
Abstract
The constant region of IgG antibodies mediates antiviral activities upon engaging host Fcγ receptors (FcγRs) expressed by a variety of immune cells, such as antibody-dependent cellullar cytotoxcity (ADCC) executed by natural killer (NK)cells. Human cytomegalovirus (HCMV) is unique among viruses by encoding also an array of several Fcγ-binding glycoproteins with cell surface disposition and concomitant incorporation into the virion. Evidence is increasing that the virus-encoded Fcγ receptors differ in their Fcγ binding mode but effectively operate as adversaries of host FcγRs since they are able to prevent IgG-mediated triggering of activating host FcγRs, i.e., FcγRI, FcγRIIA, and FcγRIIIA. Here we discuss virus-encoded FcγRs as the first known HCMV inhibitors of IgG-mediated immunity which could account for the limited efficacy of HCMV hyperimmune globulin in clinical settings. A better understanding of their molecular mode of action opens up new perspectives for improving IgG therapies against HCMV disease.
Similar content being viewed by others

Human cytomegalovirus
Cytomegaloviruses (CMVs) are prototypical members of the β-subgroup of the herpesvirus family with a relatively broad tropism for many cell types and tissues. Due to millions of years of co-evolution and co-speciation, all CMVs exhibit a close adaptation to their individual host. The best studied CMVs are the members infecting humans (HCMV), chimpanzee (CCMV), Mus musculus (MCMV), rhesus monkeys (RhCMV), Rattus norvegicus (RCMV), and guinea pigs (GPCMV). HCMV has the largest genome of all human pathogenic herpesviruses encompassing two covalently linked unique segments (large, L, and small, S), UL and US [1]. Endowed with double-stranded DNA genomes of approximately 235 kbp, traditional estimates of the coding capacity of CMVs varied between 166 to more than 200 open reading frames (ORFs) [2, 3]. However, in a recent study, unexpectedly more than 750 HCMV translational products could be detected [4]. A further hallmark of the HCMV genome is the presence of 12 multigene families that probably arose by gene duplication during virus evolution. As all herpesviruses, CMVs form enveloped virions and establish a lifelong infection with alternating phases of active replication and latency [5]. HCMV infection is highly prevalent in all human populations reaching seroprevalence rates of 50–99 %. While passing asymptomatic and mostly unnoticed in immunocompetent individuals, HCMV infection in immunocompromised individuals or newborn infants leads to massive tissue damage and often life-threatening disease manifestations [6]. However, HCMV-induced health impairment may not only be restricted to immunocompromised individuals since the infection has been also linked to further acquired disorders such as arteriosclerosis and vascular disease, immune aging [7, 8], and certain types of tumors [9].
Primary CMV infection leads to disseminated replication in multiple organs including the liver, spleen, lungs, kidney, and bone marrow before it is controlled and eventually terminated by well-orchestrated innate and adaptive immune responses. Primary immune reaction starts with the induction of innate responses comprising type I interferons (IFN) and activation of natural killer (NK) cells. Next, adaptive immune responses are initiated among which CD8+ and CD4+ T cells are essential for protection from primary and also recurrent MCMV infection [10, 11]. B-cell-dependent immunity plays a decisive role in the containment and control of recurrent infection [12]. Despite of successful termination of primary infection, CMVs invariably establish latent infection in certain cell types including CD34+ hematopoietic stem cells, from which periodic reactivation consistently occurs. Additional experimental and clinical findings suggest that humoral immunity to CMV has a protective potential. In the murine system, adoptive transfer of immune serum to naïve Rag1−/− (T and B cell deficient) mice was sufficient for effective MCMV control after challenge [13]. On the other hand, CMVs must deal with adaptive immune pressure since they replicate predominantly in the presence of primed immune responses. To dampen antiviral immunity and promote CMV persistence and replication, a very large proportion of the CMV genome is employed in manipulating immune responses [14–16]. In this way, CMV has learned to target those immunological pathways and mechanisms that mediate the most critical antiviral effects, e.g., CD8+ T cells, natural killer cells, interferons, and antibodies.
IgG antiviral effector functions
Immunoglobulins are essential mediators of the humoral immune system recognizing invading pathogens like bacteria or viruses. Immunoglobulin G (IgG) is the most abundant immunoglobulin subclass in serum and mediates immunological memory [17, 18]. Most of the antiviral IgG effector responses require the fragment crystallizable (Fc) part of the IgG molecule, Fcγ, which is designed to interact with a variety of soluble and cell-bound ligands. While a majority of IgG effector functions are both fragment antigen binding (Fab)- and Fcγ-dependent as further outlined below, neutralization of virions or toxins can be reached in a solely Fab-dependent but Fcγ-independent manner [19]. Crucial interaction partners of Fcγ are the C1q component of the complement system, and distinct classes of cellular receptors, e.g., the FcγRs (canonical or type I Fc receptors) and CD23 as well as DC-SIGN (CD209) (type II Fc receptors) [19–21]. Binding of C1q to opsonizing IgG leads to complement-dependent cytotoxicity of virally infected cells or virolysis, i.e., destruction of the viral particle [19]. The FcγRs connect the innate and the adaptive arm of the immune system and humoral and cell-mediated immunity, conferring FcγR central roles in the execution of immune responses [22]. Interaction of antigen-bound IgG with activatory FcγRs results in phagocytosis of immune complexes and opsonized pathogens, antibody-dependent cell-mediated cytotoxicity (ADCC) by NK cells or macrophages, and the release of inflammatory cytokines, chemokines, or superoxide radicals [23]. Binding of IgG to the only known inhibitory FcγR, FcγRIIB, leads to anti-inflammatory responses and downregulation of B cell activation [20, 24, 25]. The glycan structure of IgG is of critical importance for the immune response elicited via FcγRs. Certain modifications on the IgG glycan such as galactosylated IgG1 is associated with anti-inflammatory responses [26].
Diversity of host FcγRs
Ligands, expression pattern, and function of human FcγRs
Canonical FcγRs belong to the immunoglobulin receptor superfamily and constitute critical receptors of immune cell activation and deactivation that recognize Fcγ domains in an open conformation (reviewed in Pincetic et al. [21]). Due to their broad expression pattern, they mediate a large variety of pleiotropic immune effector functions upon triggering by IgG-composed immune complexes. Specifically, they mediate the clearance of pathogens and antigens, regulate many inflammatory effector mechanisms including cytokine production, induce destruction of target cells by ADCC, and control antibody production and the initiation of anti-inflammatory pathways [23, 27, 28]. While Fcγ is thought to be the prime ligand of FcγRs, other soluble factors of the innate immune system are also known to be recognized and bound by FcγRs. These ligands mainly belong to the family of pentraxins, e.g., human C-reactive protein (CRP), serum amyloid P (SAP), and pentraxin 3 (PTX3) [29, 30]. It is not excluded that pentraxins may compete with IgG for binding to FcγRs due to overlapping binding sites on FcγRs [31], thereby potentially interfering with IgG-mediated responses.
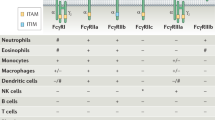
In humans, FcγRs are encoded by six genes clustered in close proximity on chromosome 1–FCGR1A, FCGR2A, FCGR2B, FCGR2C, FCGR3A, and FCGR3B. The FCGR2C gene resulted from a crossover between FCGR2A and FCGR2B [32, 33]. Some of the human FcγRs exist in different allelic variants that exhibit remarkably variable characteristics regarding IgG subclass affinity and immune cell activation (see Table 1). Such allelic variants are known as FcγRIIA, FcγRIIB, FcγRIIC, FcγRIIIA, and FcγRIIIB [20, 34–38]. Most immune cell types co-express different types of FcγRs, albeit at different levels (see Table 2). FcγRI is mainly expressed on monocytes, macrophages, dendritic cell (DC), mast cells, eosinophils, and basophils and is inducible on neutrophils [39]. Moreover, FcγRI expression is strongly inducible by IFN-γ [40]. FcγRIIA is predominantly expressed on myeloid cells and mast cells. Interestingly, FcγRIIA is the only FcγR expressed at high levels by platelets [36]. FcγRIIB has a prominent inhibitory role for circulating B cells [41], which express also FcγR-like receptors, FCRL1-6, as a separate class of surface receptors modulating their function [42]. Other immune cells found to express inhibitory FcγRIIB are basophils [43] and NK cells [44], but not mast cells [45]. Furthermore, FcγRIIB is expressed by smaller subpopulations of monocytes [46], neutrophils, as well by macrophages and DCs [41]. FcγRIIC is only expressed in about 20 % of humans due to a stop codon at position 13, but if expressed, it is regularly found on NK cells, monocytes, and neutrophils [44, 47]. FcγRIIIA is present on NK cells, monocytes, and macrophages, whereas glycosylphosphatidyl-inositol (GPI)-anchored FcγRIIIB is expressed mainly on neutrophils and a subset of basophils [48]. FcγR expression is critically modulated by cytokines [36, 49, 50]. For example, the cytokine transforming growth factor ß1 (TGF-ß1) is an immunosuppressive cytokine that reduces the expression of the FcγR-associated common γ-chain and thereby also reduces the surface expression of the activating FcγRs FcγRI and FcγRIII, while FcγRIIA seems to be unaffected. In this way, TGF-β1 may dampen inflammation [50].
