIpixel xy (t) = So (pixel xy) [1-exp(-t/T1pixel xy)] for T1 and SIpixel xy (t) = So (pixel xy) exp(-t/T2pixel xy) for T2 and T2*. T1, T2 and T2* relaxation times of cell samples were derived by ROI measurements on the resultant T1, T2 and T2* maps. Care was taken to analyze only data points with signal intensities significantly above the noise level. Relaxation rates (1/Tx) were derived, and the means and standard deviations of triplicate samples were calculated.
Spectrometry
The iron concentrations of all test samples were determined by inductively coupled plasma atomic emission spectrometry (ICP-AES) (IRIS Advantage, Thermo Jarrell-Ash, MA). Samples were dissolved in the microwave (400 W for 55 min) by adding 65% HNO3 and 30% H2O2. The solutions obtained were nebulized into an argon plasma. Collaborators from Bayer Schering Pharma AG Berlin who were blinded with respect to the content of the samples performed these analyses. Means and standard deviations of triplicate samples were derived.
Electron microscopy
Cells were treated as described and then fixed with 2.5% glutaraldehyde-cacodylate buffer overnight for electron microscopy as described in [24]. In brief, cells were post-fixed in 1% OsO4 and embedded in Epon resin. Thin sections were cut and examined with an EM 10 electron microscope (Carl Zeiss, Oberkochen, Germany). One investigator (A.M.) evaluated the cells for any structural changes as a result of the labeling process as well as the localization of the contrast agent inside the cell.
Results
Contrast agent physicochemical properties
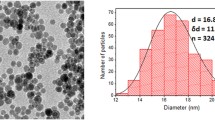
Large, microscopically and macroscopically visible precipitates were observed when ferucarbotran was mixed with protamine in DMEM alone. The quantity and size of the precipitates grew steadily with time. Stable particle formation, determined by sequential measurement of hydrodynamic diameter over a period of 24 h, was achieved with the addition of 25% of FCS to DMEM. This was subsequently used as our labeling medium. The physicochemical properties of ferucarbotran with varying concentrations of protamine are seen in Fig. 1. With increasing protamine concentrations, the hydrodynamic diameter increases, as does the width of the size distribution, that is, there are larger but also less uniform complex populations. Surface charge decreases with increasing concentrations of protamine, with the complex switching from a negative to positive surface charge between protamine concentrations of 7 and 10 μg/ml. This was expected as ferucarbotran is negatively charged, whereas protamine exhibits a positive charge.

Hydrodynamic diameters and zeta potentials of ferucarbotran (50 mg iron per ml) with increasing concentrations of protamine in DMEM with 25% FCS. With increasing concentration of protamine the complexes increase in average size and size distribution. Data points are z-average and zeta potential means ± standard deviations of five and ten measurements, respectively. Note the zeta potentials become positive with a protamine concentration between 7 and 10 μg/ml
With increasing protamine concentration, and as such, increasing complex size, the r1 and r2 relaxivities decrease, whereas the r2 * relaxivity increases (Fig. 2). This is in accordance with findings with contrast agents of different sizes [25]. There seems to be a plateau effect with higher concentrations of protamine; however, because of instable complex formation seen with concentrations above 10 μg/ml, we were unable to confirm this. We believe that a concentration of 5 μg/ml is optimal for uptake in MSCs as this combines a complex size and electrical charge that are not too large, are still negative and have favorable relaxivity properties. At a concentration of 5 μg/ml, the complexes displayed the following relaxivities (in DMEM + 25% FCS, 25°C, 1.5 T): r1 2.50 ± 0.13 s−1mM−1, r2 20.16 ± 5.04 s−1mM−1 and r2 * 574.00 ± 10.64 s−1mM−1.

Relaxivities of complexes of ferucarbotran (50 mg iron per ml) related to the molar concentration of iron and increasing concentrations of protamine [in labeling media (DMEM + 25% FCS), 1.5 T, 25°C]
Cell labeling and MR imaging
Simple incubation
On qualitative T2-weighted images, contrast effect was visible after 6 or more h of incubation with ferucarbotran in DMEM and after 24 h of incubation with ferucarbotran in DMEM + 25% FCS (Fig. 3). Quantitatively, R1, R2 and R2 * relaxation rates gradually increased with increasing incubation time (Fig. 4). The effect on relaxation rates was more pronounced with cells labeled with ferucarbotran in DMEM than with ferucarbotran in DMEM + 25% FCS.