In contrast to the above mentioned FcγRs, the neonatal FcR (FcRn) encoded by the FCGRT gene is not expressed on immune cells but certain epithelial cells and able to bind monomeric IgG with very high affinity [51] (see Table 1). The FcRn structure is similar to MHC class I molecules and allows for the association with β2m [51, 52]. FcRn expressed by syncytiotrophoblasts of the placenta mediates transport of maternal IgG to the fetus and thus controls maternal passive immunity [53] (Table 2). In addition, FcRn binds IgG at the intestinal lumen of mucosal surfaces at a slightly acidic pH and ensures efficient unidirectional transport to the basolateral side and thus controls IgG turnover [53]. Circumstantial evidence was provided supporting a significant protective role of FcRn in simian immunodeficiency virus infection [54].
Besides the canonical FcγRs that bind Fcγ in an open conformation in a 1:1 stoichiometry, type II Fc receptors that include CD23 and DC-SIGN recognize Fcγ in a closed conformation with a 2:1 stoichiometry (reviewed in Pincetic et al. [21]).
Structure of FcγRs
Canonical type I FcγRs are glycoproteins belonging to the fast expanding IgG superfamily. They are composed of an α-subunit responsible for ligand binding (e.g., Fcγ) that contains two or three C2-type extracellular domains (Ig-binding domains) depending on the FcγR type, a transmembrane domain, and an intracellular tail that may contain signaling motifs (see Fig. 1) [23, 27, 55]. The human FcγRs are all type 1 transmembrane proteins, with the exception of the GPI-anchored FcγRIIIB [48]. FcγRI is the only human FcγR possessing a unique additional third Ig-like domain (D3) that may contribute to its high-affinity Fcγ binding [39, 42], although domains 1 and 2 are sufficient to retain a weaker affinity for IgG [56]. Atomic-level structural data are available for the ectodomains D1 and D2 of FcγRIIA, FcγRIIB, and FcγRIII [57–59]. Both domains are each arranged in an identical immunoglobulin fold building a sandwich of two β-sheets. Moderate flexibility at the domain interface might allow the interdomain angle to vary slightly [55]. Host FcγRs bind Fcγ with 1:1 stoichiometry in an asymmetric manner, contacting residues in the CH2 domain and in the CH1-CH2 hinge which connects the Fab to Fcγ [59]. The cross-linking of FcγRI-bound antibodies by multivalent antigens or the recognition of preformed immune complexes by FcγRII or FcγRIII results in clustering of the FcγR and triggering a variety of effector mechanisms [55].

Structural composition of human FcRs. Canonical FcγRs contain two or three immunoglobulin-like domains in their extracellular parts to bind IgG. Signal transduction upon receptor activation occurs via immuno-tyrosine-based-activating motifs (ITAM, indicated in green) usually in combination with the ITAM-containing, dimeric subunit, the common γ-chain, or immuno-tyrosine-based inhibitory motifs (ITIM, indicated in red). The family of human FcγRs comprises several activating members (FcγRI, FcγRIIA, FcγRIIIA) but only one inhibitory FcγR (FcγRIIB). Human FcγRs are shown color-coded based on their sequence relatedness. The neonatal FcR (FcRn) is structurally related to MHC-class I molecules and is able to transport and recycle IgG and thereby increases IgG half-life. DC-SIGN (dendritic-cell-specific intercellular adhesion molecule-3-grabbing non-integrin) is a C-type lectin receptor expressed by macrophages and dendritic cells. It is known to bind directly to C1q and Fcγ in a closed conformation
In some FcγRs, the α-subunit is associated with a signaling adaptor molecule, the common γ-chain or CD3ζ. The common γ-chain and CD3ζ are dimeric signaling adaptors, containing immunoreceptor tyrosine-based activation motifs (ITAM) (Fig. 1) [42]. In contrast, FcγRIIA, FcγRIIC, and FcγRIIB have integrated activating or inhibitory signaling motifs in their own cytoplasmic tails [23, 36]. FcγRIIB is further unique because it is the only known inhibitory FcγR containing an immunoreceptor tyrosine-based inhibitory motif (ITIM) [27, 60].
IgG features influencing ligand binding
Canonical FcγRs are characterized with regard to their binding affinity toward Fcγ into high-affinity and low-affinity receptors (see Table 1). High-affinity Fcγ receptors include FcγRI and the neonatal Fc receptor (FcRn). They are not only able to bind aggregated IgG, i.e., immune complexes, but also recognize equally well monomeric IgG. This implies that such FcγRs are easily saturated with “non-immune” IgG [61]. In contrast, low-affinity FcγRs, e.g., FcγRII and FcγRIII, preferentially bind IgG complexes, although low-affinity receptors may also be associated with monomeric IgG, which is referred to as “cytophilic” IgG and discussed as an inhibitory modulating ligand for FcγRIII+ NK cells [61]. Curiously, the inhibitory FcγRIIB shows the lowest IgG affinity from all low-affinity receptors [37, 39]. In general, the human FcγRs bind to monomeric IgG of the subclasses IgG1, IgG3, and IgG4, albeit with clearly different affinities and with remarkable differences in binding affinities regarding distinct allelic variants of FcγRs. The IgG2 subclass is only poorly recognized by any canonical FcγR (see Table 1).
Binding of IgG to canonical FcγRs and downstream receptor activation is further tightly controlled by the presence and biochemical subcomposition of biantennary N-linked glycans conjugated to amino acid Asn297 in the CH2 domain of IgG, which is highly conserved between all IgG subclasses and strongly influences the structural framework of Fcγ [20, 62]. For instance, defucosylation showed enhanced binding to FcγRIIIA, thereby increasing NK-cell-mediated ADCC up to 50-fold [63–66]. Conversely, unglycosylated IgG is not able to bind to FcγRs [67]. In conjunction with the remarkable structural diversity of the protein backbone of the IgG subclasses, the Fcγ domain thus takes a considerable heterogenic shape resulting in numerous Fcγ overall structures which could perhaps widely differ in their interaction with canonical and non-canonical Fc receptors.
Microbial Fcγ-binding proteins
To avoid the powerful effector functions mediated by IgG, various pathogens evolved specific immune evasion strategies. Some of these mechanisms interfere with the function of host FcγRs by competing with their ability to bind IgG, e.g., protein A (Staphylococcus aureus), protein G (Streptococcus sp.) [68], and the Phage-encoded protein TspB from Neisseria meningitides [69]. Several viruses from the herpesvirus family [70–77] and the core protein of hepatitis C [78] were also found to express Fcγ-binding proteins in virus-infected cells and as structural proteins incorporated into virus particles. In this respect, according to current knowledge, HCMV is provided with the most extensive genetic repertoire of independently acting viral FcγRs (vFcγRs). Notably, HCMV has been shown to encode quite a number of IgG-Fc-interacting proteins, i.e., vFcγR gp34 (RL11) [71, 72], vFcγR gp68 (UL119–UL118) [71], and another identified Fc-binding protein encoded by the gene RL13 [70]. Furthermore, a fourth vFcγR, RL12, has been recently identified [70, 79] (see Table 3). Three out of four known vFcγRs belong to the RL11 multigene family of HCMV comprising 14 individual members which are characterized by the RL11D or CR1 domain in their luminal part [1, 2]. The RL11 gene family is supposed to have arisen by gene duplication before diverged by selection forces during the evolution process of primate CMVs [2]. The RL11D domain includes a characteristic key motif [1] as CXX (NQEKTY) ×4–6 (YFLI) NX (ST) XXXXGXY (alternative residues given in brackets) consisting of a region of variable length formed around three conserved amino acid residues and including potential N-linked glycosylation sites. The HCMV FcγR genes are all transcribed with a relatively delayed kinetics during the protracted viral replication cycle reaching abundant protein amounts in the late phase of infection [71]. Isolated expression of their products proved that each molecule has intrinsic Fcγ binding capabilities. All the HCMV Fcγ-binding proteins readily reach the cell surface, thus constituting genuine FcγRs [70, 79, 80] (see Fig. 2). For comparison, we will also briefly discuss relevant aspects of other herpesvirus-encoded vFcγRs, i.e., the first-described gE/gI-Fcγ receptor complex of herpes simplex virus 1 (HSV-1) [73] and the mouse cytomegalovirus (MCMV) Fc-binding protein m138 (fcr-1) [74, 76] (Table 4).