T2-weighted MR images (SE, TE 60, TR 4,000, 3 T, 5 mm) of Eppendorf tubes containing 150,000 MSCs in 400 ml Ficoll. Ferucarbotran concentration used for labeling was 50 μg iron per ml, and protamine concentration was 5 μg/ml

R1, R2 and R2 * relaxation rates of samples of 150,000 MSCs in 400 μl Ficoll labeled with ferucarbotran (solid block), ferucarbotran in 25% FCS (diamond) and ferucarbotran with protamine (solid triangle) at 3 T. The R2 * rate 0.19 ms−1 corresponds to the lowest measurable TE time of 5.2 ms. Data points are mean relaxation rates of triplicate samples ± standard deviation
Enhanced uptake
Qualitative T2-weighted images show pronounced contrast effects in cells labeled with ferucarbotran and protamine at all incubation times investigated (Fig. 3). Quantitatively, all relaxation rates were markedly increased compared with MSCs labeled without protamine. MSCs incubated for 2 h and longer showed R2 * rates higher than the corresponding lowest TE (5.2 ms or 0.19 ms−1) used for imaging (Fig. 4). One hour of incubation with protamine resulted in R2 and R2 * rates comparable with simple incubation over 24 h. R1 relaxation rates were highest after 6 h of incubation with lower R1 rates seen after 10 and 24 h of incubation.
Cell viability was >90% for all cell labeling protocols with no noteworthy differences found among the different labeling methods and incubation times.
Spectrometry
ICP-AES quantification of the amount of intracellular iron correlates with the findings of MR imaging (Fig. 5). Labeling without protamine resulted in a slow increase in intracellular iron over time. MSCs incubated with DMEM and FCS showed worse internalization of contrast agent, with noticeable changes in intracellular iron content only after 24 h of incubation. MSCs labeled with protamine displayed the highest iron uptake at an incubation time of 2 h with a decline in intracellular iron concentrations seen with 6, 10 and 24 h of incubation. This may possibly be because of lysosomal digestion of the contrast agent followed by efflux of contrast agent out of the cell. Notably, the amount of iron uptake after 1 h with protamine approaches the intracellular iron content after 24 h of incubation without protamine.

Intracellular contrast agent content in pg iron per cell as determined by ICP-AES. Data points are mean values of triplicate samples ± standard deviation
Electron microscopy
Electron microscopy of MSCs labeled with ferucarbotran alone and ferucarbotran with protamine showed accumulation of contrast agent/contrast agent complex intracellularly in the cytoplasm only. The contrast agent was not seen within the nucleus (Fig. 6). The contrast agent is located in membrane-bound structures indicative of lysosomes, a finding that is in line with those of previous studies [18].

Transmission electron microscopy of MSCs: (a) unlabeled control, (b) labeled with ferucarbotran for 24 h, (c) labeled with ferucarbotran and protamine for 2 h. Magnification is approximately 1:8,000
Discussion
We have shown that the combination of ferucarbotran and protamine is a fast, efficient and potentially clinically applicable method for labeling MSCs for MR imaging. Protamine has been previously used to improve transfection of oligonucleotides [15], DNA [14] and cellular uptake of ferumoxides [17]. Being a small protein with a net positive charge, protamine is complexed to negatively charged particles by electrostatic interaction [26]. In the application of DNA transfection, the protein enhances cellular uptake and inhibits nuclease digestion by condensing DNA [27]. In the application of improving cellular contrast agent uptake, results obtained with ferumoxides and protamine show increasing labeling efficiencies with protamine [17, 28, 29]. To our knowledge, this is the first time that cell labeling with ferucarbotran and protamine is reported.
Cell uptake mechanisms for ferucarbotran have not been studied in detail. Ferumoxide, a slightly larger SPIO with a neutral charge, is internalized primarily through scavenger receptor-mediated endocytosis [30]. Scavenger receptors are known to play a role in LDL uptake in macrophages; however, they can internalize structurally unrelated substances such as fibrillar amyloid-β, anionic polysaccharides or bacteria [31]. The cell uptake process involves clathrin, a protein that forms basket-like scaffolds around invaginations caused by receptor ligand complexes on the cell membrane [32, 33]. Although ferumoxides are neutral and ferucarbotran negatively charged, scavenger receptors encompass a number of subtypes with different properties; thus, this explanation may hold for ferucarbotran as well.
The primary problem we experienced when using protamine in combination with ferucarbotran in standard labeling solution not supplemented with 25% FCS was the formation of large aggregates within minutes. The size of the aggregates made them visible to the naked eye, and as such rendered them unsuitable for cell labeling. Similar behavior was noticed when protamine was used to complex oligonucleotides, and it was found that the addition of albumin hindered aggregation [34, 35]. When we attempted to add albumin at different concentrations to a ferucarbotran and protamine solution, no beneficial effect was noticed. However, the addition of FCS at different concentrations slowed (10%) or entirely inhibited (25%) aggregation as demonstrated by serial measurement of hydrodynamic diameter over the course of 24 h. We postulate that components of FCS other than or in addition to albumin coat the ferucarbotran protamine complexes and prevent aggregation.
The physicochemical properties of the ferucarbotran protamine complex are interesting, as particles of different sizes, surface charges and relaxivities can be created by simply varying the concentration of protamine. The MSCs we labeled were responsive to labeling with ferucarbotran and protamine at a concentration at 5 μg/ml. However, other cell lines may not be and may be more responsive to a larger or smaller particle size. While we found it difficult to create stable ferucarbotran protamine complexes with a larger hydrodynamic diameter than approximately 1,100 nm (corresponding to a protamine concentration of 10 μg/ml), the spectrum of hydrodynamic diameters from approximately 100 to 1,100 nm was easily controlled with different protamine concentrations. This may prove beneficial for cell labeling.
Protamine has been used clinically for over 30 years as an antidote for heparin anticoagulation and for slow-release formulations of insulin. One well-known complication is allergoid reactions up to anaphylactic shock [36]. The incidence is fairly low for patients using NPH insulin (0.13%) [37], but might increase with preconditioning to protamine, such as in a patient undergoing cardiovascular surgery after years of NPH insulin use [36]. If this becomes a problem for our application, low molecular weight protamine fragments — protamine that is enzymatically digested into smaller proteins — may be a solution, as these fragments seem to display lower immunogenicity while retaining most of the beneficial properties of protamine [38]. However, given protamine’s long safety record and seeing that concerns of anaphylaxis may not even be applicable in our setting as protamine is located inside the cell and is invisible to the patient’s immune system, we believe protamine is safe for use in enhancing contrast agent uptake.
Cell labeling can be achieved through a number of means. Cells can be labeled using simple incubation with contrast agent alone, an uptake-enhancing agent can be added, or other means such as electroporation can be utilized. Simple incubation has been used extensively for cell labeling with adequate results for some cell types [18]. In our case, after 24 h of incubation, we noticed contrast agent uptake that resulted in detection on qualitative imaging; however, even better results were obtained with much lower incubation times when protamine was added. Other transfection agents such as lipofectin or poly-L-lysine (PLL) can be used to expedite labeling with great success [39, 40]. However, unlike protamine, most uptake-enhancing agents are not approved by health authorities and as such will have limited usefulness for cell tracking applications in humans. Other techniques such as electroporation circumvent this problem by avoiding uptake-enhancing agents altogether [41, 42]. This method can offer almost instant labeling, but results in significantly lower cell viability after the labeling process. A method utilizing protamine with near-instant labeling but without deleterious effects on viability is preferred.
We acknowledge various limitations of our study. In particular, although such a technique is promising for in vivo cell tracking purposes, results must be validated in further preclinical testing. We realize that our enhanced labeling method relies on fetal calf serum (FCS) in order to stabilize the complexes and to culture the MSCs. FCS is not approved for human use, and as such, the tolerance and clinical applicability are unknown. Also, potential detrimental effects on long-term cell survival or differentiation capacity should be assessed.
In conclusion, we have shown that the addition of protamine greatly improves cellular labeling of MSCs with ferucarbotran. Given the high sensitivity of iron oxide-based contrast agents and the straightforward labeling method with the health authority-approved protamine, future use of this technique to label stem cells destined for cellular and molecular imaging is promising.