Structural composition of herpesviral FcγRs. In herpes simplex virus (HSV) infection, the two glycoproteins gE and gI encoded by the genes US7 and US8 form a heterodimeric receptor complex acting as a FcγR. Unlike gE, the protein gI itself is not able to interact with IgG. In human cytomegalovirus (HCMV), there were four independent FcγRs identified: gp68, encoded by UL119-118; gp34, encoded by RL11; gp95, encoded by RL12; and gpRL13, encoded by RL13. In mouse cytomegalovirus (MCMV), there is one vFcγR identified, m138-encoded fcr-1
Herpesviral-encoded vFcγRs
HCMV vFcγR gp34 (RL11/TRL11)
The HCMV-encoded FcγR gp34 [71, 72] is a single-chain type 1 transmembrane glycoprotein transcribed from the RL11 gene and its duplication, TRL11, which is present only in the ULb’ negative HCMV laboratory strains Towne and AD169varATCC [81]. Like all other HCMV vFcγRs, RL11 is dispensable for viral replication in vitro [71, 72]. gp34 consists of 234 aa with a N-terminal signal peptide and an extracellular region with three N-glycosylation sites, a transmembrane domain, and a C-terminal cytoplasmic tail of 31 aa. The cytoplasmic tail contains a dileucine consensus motif (DXXXLL, where X is unknown) indicating a potential function in intracellular targeting of the protein to the endocytic route [71]. The glycoprotein gp34 is synthesized with early and late kinetics reaching continuously increasing levels during the course of HCMV replication. Analysis of purified soluble gp34 (aa 24–182) revealed that a large majority of the protein forms dimers of an apparent molecular mass of 60 kDa [75]. Thus, a disulfide-linked gp34 homodimer [gp34 (24–182) contains five cysteins] may be present in the non-reducing environment of the extracellular milieu when the intact protein is present at the cell surface. Its ability to bind to the Fcγ fragment of IgG comprises not only all human IgG subclasses (IgG1, IgG2, IgG3 and IgG4) but also rabbit IgG and also, to a lesser extent, rat IgG (see Table 3) [71]. Importantly, gp34 as well as HCMV vFcγR gp68 exhibit a glycan-independent binding mode to Fcγ [75], implying that their binding characteristics must fundamentally differ from host FcγRs.
HCMV vFcγR gp68 (UL119–118)
Like gp34, the vFcγR gp68 is composed of a single-chain type 1 transmembrane glycoprotein that is transcribed from the spliced mRNA encoded by the open reading frames UL119 and UL118 [71]. The resulting protein consists of 347 aa and has been detected as a structural component in preparations of purified virions [82]. The gp68 polypeptide includes a N-terminal signal peptide followed by the extracellular region predicted to form a single immunoglobulin superfamily (IgSF)-like domain, residues 71–289 of which were demonstrated to be required for Fcγ binding [75]. The luminal domain of gp68 is joined by a transmembrane domain and a C-terminal cytoplasmic tail displaying a potential intracytoplasmic immunoreceptor tyrosine-based-like inhibition motif (consensus sequence I/V/L/SxYxxL). gp68 undergoes extensive glycan modification: the ectodomain exhibits 12 putative N-glycosylation sites and 8 putative O-glycosylation sites, most of which are obviously used thus increasing the molecular weight of the 33-kDa protein backbone to an 68-kDa endoglycosidase-H-sensitive intermediate which matures further to more than 100 kDa weighing endoglycosidase-H-resistant forms [71]. The vFcγR gp68 is co-expressed with vFcγR gp34 and synthesized during the early and late phases of the HCMV replication cycle [71]. Although it has not yet been possible to obtain a crystal structure for HCMV vFcγR gp68, at least detailed biochemical evidence was generated of how HCMV FcγR gp68 recognizes Fcγ. Gel filtration and biosensor binding experiments revealed that, unlike host FcγRs but similar to the HSV-1 Fc receptor gE-gI, gp68 binds to the CH2-CH3 interdomain interface of the Fcγ dimer with high affinity in the nanomolar range and a 2:1 stoichiometry [75]. gp68 binds to all human IgG subclasses, i.e., IgG1, IgG2, IgG3, and IgG4 (see Table 3) [71].
HCMV RL13-encoded vFcγR
Only recently, a third vFcγRs was identified also belonging to the RL11 gene family of HCMV which is expressed by RL13, a member of a restricted set of hypervariable genes found in the HCMV genome [70]. The ORF RL13 is present in all primary isolates of HCMV, but it is very rapidly mutated when HCMV is adapted to cell culture growth, indicating that the intact RL13 glycoprotein (gpRL13) is detrimental to virus replication in various types of cells including fibroblasts and endothelial and epithelial cells [83]. This unique feature of gpRL13 selects for truncated and defect versions of the protein in HCMV strains propagated in cell culture [83–85]. The primary translation product of RL13 is predicted to result into a 35-kDa protein backbone which is subjected to extensive glycosylation [86]. The glycoproteins found in RL13-transfected cells exhibit a molecular weight of approximately 100 and 55 kDa [70]. The glycosylated protein was found in the envelope of HCMV at relatively high densities and on the surface of transfected cells [86]. In its C-terminal cytoplasmic tail, gpRL13 harbors a YxxL endocytic motif which was shown to mediate the internalization of the Fcγ ligand [70]. RL13 displays a selective preference for IgG binding to the human IgG1 and IgG2 subclasses while ignoring IgG3 and IgG4 (see Table 3) [70]. Rough mutational analysis of the RL13 extracellular region revealed that the putative Ig-like region including further membrane-proximal sequences was required for IgG binding [70].
HCMV RL12-encoded vFcγR
A fourth recently discovered HCMV vFcγR is encoded by RL12, a further RL11 gene family member [70] and thus displaying the same basic molecular architecture as the other type 1 transmembrane glycoproteins of HCMV which represent vFcγRs. According to its RL13 relatedness, it is predicted to contain one IgSF-like domain in its extracellular domain, followed by a transmembrane domain and a C-terminal cytoplasmic tail, altogether yielding a molecular backbone of approximately 45–48 kDa, depending on the HCMV strain. Predictions for N- and O-linked glycans confirm the presence of numerous putative sites throughout the sequence resulting in a high molecular weight of about 95 kDa prior to endoglycosidase H treatment. The RL12 sequence is, in contrast to gp34 and gp68, but similarly as noted for RL13, remarkably divergent among various HCMV strains. gp95 selectively binds only to monomeric human IgG1 and IgG2 but does not recognize IgG3 and IgG4 (see Table 3) [70].
MCMV vFcγR m138 (fcr-1)
So far, only one MCMV-encoded vFcγR could be identified which is transcribed from the early gene m-138/fcr-1 [76]. However, this vFcγR allowed a detailed analysis not only in vitro but also in infected mice and may thus exemplify the exciting and indispensable potential of vFcγRs for cytomegalovirus fitness in vivo. The MCMV-encoded vFcγR was identified as a 569 aa type 1 transmembrane glycoprotein of 65 kDa that is further processed into a highly glycosylated form of 105 kDa detected on the plasma membrane of MCMV-infected cells [74] (Fig. 2). Deletion of this gene from the MCMV genome resulted in a dramatic virus attenuation in vivo irrespective of the presence of B cells and antibodies, demonstrating that the observed alleviation of MCMV replication in mice was not only dependent on the m138/fcr-1 property to bind IgG [87], but rather suggested the existence of further dominant targets of m138/fcr-1 (see Table 3). fcr-1 is predicted to preserve three FcγR-related putative IgSF-like domains termed Ig1, Ig2, and Ig3, displaying a relatively low but still significant sequence homology with IgSF domains of murine cellular FcγRs CD16/FcγRIII and CD32/FcγRII, reaching 17 % identity and 24 % similarity, and are adjacent to a top N-glycan-rich stabilizing domain [74]. m138/fcr-1 not only binds to Fcγ but was also shown to impair NK cell functions by downmodulation of the NKG2D ligands MULT-1, H60, and RAE-ε [74, 88] as well as the B7-1 molecule CD80 [89], readily explaining the strong attenuation of replication in mice upon deletion of the m138 gene [87]. The same N-terminal part of the m138/fcr-1 ectodomain, Ig1, which is sufficient for MULT-1 function, is also needed to complex with the Fc part of IgG. In fact, soluble Fc fragments of IgG are able to inhibit fcr-1-mediated downmodulation of MULT-1, presumably by competing for the binding to the same fcr-1 domain [74]. In conclusion, m138/fcr-1 represents an excitingly versatile CMV inhibitor which simultaneously interacts with IgG as well as NKG2D and T cell ligands to counteract several concurrent immune responses.
HSV vFcγR complex gE/gI
The herpes simplex virus (HSV)-expressed vFcγR consisting of the virion structural proteins gE and gI represents the first described herpesviral vFcγR [90–92]. The glycoproteins gE and gI are transcribed from the HSV genes US7 and US8, respectively. They are both displayed on the surface of infected cells and are also incorporated into the virus envelope [73, 93, 94] (Fig. 2). gE shows slight sequence homologies to the second Ig domain of host FcγRs [93]. The glycoprotein gE but not gI shows Fc binding function when expressed alone [73]. Specifically, gE is able to bind weakly aggregated IgG with low affinity, while when acting in conjunction with gI, the gE/gI complex is able to recognize also monomeric IgG with high affinities. Biochemical and ultra-structural analyses of gE-gI binding to Fcγ revealed that gE-gI interacts with the Fcγ CH2-CH3 interdomain junction with a stoichiometry of two molecules of gE-gI per Fcγ [95]. In contrast to the HCMV vFcγRs, it binds human IgG1, IgG2, and IgG4, but it is surprisingly not able to recognize IgG3 (Table 3) [96]. Studies with HSV-1 have demonstrated that simultaneous binding of human anti-HSV IgG to a HSV antigen with its Fab arms and to gE-gI with its Fcγ region, a phenomenon referred to as “antibody bipolar bridging,” protects the virion and infected cells from IgG-mediated immune responses [73], i.e., gE and gI cooperate to protect infected cells from ADCC [112, 113].
Singular Fcγ binding modalities
Sequence alignments of cellular FcγRs revealed that they do not share obvious structural commonalities with HCMV or bacterial FcγRs. While all FcγR structures consistently bind Fcγ, a range of observations indicate that they indeed differ in their individual binding mode. These differences may be crucial for the immune effector functions executed by host FcγRs on the one hand and antagonistic consequences of microbial FcγRs on the other hand. While all canonical host FcγRs bind IgG molecules in the upper hinge region between the Fab arms and the CH2 domain of the Fc part [59, 97], some of the herpesviral FcγRs are known to approach other contact sites on the antibody, e.g., the CH2-CH3 interdomain interface (see Table 3). This binding site was mapped for the HCMV-encoded vFcγR gp68 as well as the HSV FcγR gE [75]. Surprisingly, experiments with mutated IgG variants indicated that the precise binding sites are nevertheless not congruent [75]. Moreover, there is an additional clear difference between both FcγRs: the heterodimeric receptor complex gE/gI binds IgG only at slightly basic pH values in the extracellular milieu but not at acidic pH (e.g., in the endosomes upon internalization). In contrast, gp68 IgG-Fc binding is stable at pH values from pH 5.6 to pH 8.1 [75]. Another significant hint is provided by the fact that HCMV FcγRs and HSV gE differ in their potential to bind to IgG subclasses (see Table 3). While gp34 and gp68 bind all human IgG subclasses, the HSV gE FcγR fails to recognize IgG3 [71]. Similar differences extend to the HCMV vFcγR encoded by the HCMV genes RL13 and RL12 which only bind to IgG1 and IgG2, but not to IgG3 and IgG4 [70]. Such a preference in IgG subclass binding contrasts also with all human activating FcγRs (with the exception that FcγRI does not bind IgG2) [35–37]. Last but not least, host and the HCMV FcγRs gp34 and gp68 seem to differ regarding the conformational requirements of the Fcγ ligand. This notion was deduced from the observation that N-linked glycosylation leading to an open conformation is mandatory for the recognition by human FcγRs, while the HCMV-encoded FcγR gp68 and HSV gE/gI readily recognize and bind deglycosylated IgG in a closed configuration [75].
HCMV vFcγRs gp34 and gp68 inhibit activation of FcγRI, FcγRIIA, and FcγRIIIA
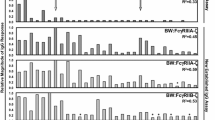
Cytomegaloviruses frequently reactivate or super-infect successfully CMV-infected hosts in the presence of high levels of CMV-specific IgG [98–100], suggesting that HCMV must have mechanisms to circumvent the antiviral effect of nevertheless protective antibodies. Despite the evidently different Fcγ binding modes between HCMV FcγRs and host FcγRs as outlined above, a systematic study was pursued to assess the potential interferences between both types of Fcγ binding devices [80]. The methodological basis of this comprehensive approach was provided by a newly developed BW5147 hybridoma-based FcγR activation assay which allows precise quantitative measurement of FcγR responses in vitro [101]. This procedure identified various members of the human FcγR family, i.e., FcγRI/CD64, FcγRIIA/CD32A, and FcγRIIIA/CD16A, to be targeted by the HCMV FcγRs gp34 and gp68 acting as effective antagonists of Fcγ ligand induced receptor responses [80]. The functional impact of both antagonists was first demonstrated by comparing gp34- and gp68-deficient HCMV mutants derived from various HCMV strains with wild-type viruses and RL11 and UL119–118 gene-revertant mutants, respectively, using polyclonal human HCMV-IgG preparations. Primary human FcγRIII/CD16+ NK cells precisely traced the results obtained before with FcγRIII expressing BW5147 reporter cells, confirming the predictive value of the innovative surrogate test.
Next, the findings were verified by gain-of-function experiments based on humanized monoclonal antibodies (e.g., trastuzumab and rituximab) and isotypes thereof allowing testing of different IgG subclasses in the presence of ectopically expressed gp34 and gp68. The humanized monoclonal antibodies also facilitated surface immune-precipitation studies to show that both HCMV-encoded Fcγ binding proteins possess the capacity to bind tightly to trastuzumab when fixing its antigen, HER2, on the plasma membrane. This result demonstrated simultaneous linkage of immune IgG with antigen and the HCMV inhibitors on the plasma membrane (see Fig. 3), compatible with the model of “antibody bipolar bridging” which was suggested for HSV gE/gI [73, 95].

Scenarios of interference of HCMV encoded FcγRs with antiviral IgG and host FcγR activation. State 1: Upon opsonization of viral antigen on the cell surface, IgG is competent to activate host FcγRs, e.g., FcγRIIIA (CD16) on NK cells, leading to immune effector cell responses (e.g., antibody-dependent cellular cytotoxicity (ADCC)). State 2: HCMV expresses four viral FcγRs with cell surface disposition, i.e., gp34, gp68, gp95, and gpRL13. Immune IgG bound to viral antigen on the cell surface (state 3) is recognized by a HCMV FcγR (state 4) forming ternary heterocomplexes, preventing the activation of host FcγRs (state 5). HCMV vFcγRs may internalize bound IgG from the cell surface and transport IgG to endolysosomal compartments for subsequent degradation of IgG (state 6) or incorporation of IgG into viral progeny at sites of virion maturation and secondary envelopment (state 7a) before vFcγR containing HCMV particles are released from infected cells (state 7b)
Favorably, the BW5147 test system allowed a direct functional comparison of the established HSV gE/gI inhibitor with the HCMV candidates, gp34 and gp68. In relation to HSV gE, both gp34 and gp68 demonstrated at least equivalent if not superior forces to impair FcγRIIIA and FcγRIIA-mediated responses. Unexpectedly and contrasting with both HCMV vFcγRs, HSV gE enhanced rather than weakened FcγRI triggering. This finding warrants further experiments exploring to which end IgG-coated HSV-infected cells can stimulate FcγRI bearing immune cells. Another difference between gE/gI and HCMV vFcγRs is the preference for varying IgG subclasses. HSV gE/gI does not bind to IgG3 [90] which differs from the other subclasses by its unique extended hinge region [97]. Unlike gE, both of the HCMV vFcγRs were able to efficiently block IgG3 immune complexes [80], corresponding to the fact that HCMV-specific IgG responses are constituted primarily by IgG1 and IgG3 antibodies [102, 103].
Which HCMV antigens are exposed on the plasma membrane of infected target cells and recognized by cytotoxic IgG triggering ADCC and further FcγR-mediated responses? It was demonstrated that HCMV antigens eliciting efficient ADCC responses become exposed only in the late phase of the replication cycle on the cell surface, while IgG-opsonized cells arrested in the early phase constituted only very poor target cells [80]. This observation suggests (i) that structural HCMV glycoproteins known to become exposed on the cell surface as the HCMV replication cycle progresses, e.g., gB [67], gH [68], and UL128 [69], are prime candidate targets of relevant IgG and (ii) that the expression kinetics of the HCMV FcγRs has closely adapted to the abundance of surface resident HCMV antigens yielding maximal levels in the late phase of infection [71].