References
Soria B, Roche E, Berna G, Leon-Quinto T, Reig JA, Martin F (2000) Insulin-secreting cells derived from embryonic stem cells normalize glycemia in streptozotocin-induced diabetic mice. Diabetes 49:157–162
Freed CR, Greene PE, Breeze RE et al (2001) Transplantation of embryonic dopamine neurons for severe Parkinson’s disease. N Engl J Med 344:710–719
Yoon YS, Lee N, Scadova H (2005) Myocardial regeneration with bone-marrow-derived stem cells. Biol Cell 97:253–263
Gregory CA, Prockop DJ, Spees JL (2005) Non-hematopoietic bone marrow stem cells: molecular control of expansion and differentiation. Exp Cell Res 306:330–335
Chen SL, Fang WW, Ye F et al (2004) Effect on left ventricular function of intracoronary transplantation of autologous bone marrow mesenchymal stem cell in patients with acute myocardial infarction. Am J Cardiol 94:92–95
Jain M, Pfister O, Hajjar RJ, Liao R (2005) Mesenchymal stem cells in the infarcted heart. Coron Artery Dis 16:93–97
Frangioni JV, Hajjar RJ (2004) In vivo tracking of stem cells for clinical trials in cardiovascular disease. Circulation 110:3378–3383
Bulte JW, Kraitchman DL (2004) Iron oxide MR contrast agents for molecular and cellular imaging. NMR Biomed 17:484–499
Daldrup-Link HE, Rudelius M, Piontek G et al (2005) Migration of iron oxide-labeled human hematopoietic progenitor cells in a mouse model: in vivo monitoring with 1.5-T MR imaging equipment. Radiology 234:197–205
Arbab AS, Yocum GT, Wilson LB et al (2004) Comparison of transfection agents in forming complexes with ferumoxides, cell labeling efficiency, and cellular viability. Mol Imaging 3:24–32
Franklin RJ, Blaschuk KL, Bearchell MC et al (1999) Magnetic resonance imaging of transplanted oligodendrocyte precursors in the rat brain. Neuroreport 10:3961–3965
Josephson L, Tung CH, Moore A, Weissleder R (1999) High-efficiency intracellular magnetic labeling with novel superparamagnetic-Tat peptide conjugates. Bioconjug Chem 10:186–191
Bulte JW, Douglas T, Witwer B et al (2001) Magnetodendrimers allow endosomal magnetic labeling and in vivo tracking of stem cells. Nat Biotechnol 19:1141–1147
Sorgi FL, Bhattacharya S, Huang L (1997) Protamine sulfate enhances lipid-mediated gene transfer. Gene Ther 4:961–968
Lochmann D, Jauk E, Zimmer A (2004) Drug delivery of oligonucleotides by peptides. Eur J Pharm Biopharm 58:237–251
Dinauer N, Lochmann D, Demirhan I et al (2004) Intracellular tracking of protamine/antisense oligonucleotide nanoparticles and their inhibitory effect on HIV-1 transactivation. J Control Release 96:497–507
Arbab AS, Yocum GT, Kalish H et al (2004) Efficient magnetic cell labeling with protamine sulfate complexed to ferumoxides for cellular MRI. Blood 104:1217–1223
Metz S, Bonaterra G, Rudelius M, Settles M, Rummeny EJ, Daldrup-Link HE (2004) Capacity of human monocytes to phagocytose approved iron oxide MR contrast agents in vitro. Eur Radiol 14:1851–1858
Reimer P, Balzer T (2003) Ferucarbotran (Resovist): a new clinically approved RES-specific contrast agent for contrast-enhanced MRI of the liver: properties, clinical development, and applications. Eur Radiol 13:1266–1276
Rohrer M, Bauer H, Mintorovitch J, Requardt M, Weinmann HJ (2005) Comparison of magnetic properties of MRI contrast media solutions at different magnetic field strengths. Invest Radiol 40:715–724
In den Kleef JJ, Cuppen JJ (1987) RLSQ: T1, T2, and rho calculations, combining ratios and least squares. Magn Reson Med 5:513–524
Wang ML, Nesti LJ, Tuli R et al (2002) Titanium particles suppress expression of osteoblastic phenotype in human mesenchymal stem cells. J Orthop Res 20:1175–1184
Graziani-Bowering GM, Graham JM, Filion LG (1997) A quick, easy and inexpensive method for the isolation of human peripheral blood monocytes. J Immunol Methods 207:157–168. doi:S0022-1759(97)00114-2 [pii]
Albrecht M, Ramsch R, Kohn FM, Schwarzer JU, Mayerhofer A (2006) Isolation and cultivation of human testicular peritubular cells: a new model for the investigation of fibrotic processes in the human testis and male infertility. J Clin Endocrinol Metab 91:1956–1960