Do HCMV FcγRs affect IgG and complement mediated virion neutralization?
gE/gI complexes are constitutively incorporated into HSV virions. There is evidence that gE/gI protect viral particles from virolysis, i.e., neutralization by immune IgG and complement [73, 94, 104]. Likewise, the FcγRs gp68 and gp34 were found in HCMV virions ([82] and unpublished data). These features of the prototypic vFcγR prompted us to test equivalent functions of the HCMV vFcγRs. Interestingly, gp34 as well as gp68 did not affect the potency of polyclonal or monoclonal neutralizing IgG, independent on the presence of complement (Henrike Reinhard, Hartmut Hengel, unpublished data). The fact that HCMV virion entry was insensitive to the vFcγRs in the presence of HCMV immune IgG in fibroblast as well as endothelial cell cultures indicates that the two known routes of HCMV entry, i.e., via gH-gL-gO and via the pentameric gH-gL-UL128-131 complex [105], cannot be affected by Fcγ domains bound to the vFcγRs but only by the Fab part of IgG bound to the relevant virion glycoproteins. Furthermore, gp68 and gp34 did not influence complement-mediated cell lysis when tested in the context of HCMV-infected cells (Eugenia Corrales-Aguilar, Hartmut Hengel, unpublished data). The lack of lysis in HCMV-infected fibroblasts can be explained by findings of Spiller et al. [106], demonstrating that transcription of CD46 and CD55, two complement control proteins (CCPs), is drastically upregulated during HCMV infection. These two CCPs prevent the activation of the C3 convertase; thus, complement-mediated lysis of cells is abolished.
Implications for immunotherapy
Virus-specific hyperimmune globulins from human donors are often used to treat or prevent threatening virus infections. Traditionally, neutralizing IgG is considered to be the primary correlate of seroprotection. In many afflicted tissues, however, HCMV is known to spread intracellularly (e.g., via infected endothelial cells and leukocytes) and via cell to cell, i.e., without diffusing to the extracellular space as a cell-free virion [70], thus avoiding virion neutralization by antibodies. Given such a scenario, non-neutralizing but FcγR-activating and particularly ADCC-inducing IgG is plausible to represent an effective component of humoral immunity against HCMV. Several findings are compatible with this notion. Only recently, a novel phenotype of human NK cells transcriptionally deficient for the FcR signaling transmembrane adaptor γ chain (Fig. 1) has been identified which instead expresses CD3ζ [107], another signaling adaptor associated with FcγRIII/CD16 containing even three ITAM motifs at fairly high levels. This NK cell population was characterized to exhibit a reduced natural cytotoxicity via the NK cell receptors NKp46 and NKp30 but a remarkably vigorous responsiveness via CD16/FcγRIII [108]. Even more excitingly, this NK cell phenotype (g− NK cells) was associated with prior HCMV infection and confirmed to have clearly enhanced ADCC capabilities compared to conventional NK cells [108]. Independently, NKG2Chigh CD57high NK cells which are expanded by HCMV in vivo and in vitro [109, 110] were demonstrated to act as prominent effectors of ADCC against HCMV [111], which is tempting to speculate that the HCMV-imprinted NK cell populations g− NK cells and NKG2Chigh CD57high may overlap. Combining these findings, ADCC-inducing IgG is plausible to represent a primary effective component of humoral immunity, which becomes only secondary attenuated by gp34 and gp68. Both immunoevasins could thus contribute to the disappointingly poor therapeutic performance of HCMV-specific hyperimmune globulin observed in a variety of clinical settings [112–115] since they enable HCMV to evade from IgG effector responses and take direct proviral effects in scenarios of post-acute and recurrent infection when HCMV-IgG antibodies are present at higher concentrations [116]. In this context, it is worth mentioning that the optimal range of gp34- and gp68-mediated IgG inhibition is below the HCMV-IgG serum concentration of humans. Replicating mainly at organ sites with IgG concentrations that are lower than those in the serum, it is thus conceivable that the inhibitory effects detected in vitro are indeed physiologically relevant. These insights offer new perspectives to improve the efficacy of HCMV-specific IgGs. One conceivable approach could aim at an enrichment of gp34- and gp68-specific IgG within HCMV hyperimmune globulin preparations since those antibodies should override the inhibitory effect of the HCMV vFcγRs and strongly enhance immune cell signaling via host FcγRs.
Further perspectives and open questions
A newly established murine BW 5147 reporter cell based methodology has provided evidence that the vFcγRs gp34 and gp68 possess a broad immune evasion potential by impairing the activation of all canonical activating human FcγRs, i.e., FcγRI, FcγRIIA, and FcγRIIIA [80]. Since the predictions of the reporter cell assays were accurately verified by the subsequently demonstrated attenuation of ADCC responses of analyzed FcγRIII+ human NK cells [80], further ramifications of gp34 and gp68 on HCMV immune responses mediated by FcγRI+ and FcγRIIA+ cells are plausible to assume. The mechanistic insight into the inhibition process of gp34 and gp68 is still at the very beginning. We showed that HCMV gp34 and gp68 are able to form ternary heterocomplexes on the surface of infected cells containing IgG, the IgG-bound antigen, and the vFcγR, suggesting a molecular configuration compatible with the requirements of the concept of “bipolar bridging” [73, 95]. While the Bjorkman laboratory has demonstrated on the ultra-structural level that the architecture of the prototypical HSV FcγR gE/gI-IgG complex allows indeed bipolar bridging of IgG [95], additional interaction models may be required for the HCMV FcγRs—merely due to the fact that at least four different HCMV binding proteins are competitively expressed on the surface of HCMV-infected cells. The immunological selection pressure may support their functional diversification rather than molecular redundancy in their protein-protein interactions. The presence of independent but functionally related immunoevasins jointly targeting immune control mechanism is a characteristic feature of cytomegalovirus evasion from CD8+ T cell responses by manipulating the MHC I pathway of antigen presentation or from NK cells by preventing activating NK receptor interactions [14, 117–121]. Nevertheless, combined removal of gp34 as well as gp68 inhibitors from the surface of HCMV infected cells reveals hardly additive or even synergistic effects [80], suggesting that these two factors do not act in an obvious cooperative manner and leaves an open question if removal of additional HCMV vFcγRs will have additive or synergistic effects. Furthermore, it will be interesting to test if HCMV vFcγRs are also able to act in a cis-inhibiting manner when HCMV is infecting myelomonocytic target cells bearing host FcγRs.
Recently, Manley et al. [122] showed that MSL-109, a neutralizing human monoclonal IgG isolated from a CMV seropositive individual that recognizes the viral glycoprotein H (gH), is selectively taken up by infected cells and incorporated into assembling virions. However, HCMV progeny generated in the presence of MSL-109 acquires rapid resistance. Their data showed facilitation of a MSL-109-resistant virus infection of naive cells by a model in which the Fcγ domain of virus-attached MSL-109 aids to the entry of the virus. The authors suggested that the role of the Fcγ domain may be twofold—first, in the uptake of the antibody into infected cells to generate resistance and, second, the subsequent infection of naive cells by resistant virus. Yet, the precise mechanism of how this resistant virus enters cells and the role the Fc domain plays in mediating this process remain still unclear. The antibody uptake into HCMV-infected cells and transportation to sites of virion membrane assembly and maturation may be explained by a possible Fcγ-mediated interaction through the HCMV vFcγRs (see Fig. 3). For example, gp34 may mediate this IgG uptake since its cytoplasmic tail contains a dileucine consensus motif (DXXXLL, where X is unknown) indicating a potential function in intracellular targeting of the protein to the endocytic route [71, 123]. Taken together, HCMV FcγRs could thus be implicated in further host FcγR-independent mechanism of evasion from antiviral IgG and support Fcγ exploitation to promote HCMV entry and dissemination.
Lastly, all of the HCMV FcγRs may fulfill further Fcγ independently executed proviral functions. This is exemplified by the impressive multitasking abilities of the MCMV m138/fcr-1 molecule which downregulates the NKG2D ligands MULT-1, H60, and RAE-ε [74, 88] as well as the B7-1 molecule CD80 [89] beyond its Fcγ binding activity.
References
Chee MS, Bankier AT, Beck S et al (1990) Analysis of the protein-coding content of the sequence of human cytomegalovirus strain AD169. Curr Top Microbiol Immunol 154:125–69
Davison AJ, Dolan A, Akter P et al (2003) The human cytomegalovirus genome revisited: comparison with the chimpanzee cytomegalovirus genome. J Gen Virol 84:17–28
Murphy E, Yu D, Grimwood J et al (2003) Coding potential of laboratory and clinical strains of human cytomegalovirus. Proc Natl Acad Sci U S A 100:14976–81. doi:10.1073/pnas.2136652100
Stern-Ginossar N, Gur C, Biton M et al (2008) Human microRNAs regulate stress-induced immune responses mediated by the receptor NKG2D. Nat Immunol 9:1065–73. doi:10.1038/ni.1642
Goodrum F, Reeves M, Sinclair J et al (2007) Human cytomegalovirus sequences expressed in latently infected individuals promote a latent infection in vitro. Blood 110:937–45. doi:10.1182/blood-2007–01–070078
Gandhi MK, Khanna R (2004) Human cytomegalovirus: clinical aspects, immune regulation, and emerging treatments. Lancet Infect Dis 4:725–38. doi:10.1016/S1473–3099(04)01202–2
Ji Y-N, An L, Zhan P, Chen X-H (2012) Cytomegalovirus infection and coronary heart disease risk: a meta-analysis. Mol Biol Rep 39:6537–46. doi:10.1007/s11033–012–1482–6
Pawelec G, McElhaney JE, Aiello AE, Derhovanessian E (2012) The impact of CMV infection on survival in older humans. Curr Opin Immunol 24:507–11. doi:10.1016/j.coi.2012.04.002
Cobbs CS (2013) Cytomegalovirus and brain tumor: epidemiology, biology and therapeutic aspects. Curr Opin Oncol 25:682–8. doi:10.1097/CCO.0000000000000005
Reddehase MJ, Mutter W, Münch K et al (1987) CD8-positive T lymphocytes specific for murine cytomegalovirus immediate-early antigens mediate protective immunity. J Virol 61:3102–8
Polić B, Hengel H, Krmpotić A et al (1998) Hierarchical and redundant lymphocyte subset control precludes cytomegalovirus replication during latent infection. J Exp Med 188:1047–54
Jonjić S, Pavić I, Polić B et al (1994) Antibodies are not essential for the resolution of primary cytomegalovirus infection but limit dissemination of recurrent virus. J Exp Med 179:1713–7
Klenovsek K, Weisel F, Schneider A et al (2007) Protection from CMV infection in immunodeficient hosts by adoptive transfer of memory B cells. Blood 110:3472–9. doi:10.1182/blood-2007–06–095414
Hengel H, Brune W, Koszinowski UH (1998) Immune evasion by cytomegalovirus-survival strategies of a highly adapted opportunist. Trends Microbiol 6:190–7
Mocarski ES (2004) Immune escape and exploitation strategies of cytomegaloviruses: impact on and imitation of the major histocompatibility system. Cell Microbiol 6:707–717. doi:10.1111/j.1462–5822.2004.00425.x
Scalzo A, Corbett A (2007) The interplay between host and viral factors in shaping the outcome of cytomegalovirus infection. Immunol cell … 46–54. doi: 10.1038/sj.icb.7100013
Burton DR (2002) Antibodies, viruses and vaccines. Nat Rev Immunol 2:706–13. doi:10.1038/nri891
Corti D, Lanzavecchia A (2013) Broadly neutralizing antiviral antibodies. Annu Rev Immunol 31:705–42. doi:10.1146/annurev-immunol-032712–095916
Parren PW, Burton DR (2001) The antiviral activity of antibodies in vitro and in vivo. Adv Immunol 77:195–262
Nimmerjahn F, Ravetch JV (2007) Fc-receptors as regulators of immunity. Adv Immunol 96:179–204. doi:10.1016/S0065–2776(07)96005–8
Pincetic A, Bournazos S, DiLillo DJ et al (2014) Type I and type II Fc receptors regulate innate and adaptive immunity. Nat Immunol 15:707–716. doi:10.1038/ni.2939
Nimmerjahn F, Ravetch JV (2010) Antibody-mediated modulation of immune responses. Immunol Rev 236:265–75. doi:10.1111/j.1600–065×.2010.00910.x
Nimmerjahn F, Ravetch JV (2008) Fcgamma receptors as regulators of immune responses. Nat Rev Immunol 8:34–47. doi:10.1038/nri2206
Takai T, Ono M, Hikida M et al (1996) Augmented humoral and anaphylactic responses in Fc gamma RII-deficient mice. Nature 379:346–9. doi:10.1038/379346a0
Bolland S, Ravetch JV (1999) Inhibitory pathways triggered by ITIM-containing receptors. Adv Immunol 72:149–77
Karsten CM, Pandey MK, Figge J et al (2012) Anti-inflammatory activity of IgG1 mediated by Fc galactosylation and association of FcγRIIB and dectin-1. Nat Med 18:1401–6. doi:10.1038/nm.2862
Nimmerjahn F, Ravetch JV (2011) FcγRs in health and disease. Curr Top Microbiol Immunol 350:105–25. doi:10.1007/82_2010_86
Ravetch JV, Bolland S (2001) IgG Fc receptors. Annu Rev Immunol 19:275–90. doi:10.1146/annurev.immunol.19.1.275
Lu J, Marjon KD, Mold C et al (2012) Pentraxins and Fc receptors. Immunol Rev 250:230–8. doi:10.1111/j.1600–065×.2012.01162.x
Du Clos TW (2013) Pentraxins: structure, function, and role in inflammation. ISRN Inflamm 2013:379040. doi:10.1155/2013/379040
Lu J, Marnell LL, Marjon KD et al (2008) Structural recognition and functional activation of FcgammaR by innate pentraxins. Nature 456:989–92. doi:10.1038/nature07468
Ernst LK, van de Winkel JG, Chiu IM, Anderson CL (1992) Three genes for the human high affinity Fc receptor for IgG (Fc gamma RI) encode four distinct transcription products. J Biol Chem 267:15692–700
Warmerdam PA, Nabben NM, van de Graaf SA et al (1993) The human low affinity immunoglobulin G Fc receptor IIC gene is a result of an unequal crossover event. J Biol Chem 268:7346–9
Ory PA, Clark MR, Kwoh EE et al (1989) Sequences of complementary DNAs that encode the NA1 and NA2 forms of Fc receptor III on human neutrophils. J Clin Invest 84:1688–91. doi:10.1172/JCI114350
Guilliams M, Bruhns P, Saeys Y et al (2014) The function of Fcγ receptors in dendritic cells and macrophages. Nat Rev Immunol 14:94–108. doi:10.1038/nri3582
Hogarth PM, Pietersz GA (2012) Fc receptor-targeted therapies for the treatment of inflammation, cancer and beyond. Nat Rev Drug Discov 11:311–31. doi:10.1038/nrd2909
Bruhns P, Iannascoli B, England P et al (2009) Specificity and affinity of human Fcgamma receptors and their polymorphic variants for human IgG subclasses. Blood 113:3716–25. doi:10.1182/blood-2008–09–179754
Bruhns P (2012) Properties of mouse and human IgG receptors and their contribution to disease models. Blood 119:5640–9. doi:10.1182/blood-2012–01–380121
Ravetch JV, Kinet JP (1991) Fc receptors. Annu Rev Immunol 9:457–92. doi:10.1146/annurev.iy.09.040191.002325
Pan LY, Mendel DB, Zurlo J, Guyre PM (1990) Regulation of the steady state level of Fc gamma RI mRNA by IFN-gamma and dexamethasone in human monocytes, neutrophils, and U-937 cells. J Immunol 145:267–75
Veri M-C, Gorlatov S, Li H et al (2007) Monoclonal antibodies capable of discriminating the human inhibitory Fcgamma-receptor IIB (CD32B) from the activating Fcgamma-receptor IIA (CD32A): biochemical, biological and functional characterization. Immunology 121:392–404. doi:10.1111/j.1365–2567.2007.02588.x
Ehrhardt GRA, Cooper MD (2011) Immunoregulatory roles for fc receptor-like molecules. Curr Top Microbiol Immunol 350:89–104. doi:10.1007/82_2010_88
Cassard L, Jönsson F, Arnaud S, Daëron M (2012) Fcγ receptors inhibit mouse and human basophil activation. J Immunol 189:2995–3006. doi:10.4049/jimmunol.1200968
Van der Heijden J, Breunis WB, Geissler J et al (2012) Phenotypic variation in IgG receptors by nonclassical FCGR2C alleles. J Immunol 188:1318–24. doi:10.4049/jimmunol.1003945
Zhao W, Kepley CL, Morel PA et al (2006) Fc gamma RIIa, not Fc gamma RIIb, is constitutively and functionally expressed on skin-derived human mast cells. J Immunol 177:694–701
Magnusson SE, Engström M, Jacob U et al (2007) High synovial expression of the inhibitory FcgammaRIIb in rheumatoid arthritis. Arthritis Res Ther 9:R51. doi:10.1186/ar2206