Muller RN, Gillis P, Moiny F, Roch A (1991) Transverse relaxivity of particulate MRI contrast media: from theories to experiments. Magn Reson Med 22:178–182 discussion 195–176
Balhorn R (2007) The protamine family of sperm nuclear proteins. Genome Biol 8:227
Junghans M, Kreuter J, Zimmer A (2001) Phosphodiester and phosphorothioate oligonucleotide condensation and preparation of antisense nanoparticles. Biochim Biophys Acta 1544:177–188
Mani V, Adler E, Briley-Saebo KC et al (2008) Serial in vivo positive contrast MRI of iron oxide-labeled embryonic stem cell-derived cardiac precursor cells in a mouse model of myocardial infarction. Magn Reson Med 60:73–81
Sadek H, Latif S, Collins R, Garry MG, Garry DJ (2008) Use of ferumoxides for stem cell labeling. Regen Med 3:807–816
Raynal I, Prigent P, Peyramaure S, Najid A, Rebuzzi C, Corot C (2004) Macrophage endocytosis of superparamagnetic iron oxide nanoparticles: mechanisms and comparison of ferumoxides and ferumoxtran-10. Invest Radiol 39:56–63
Husemann J, Loike JD, Anankov R, Febbraio M, Silverstein SC (2002) Scavenger receptors in neurobiology and neuropathology: their role on microglia and other cells of the nervous system. Glia 40:195–205
Kosswig N, Rice S, Daugherty A, Post SR (2003) Class A scavenger receptor-mediated adhesion and internalization require distinct cytoplasmic domains. J Biol Chem 278:34219–34225
Rejman J, Oberle V, Zuhorn IS, Hoekstra D (2004) Size-dependent internalization of particles via the pathways of clathrin- and caveolae-mediated endocytosis. Biochem J 377:159–169
Lochmann D, Weyermann J, Georgens C, Prassl R, Zimmer A (2005) Albumin-protamine-oligonucleotide nanoparticles as a new antisense delivery system. Part 1: physicochemical characterization. Eur J Pharm Biopharm 59:419–429
Weyermann J, Lochmann D, Georgens C, Zimmer A (2005) Albumin-protamine-oligonucleotide-nanoparticles as a new antisense delivery system. Part 2: cellular uptake and effect. Eur J Pharm Biopharm 59:431–438
Nybo M, Madsen JS (2008) Serious anaphylactic reactions due to protamine sulfate: a systematic literature review. Basic Clin Pharmacol Toxicol 103:192–196
Levy JH, Zaidan JR, Faraj B (1986) Prospective evaluation of risk of protamine reactions in patients with NPH insulin-dependent diabetes. Anesth Analg 65:739–742
Kharidia R, Friedman KA, Liang JF (2008) Improved gene expression using low molecular weight peptides produced from protamine sulfate. Biochemistry (Mosc) 73:1162–1168
Anderson SA, Glod J, Arbab AS et al (2005) Noninvasive MR imaging of magnetically labeled stem cells to directly identify neovasculature in a glioma model. Blood 105:420–425
Daldrup-Link HE, Rudelius M, Oostendorp RA et al (2005) Comparison of iron oxide labeling properties of hematopoietic progenitor cells from umbilical cord blood and from peripheral blood for subsequent in vivo tracking in a xenotransplant mouse model XXX. Acad Radiol 12:502–510
Daldrup-Link HE, Meier R, Rudelius M et al (2005) In vivo tracking of genetically engineered, anti-HER2/neu directed natural killer cells to HER2/neu positive mammary tumors with magnetic resonance imaging. Eur Radiol 15:4–13
Walczak P, Kedziorek DA, Gilad AA, Lin S, Bulte JW (2005) Instant MR labeling of stem cells using magnetoelectroporation. Magn Reson Med 54:769–774
Acknowledgements
We would like to thank Mrs. Gabriele Terfloth for her technical assistance with EM imaging. Additionally, we would like to thank Dr. Christian Plank and Mrs. Olga Mykhaylyk for their assistance with the DLS equipment. This work was supported by a seed grant from the Department of Radiology, University of California of San Francisco, and a grant from the National Institute of Arthritis and Musculoskeletal and Skin Diseases (grant no. NIH R01AR054458).
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License (https://creativecommons.org/licenses/by-nc/2.0), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
Golovko, D.M., Henning, T., Bauer, J.S. et al. Accelerated stem cell labeling with ferucarbotran and protamine. Eur Radiol 20, 640–648 (2010). https://doi.org/10.1007/s00330-009-1585-1
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00330-009-1585-1