Metes D, Manciulea M, Pretrusca D et al (1999) Ligand binding specificities and signal transduction pathways of Fc gamma receptor IIc isoforms: the CD32 isoforms expressed by human NK cells. Eur J Immunol 29:2842–52. doi:10.1002/(SICI)1521–4141(199909)29:09<2842::AID-IMMU2842>3.0.CO;2–5
Meknache N, Jönsson F, Laurent J et al (2009) Human basophils express the glycosylphosphatidylinositol-anchored low-affinity IgG receptor FcgammaRIIIB (CD16B). J Immunol 182:2542–50. doi:10.4049/jimmunol.0801665
Flinsenberg TWH, Compeer EB, Koning D et al (2012) Fcγ receptor antigen targeting potentiates cross-presentation by human blood and lymphoid tissue BDCA-3+ dendritic cells. Blood 120:5163–72. doi:10.1182/blood-2012–06–434498
Tridandapani S, Wardrop R, Baran CP et al (2003) TGF-beta 1 suppresses [correction of supresses] myeloid Fc gamma receptor function by regulating the expression and function of the common gamma-subunit. J Immunol 170:4572–7
Roopenian DC, Akilesh S (2007) FcRn: the neonatal Fc receptor comes of age. Nat Rev Immunol 7:715–25. doi:10.1038/nri2155
Ghetie V, Ward ES (1997) FcRn: the MHC class I-related receptor that is more than an IgG transporter. Immunol Today 18:592–8
Rath T, Kuo TT, Baker K et al (2013) The immunologic functions of the neonatal Fc receptor for IgG. J Clin Immunol 33(Suppl 1):S9–17. doi:10.1007/s10875–012–9768-y
Ko S-Y, Pegu A, Rudicell RS et al (2014) Enhanced neonatal Fc receptor function improves protection against primate SHIV infection. Nature. doi:10.1038/nature13612
Woof JM, Burton DR (2004) Human antibody-Fc receptor interactions illuminated by crystal structures. Nat Rev Immunol 4:89–99. doi:10.1038/nri1266
Hulett MD, Osman N, McKenzie IF, Hogarth PM (1991) Chimeric Fc receptors identify functional domains of the murine high affinity receptor for IgG. J Immunol 147:1863–8
Zhang Y, Boesen CC, Radaev S et al (2000) Crystal structure of the extracellular domain of a human Fc gamma RIII. Immunity 13:387–95
Sondermann P, Huber R, Jacob U (1999) Crystal structure of the soluble form of the human fcgamma-receptor IIb: a new member of the immunoglobulin superfamily at 1.7 A resolution. EMBO J 18:1095–103. doi:10.1093/emboj/18.5.1095
Sondermann P, Huber R, Oosthuizen V, Jacob U (2000) The 3.2-A crystal structure of the human IgG1 Fc fragment-Fc gammaRIII complex. Nature 406:267–73. doi:10.1038/35018508
Hamerman JA, Lanier LL (2006) Inhibition of immune responses by ITAM-bearing receptors. Sci STKE 2006:re1. doi: 10.1126/stke.3202006re1
Sulica A, Herberman RB (1996) Cytophilic immunoglobulins revisited via natural killer cells. FASEB J 10:1495–504
Lux A, Yu X, Scanlan CN, Nimmerjahn F (2013) Impact of immune complex size and glycosylation on IgG binding to human FcγRs. J Immunol 190:4315–23. doi:10.4049/jimmunol.1200501
Shields RL, Lai J, Keck R et al (2002) Lack of fucose on human IgG1 N-linked oligosaccharide improves binding to human Fcgamma RIII and antibody-dependent cellular toxicity. J Biol Chem 277:26733–40. doi:10.1074/jbc.M202069200
Niwa R, Natsume A, Uehara A et al (2005) IgG subclass-independent improvement of antibody-dependent cellular cytotoxicity by fucose removal from Asn297-linked oligosaccharides. J Immunol Methods 306:151–60. doi:10.1016/j.jim.2005.08.009
Niwa R, Sakurada M, Kobayashi Y et al (2005) Enhanced natural killer cell binding and activation by low-fucose IgG1 antibody results in potent antibody-dependent cellular cytotoxicity induction at lower antigen density. Clin Cancer Res 11:2327–36. doi:10.1158/1078–0432.CCR-04–2263
Okazaki A, Shoji-Hosaka E, Nakamura K et al (2004) Fucose depletion from human IgG1 oligosaccharide enhances binding enthalpy and association rate between IgG1 and FcgammaRIIIa. J Mol Biol 336:1239–49. doi:10.1016/j.jmb.2004.01.007
Radaev S, Motyka S, Fridman WH et al (2001) The structure of a human type III Fcgamma receptor in complex with Fc. J Biol Chem 276:16469–77. doi:10.1074/jbc.M100350200
Langone JJ (1982) Protein A of Staphylococcus aureus and related immunoglobulin receptors produced by streptococci and pneumonococci. Adv Immunol 32:157–252
Müller MG, Ing JY, Cheng MK-W et al (2013) Identification of a phage-encoded Ig-binding protein from invasive Neisseria meningitidis. J Immunol 191:3287–96. doi:10.4049/jimmunol.1301153
Cortese M, Calò S, D’Aurizio R et al (2012) Recombinant human cytomegalovirus (HCMV) RL13 binds human immunoglobulin G Fc. PLoS One 7:e50166. doi:10.1371/journal.pone.0050166
Atalay R, Zimmermann A, Wagner M et al (2002) Identification and expression of human cytomegalovirus transcription units coding for two distinct Fcgamma receptor homologs. J Virol 76:8596–608
Lilley BN, Ploegh HL, Tirabassi RS (2001) Human cytomegalovirus open reading frame TRL11/IRL11 encodes an immunoglobulin G Fc-binding protein. J Virol 75:11218–21. doi:10.1128/JVI.75.22.11218–11221.2001
Frank I, Friedman HM (1989) A novel function of the herpes simplex virus type 1 Fc receptor: participation in bipolar bridging of antiviral immunoglobulin G. J Virol 63:4479–88
Lenac T, Budt M, Arapovic J et al (2006) The herpesviral Fc receptor fcr-1 down-regulates the NKG2D ligands MULT-1 and H60. J Exp Med 203:1843–50. doi:10.1084/jem.20060514
Sprague ER, Reinhard H, Cheung EJ et al (2008) The human cytomegalovirus Fc receptor gp68 binds the Fc CH2-CH3 interface of immunoglobulin G. J Virol 82:3490–9. doi:10.1128/JVI.01476–07
Thäle R, Lucin P, Schneider K et al (1994) Identification and expression of a murine cytomegalovirus early gene coding for an Fc receptor. J Virol 68:7757–65
Budt M, Reinhard H, Bigl A, Hengel H (2004) Herpesviral Fcgamma receptors: culprits attenuating antiviral IgG? Int Immunopharmacol 4:1135–48. doi:10.1016/j.intimp.2004.05.020
Namboodiri AM, Budkowska A, Nietert PJ, Pandey JP (2007) Fc gamma receptor-like hepatitis C virus core protein binds differentially to IgG of discordant Fc (GM) genotypes. Mol Immunol 44:3805–8. doi:10.1016/j.molimm.2007.03.022
Mercé-Maldonado E (2011) Identification and functional characterization of a novel HCMV-encoded Fcγ receptor blocking CD16-mediated activation of human natural killer cells. Heinrich-Heine-Universität, Düsseldorf
Corrales-Aguilar E, Trilling M, Hunold K et al (2014) Human cytomegalovirus Fcγ binding proteins gp34 and gp68 antagonize Fcγ receptors I, II and III. PLoS Pathog 10:e1004131. doi:10.1371/journal.ppat.1004131
Cha TA, Tom E, Kemble GW et al (1996) Human cytomegalovirus clinical isolates carry at least 19 genes not found in laboratory strains. J Virol 70:78–83
Varnum SM, Streblow DN, Monroe ME et al (2004) Identification of proteins in human cytomegalovirus (HCMV) particles: the HCMV proteome. J Virol 78:10960–6. doi:10.1128/JVI.78.20.10960–10966.2004
Dargan DJ, Douglas E, Cunningham C et al (2010) Sequential mutations associated with adaptation of human cytomegalovirus to growth in cell culture. J Gen Virol 91:1535–46. doi:10.1099/vir.0.018994–0
Dolan A, Cunningham C, Hector RD et al (2004) Genetic content of wild-type human cytomegalovirus. J Gen Virol 85:1301–12
Akter P, Cunningham C, McSharry BP et al (2003) Two novel spliced genes in human cytomegalovirus. J Gen Virol 84:1117–22
Stanton RJ, Baluchova K, Dargan DJ et al (2010) Reconstruction of the complete human cytomegalovirus genome in a BAC reveals RL13 to be a potent inhibitor of replication. J Clin Invest 120:3191–208. doi:10.1172/JCI42955
Crnković-Mertens I, Messerle M, Milotić I et al (1998) Virus attenuation after deletion of the cytomegalovirus Fc receptor gene is not due to antibody control. J Virol 72:1377–82
Arapovic J, Lenac T, Antulov R et al (2009) Differential susceptibility of RAE-1 isoforms to mouse cytomegalovirus. J Virol 83:8198–207. doi:10.1128/JVI.02549–08
Mintern JD, Klemm EJ, Wagner M et al (2006) Viral interference with B7–1 costimulation: a new role for murine cytomegalovirus fc receptor-1. J Immunol 177:8422–31
Westmoreland D, Watkins JF (1974) The IgG receptor induced by herpes simplex virus: studies using radioiodinated IgG. J Gen Virol 24:167–78
Yasuda J, Milgrom F (1968) Hemadsorption by herpes simplex-infected cell cultures. Int Arch Allergy Appl Immunol 33:151–70
Watkins JF (1964) Asorption of sensitized sheep erytrocytes to Hela cells infected with herpes simplex virus. Nature 202:1364–5
Dubin G, Socolof E, Frank I, Friedman HM (1991) Herpes simplex virus type 1 Fc receptor protects infected cells from antibody-dependent cellular cytotoxicity. J Virol 65:7046–50
Nagashunmugam T, Lubinski J, Wang L et al (1998) In vivo immune evasion mediated by the herpes simplex virus type 1 immunoglobulin G Fc receptor. J Virol 72:5351–9
Sprague ERE, Wang C, Baker D, Bjorkman PJP (2006) Crystal structure of the HSV-1 Fc receptor bound to Fc reveals a mechanism for antibody bipolar bridging. PLoS Biol 4:e148. doi:10.1371/journal.pbio.0040148
Wiger D, Michaelsen TE (1985) Binding site and subclass specificity of the herpes simplex virus type 1-induced Fc receptor. Immunology 54:565–72
Sondermann P, Oosthuizen V (2002) X-ray crystallographic studies of IgG-Fc gamma receptor interactions. Biochem Soc Trans 30:481–486
Kutza AS, Muhl E, Hackstein H et al (1998) High incidence of active cytomegalovirus infection among septic patients. Clin Infect Dis 26:1076–82
Ross SA, Arora N, Novak Z et al (2010) Cytomegalovirus reinfections in healthy seroimmune women. J Infect Dis 201:386–9. doi:10.1086/649903
Hansen SG, Powers CJ, Richards R et al (2010) Evasion of CD8+ T cells is critical for superinfection by cytomegalovirus. Science 328:102–6. doi:10.1126/science.1185350
Corrales-Aguilar E, Trilling M, Reinhard H et al (2013) A novel assay for detecting virus-specific antibodies triggering activation of Fcγ receptors. J Immunol Methods 387:21–35. doi:10.1016/j.jim.2012.09.006
Linde GA, Hammarström L, Persson MA et al (1983) Virus-specific antibody activity of different subclasses of immunoglobulins G and A in cytomegalovirus infections. Infect Immun 42:237–44
Gupta CK, Leszczynski J, Gupta RK, Siber GR (1996) IgG subclass antibodies to human cytomegalovirus (CMV) in normal human plasma samples and immune globulins and their neutralizing activities. Biologicals 24:117–24. doi:10.1006/biol.1996.0015
Lubinski JM, Lazear HM, Awasthi S et al (2011) The herpes simplex virus 1 IgG fc receptor blocks antibody-mediated complement activation and antibody-dependent cellular cytotoxicity in vivo. J Virol 85:3239–49
Ryckman BJ, Chase MC, Johnson DC (2008) HCMV gH/gL/UL128–131 interferes with virus entry into epithelial cells: evidence for cell type-specific receptors. Proc Natl Acad Sci U S A 105:14118–23. doi:10.1073/pnas.0804365105
Spiller OB, Morgan BP, Tufaro F, Devine DV (1996) Altered expression of host-encoded complement regulators on human cytomegalovirus-infected cells. Eur J Immunol 26:1532–8. doi:10.1002/eji.1830260719
Hwang I, Zhang T, Scott JM et al (2012) Identification of human NK cells that are deficient for signaling adaptor FcRγ and specialized for antibody-dependent immune functions. Int Immunol 24:793–802. doi:10.1093/intimm/dxs080
Zhang T, Scott JM, Hwang I, Kim S (2013) Cutting edge: antibody-dependent memory-like NK cells distinguished by FcRγ deficiency. J Immunol 190:1402–6. doi:10.4049/jimmunol.1203034
Gumá M, Angulo A, Vilches C et al (2004) Imprint of human cytomegalovirus infection on the NK cell receptor repertoire. Blood 104:3664–71. doi:10.1182/blood-2004–05–2058
Gumá M, Budt M, Sáez A et al (2006) Expansion of CD94/NKG2C + NK cells in response to human cytomegalovirus-infected fibroblasts. Blood 107:3624–31. doi:10.1182/blood-2005–09–3682
Wu Z, Sinzger C, Frascaroli G et al (2013) Human cytomegalovirus-induced NKG2C (hi) CD57 (hi) natural killer cells are effectors dependent on humoral antiviral immunity. J Virol 87:7717–25. doi:10.1128/JVI.01096–13
Boeckh M, Bowden RA, Storer B et al (2001) Randomized, placebo-controlled, double-blind study of a cytomegalovirus-specific monoclonal antibody (MSL-109) for prevention of cytomegalovirus infection after allogeneic hematopoietic stem cell transplantation. Biol Blood Marrow Transplant 7:343–51
Hodson EM, Jones CA, Strippoli GFM, et al. (2007) Immunoglobulins, vaccines or interferon for preventing cytomegalovirus disease in solid organ transplant recipients. Cochrane database Syst Rev CD005129. doi: 10.1002/14651858.CD005129.pub2
Raanani P, Gafter-Gvili A, Paul M et al (2009) Immunoglobulin prophylaxis in hematopoietic stem cell transplantation: systematic review and meta-analysis. J Clin Oncol 27:770–81. doi:10.1200/JCO.2008.16.8450
Revello MG, Lazzarotto T, Guerra B et al (2014) A randomized trial of hyperimmune globulin to prevent congenital cytomegalovirus. N Engl J Med 370:1316–26. doi:10.1056/NEJMoa1310214
Schoppel K, Kropff B, Schmidt C et al (1997) The humoral immune response against human cytomegalovirus is characterized by a delayed synthesis of glycoprotein-specific antibodies. J Infect Dis 175:533–44
Powers C, Früh K (2008) Rhesus CMV: an emerging animal model for human CMV. Med Microbiol Immunol 197:109–15. doi:10.1007/s00430–007–0073-y
Mocarski ES (2002) Immunomodulation by cytomegaloviruses: manipulative strategies beyond evasion. Trends Microbiol 10:332–9
Babic M, Polic B, Krmpotic A et al (2008) Immune evasion of natural killer cells by viruses. Curr Opin Immunol 20:30–38. doi:10.1016/j.coi.2007.11.002
Hengel H, Koszinowski UH (1997) Interference with antigen processing by viruses. Curr Opin Immunol 9:470–6
Halenius A, Gerke C, Hengel H (2014) Classical and non-classical MHC I molecule manipulation by human cytomegalovirus: so many targets—but how many arrows in the quiver? Cell. Mol, Immunol
Manley K, Anderson J, Yang F et al (2011) Human cytomegalovirus escapes a naturally occurring neutralizing antibody by incorporating it into assembling virions. Cell Host Microbe 10:197–209
Letourneur F, Klausner RD (1992) A novel di-leucine motif and a tyrosine-based motif independently mediate lysosomal targeting and endocytosis of CD3 chains. Cell 69:1143–57
Acknowledgments
We would like to thank Mirko Trilling for continuous inspiring intellectual exchange. Our work is supported by funds of the DFG through He 2,526/6–2, the European Commission through QLRT-2001–01112 and MRTN-CT-2005–019248, and the Helmholtz Association through VISTRIE VH-VI-242. ECA was supported by the German Academic Exchange Service (DAAD); the authors do not have any conflict of interests.
Author information
Authors and Affiliations
Corresponding author
Additional information
Eugenia Corrales-Aguilar and Katja Hoffmann contributed equally.
This article is a contribution to the special issue on Immune Modulation, properties and models of CMV - Guest Editor: Ofer Mandelboim
Rights and permissions
About this article

Cite this article
Corrales-Aguilar, E., Hoffmann, K. & Hengel, H. CMV-encoded Fcγ receptors: modulators at the interface of innate and adaptive immunity. Semin Immunopathol 36, 627–640 (2014). https://doi.org/10.1007/s00281-014-0448-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00281-014-0448-2